

Abstract
Prion diseases are fatal, neurodegenerative illnesses caused by the accumulation of PrPSc, an aberrantly folded isoform of the normal, cellular prion protein (PrPC). Detection of PrPSc commonly relies on immunochemical methods, a strategy hampered by the lack of antibodies specific for this disease-causing isoform. Here, we report the generation of 8 mAbs against PrP following immunization of Prnp-null mice with recombinant (rec) PrP. The 8 mAbs exhibited distinct differential binding to PrPC and PrPSc from different species as well as PrP-derived synthetic peptides. Five of the 8 mAbs exhibited binding discontinuous PrP epitopes, all of which were disrupted by addition of β-mercaptoethanol or dithiothreitol, which reduced the single disulfide bond found in PrP. One mAb F20-29 reacted only with human PrP while the F4- 31 mAb bound bovine PrP; the KD values for both mAbs F4-31 and F20-29 were ~500 pM. Binding of all five conformation-dependent mAbs to PrP was inhibited by β-mercaptoethanol in ELISA, Western blots and histoblots. One conformation-dependent mAb F4-31 was found to increase the sensitivity of an ELISA-based test by nearly 500-fold when it was used as the capture antibody. These new conformation-dependent mAbs were found be particularly useful in histoblotting studies, in which the low backgrounds after treatment with β-mercaptoethanol created unusually high signal-to-noise ratios.
Keywords: Prion, monoclonal antibody, histoblot, bovine spongiform encephalopathy, conformation-dependent antibody, disulfide reduction
INTRODUCTION
Accumulation of alternatively folded isoform (PrPSc) of the normal, cellular prion protein (PrPC) causes fatal neurodegeneration in humans and animals (1–3). The prion diseases include Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep, chronic wasting disease (CWD) in elk and deer, bovine spongiform encephalopathy (BSE), transmissible mink encephalophy, and feline spongiform encephalopathy. Conversion of PrPC into PrPSc results in a profound conformational change, including a reduction of the α-helical content and an increase in the β-sheet content of the molecule (4–7). Initially, PrPSc was distinguished from PrPC by its resistance to limited proteolysis.
Discovery of PrPSc and the subsequent production of antibodies to the protein made possible the definitive identification of BSE in cattle and variant (v) CJD in humans (8, 9). More importantly, immunoassays of brainstems from cattle destined for the human food supply have helped reduce exposure of people to bovine prions (10). Compelling evidence has accumulated that vCJD results from the consumption of prion-infected bovine tissue (11–13), but fortunately, cases of vCJD are dwindling. Notably, four vCJD cases have been traced to transfused blood from donors who later developed vCJD (14, 15).
Despite numerous attempts to produce PrPSc-specific antibodies that can be used in rapid immunoassays, none have been identified to date. Such PrPSc-specific antibodies would react with native PrPSc and not with PrPC; several antibodies that immunoprecipitate native PrPSc have been reported (16, 17). In addition, antibodies engineered with peptide grafts corresponding to PrP(89–112) or PrP(136–158) have been reported to selectively bind native PrPSc in prion-infected mouse, human and hamster tissues (18, 19). The need for PrPSc-specific antibodies has increased with the discovery that not all forms of PrPSc are resistant to limited proteolysis. Protease-sensitive PrPSc has been identified in humans, domestic animals and laboratory rodents.
In the work reported here, we describe studies designed to generate conformation-dependent antibodies. PrP-ablated (Prnp0/0) FVB mice were immunized with recombinant (rec) bovine or human PrP. After multiple immunizations, spleens were harvested from the Prnp0/0/FVB mice with detectable serum anti-PrP antibodies. Fusions of splenocytes with mouse myeloma cells were performed to produce hybridomas, from which 8 clones producing IgG mAbs were isolated. Five of the 8 mAbs exhibited binding to discontinuous PrP epitopes, all of which were disrupted upon reduction of the single disulfide bond with β-mercaptoethanol or DTT. The F20-29 mAb exhibited high-avidity binding to human PrP with a Kd of ~500 pM; the F4-31 mAb bound bovine PrP with similar avidity. The binding of all five conformation-dependent mAbs to PrP was inhibited by β-mercaptoethanol or DTT in ELISA, Western blots and histoblots. F4-31 was found to increase the sensitivity of an ELISA-based test by nearly 500-fold when it was used as the capture antibody. In histoblotting studies, these new conformation-dependent mAbs were found be particularly useful, when the low backgrounds after treatment with β-mercaptoethanol created unusually high signal-to-noise ratios.
MATERIALS AND METHODS
Recombinant protein
Recombinant bovine PrP [recBoPrP(102–241)] and human PrP [recHuPrP(90–231)] were purified as previously published, with slight modifications (20, 21). We used pPh41 secretion vector employing the alkaline-phosphatase (AP) promoter and the periplasmic bacterial signal peptide from Shigella toxin II. The transfected bacteria were grown in 2-L or 10-L fermenters and harvested 24 h after inoculating the fermentation media. The recombinant protein was released by disruption of bacteria in 8 M GdnHCl/100 mM DTT, pH 8. Protein was purified by size exclusion chromatography on a HiLoad 26/60-cm Superdex 200 column (GE Healthcare), followed by reverse phase–HPLC, using a Jupiter C-4 preparative column (25/250 mm, Phenomenex) and linear acetonitrile gradients.
Brain homogenates
Brain homogenates (10% wt/vol) were prepared from cattle (Bo), human (Hu), sheep (Ov), mouse (Mo), Syrian hamster (SHa), and mule deer (MD) in calcium- and magnesium-free PBS. Brain tissue was homogenized with a PowerGen homogenizer (Fisher Scientific) on ice by three 15-second strokes. The homogenate was cleared by a short centrifugation at 500 × g for 5 min and the supernatant was used for the preparation of the samples.
Antibody production
Six- to eight-week-old, Prnp0/0 female FVB mice were immunized with recBoPrP(102–241) or recHuPrP(90–231) prepared in RIBI adjuvant (Ribi, Inc.) at 1 mg/mL. The amino acid numbering used in this manuscript was based on the alignment described by Giles and Prusiner (22). Animals were immunized by subcutaneous injection of 100 μL of antigen followed by two booster immunizations at 14-day intervals. Hybridomas were generated as described (16) with the following modifications. Myeloma cells, P3X63AG8U.1, were maintained in Dulbecco’s modified Eagles media with high glucose and L-glutamine (without sodium pyruvate) (Gibco-BRL #11965-092) supplemented with 10% fetal calf serum (Gibco-BRL #10082-147), 2 mM Na pyruvate, and penicillin/streptomycin. Myeloma cells (viability greater than 99%) were mixed with the splenocytes (1:5 ratio) and the mixture centrifuged at 2000 rpm for 5 min. The supernatant was removed, the cell pellet gently disrupted, and 1 ml of a 50% polyethylene glycol solution per 108 cells was slowly added with stirring. The cell mixture was then centrifuged (500 × g, 5 min). The cell pellet was slowly suspended in serum-free growth medium and incubated for 20 min at 37 °C. Next, the cells were centrifuged (500 × g, 5 min), suspended in complete growth medium adjusted to 15% FCS and 1 mM nonessential amino acids, and incubated overnight at 37 °C in a 7% CO2 atmosphere. The next day, the cells were suspended in fresh growth medium containing FCS, non-essential amino acids, hypoxanthine, aminoptrin and thymidine (HAT-medium) and then immediately dispersed over 10 microplates (96-well format) containing irradiated 3T3 fibroblast cells. After 10–21days, media was screened for anti-PrP antibodies.
Screening
A multi-step screening strategy was employed: first, a direct binding ELISA was performed, followed by a double sandwich (ds) DELFIA, and finally Western immunoblotting using normal brain homogenates from various species. Hybridoma cells from wells containing medium that tested positive in all three screening steps were expanded and cloned (at least 3×), as described (23). Ascites tumors were generated by interperitoneal injection (IP) of approximately 5 × 106 hybridoma cells into Prnp0/0, SCID mice (McLaughlin Research Institute, Great Falls, MT), and antibodies purified by protein-G affinity chromatography.
ELISA methods
The direct binding ELISA used either recHuPrP(90–231) or recBoPrP(102–241) peptide absorbed onto the bottom of 96-well microtiter plates (Dynex/Thermo Immulon 2HB, #3455). Supernatants or other test solutions were applied and incubated for 2 h at 37 °C. The plates were washed and AP-conjugated, anti-mouse F(ab’)2 (Pierce #31324) immunoglobulin added. After a final set of washes, substrate pNPP (Sigma # N-9389) was added and the plate read in a 96-well spectrophotometer at 405 nm.
The dsDELFIA was used at an early stage in the screening process (analysis of media from the original 96-well fusion cultures) in order to identify antibodies that might function as capture reagents in the CDI. Europium-labeled, chimeric human-mouse (HuM), recFab P (24) was used as the reporting antibody in the sandwich. Fluoronunc Maxisorp 96-well black plates (Nunc #43711) were coated with anti-Fc specific goat anti-mouse IgG antibody (Chemicon #AP127) by adding 100 μL/well of a 2 μg/ml solution in carbonate coating buffer (0.1 M NaHCO3, pH 8.6) and incubating the plates at 37 °C for 2 h. Reactive sites on the plastic were then masked by incubating the plates for 1 h at 37 °C in blocking buffer [5% nonfat dry milk (NFDM) prepared in TBST. The blocking solution was then removed, test samples (hybridoma supernatants) were added and the plate incubated overnight at 4 °C. The hybridoma supernatant was aspirated; the plate washed and blocked again using 250 μL/well of a 1:1000 dilution of pure mouse IgG (Chemicon #PP54) in blocking buffer. This second blocking step was critical in order to reduce background fluorescence, presumably resulting from binding of subsequent reagents to any unoccupied combining sites of the goat-anti mouse Fc antibody; while the HuMP reporter antibody is a humanized recombinant Fab, it does contain mouse antibody sequences in the combining site. The plates were then decanted, recHuPrP(90–231) or recBoPrP(102–241) at 2 μg/ml in blocking buffer was added, the plates sealed and incubated at 37 °C for 2 h. The plates were then washed 3×, incorporating a 1 min soak per cycle and patted dry. Next, 200 μL/well of Eu-conjugated HuM-P (HuM-P-Eu) at 0.25 μg/mL in DELFIA assay buffer (Perkin Elmer 4002-0010) was added and the plates incubated for 2 h at 37 °C. Finally, the plates were washed 7× with a 1 min soak per cycle, and 200 μL/well of DELFIA enhancement solution (Perkin Elmer #1244-105) was added and incubated for 10 min at room temperature (RT). The time-resolved, fluorescence intensity was measured using a Packard Discovery fluorometer. In some cases, a dsELISA was used when HRP-conjugated HuM-P was substituted for HuM-P-Eu, and the assay was developed with a colormetric substrate. Antibody isotype analysis was performed using isotype-specific, AP-conjugated anti-mouse immunoglobulins (Southern Biotechnologies) in an ELISA format and using a one-step, lateral-flow mouse isotype assay (Roche). The CDI was as described (24).
Western blots
Antigens for Western blots were 10% (w/v) brain homogenates prepared from prion-diseased hamsters, mice, humans, cattle, sheep, and deer. Weanling Syrian hamsters and CD1 mice were inoculated with 50 μL of scrapie isolate Sc237 and 30 μL of mouse-passaged Rocky Mountain Laboratory (RML) prions, respectively. Both hamsters and mice were monitored for signs of neurologic dysfunction, then euthanized when disease was imminent (25, 26). Human samples were from patients diagnosed and confirmed postmortem by histopathology and immunochemistry. Brain tissue from BSE-infected cattle was received from Veterinary Laboratory Agency, New Haw, UK. Sheep scrapie, isolate no. 027 derived from a Suffolk sheep with the ARQ genotype, was obtained from the USDA. Brains from animals with CWD were collected at the Wildlife Research Center in Fort Collins, Colorado. For each species, brain tissue was homogenized in PBS without calcium and magnesium chloride (GIBCO, Invitrogen), by three, 15-sec strokes of a PowerGen homogenizer (Fisher Scientific International, Tustin, CA). The homogenate was centrifuged at 500 × g for 5 min and the supernatant was used for the preparation of the sample.
Samples subjected to limited PK digestion and Western immunoblotting were diluted to 0.5 mg/ml total protein in lysis buffer (100 mM Tris, 150 mM NaCl, 0.5% DOC, 0.5% NP-40). PK was added (40 μg/mg PK:protein) and incubated for 1 h at 37°C. The reaction was stopped with 2 mM PMSF. Samples were centrifuged at 100,000 × g for 1 h. The pellet was suspended in lysis buffer and diluted with an equal volume of 2× SDS sample buffer without reducing agent.
The antigen was electrophoresed using 15% Tris-HCl, SDS-PAGE gels (BioRad). Samples were boiled for 5 min, and the equivalent of 10 μg (5 μg for the RML-infected mouse samples) of total protein was applied to the gel. SDS-PAGE was performed in 1-mm-thick, 15% polyacrylamide gels and then, electrotransferred to Immobilon-p Transfer Membrane (Millipore). The membrane was blocked for 1 h with 5% NFDM in TBST at RT. We developed the blot with different purified primary mAbs at 1 μg/ml, followed by a secondary antibody, ImmunoPure goat anti-mouse IgG (Fc), peroxidase conjugated (Pierce), diluted 1:5000 in TBST. ECL detection reagent (GE Healthcare/Amersham) was used as substrate. The blots were exposed to Amersham Hyper-Film ECL (GE Healthcare).
Immunoprecipitation
Protein A or protein G-Sepharose (100 μl; Invitrogen cat no. 10-1041 and 10-1242) was mixed with 2 μg of antibody in a final volume of 0.5 ml in TBS. The pH values for protein A and protein G were 9 and 7.5, respectively. The binding of the antibody to Sepharose was done on a rugged rotator for 1 h at 4 °C. After three washes with TBS, pH 7.5, the mixture was blocked with 10% BSA in TBS, pH 7.5. The antigens were 10% brain homogenates prepared with or without PK from uninfected mice, RML-infected mice, uninfected humans or CJD cases, as previously described. We added 5 ul of the antigen to the Sepharose/antibody mixture, in 1 ml TBS, pH 7.5, and 0.3% Sarkosyl, then incubated the mixture on a rugged rotator for 1 h at 4 °C. After three consecutive washes with TBS, pH 7.5, and 0.25% Sarkosyl, the samples were boiled in sample buffer without reducing agent and then analyzed by Western blot. The mouse samples were detected with recombinant Fab HuM D18–HRP antibody (27); the human samples were detected with HuM P–HRP (24).
Epitope determination
Two series of peptides were synthesized: (1) one series spanning BoPrP(95–241), of 12 amino acids in length with an 8-residue overlap and (2) one series spanning HuPrP(88–226), 10 amino acids in length with a 7-residue overlap (Sigma Genysis, Woodlands, TX) (Supplemental Tables I–II4). Three lysine residues were added to the N-termini to improve solubility; a single biotin molecule was also added to the N-terminal lysine residue. The C-termini were amidated. Peptides were dissolved in 10% acetonitrile and diluted to 1 mM in 10% ethyl alcohol.
On the day of the experiment, peptides were diluted 200-fold in TBST, pH 7.4 to give a final peptide concentration of 5 μM. Streptavidin-coated microtiter wells (Pierce Biotechnology Inc, Rockford, IL #15501) were blocked for 60 min at RT by adding 300 μL/well of 5% blocking buffer [either NFDM or BSA with TBST]. The blocking solution was decanted, 50 μL of the 5 μM peptide solution added, and the plates incubated for 1 h at RT. Anti-PrP antibodies were added and the plate incubated for 1 h at 37 °C. Following three washes with 200 μL/well of TBST, 100 μL/well of horseradish peroxidase–conjugated, goat anti-mouse IgG+A+M (Zymed Laboratories #65-6420, Invitrogen, Carlsbad, CA), diluted 1:2000 in 5% NFDM-TBST buffer, was added and the plates incubated for 60 min at RT. Finally, the plates were washed 3× with TBST, then 100 μL/well of K-blue substrate (Neogen, Lansing, MI) was added and the reaction stopped with 0.1 N HCl. Color development was recorded at 650 nm in a 96-well plate reader. Alternatively, we used an AP-labeled secondary antibody and the substrate was pNPP.
Evaluation of recFab HuM-P antibody binding to peptides necessitated substitution of 100 μL of a 1:5000 dilution of HRP-conjugated anti-human IgG (Fab) (Pierce, #31414) for the HRP-conjugated anti-mouse IgG.
ELISA of reduced PrP
PrP at 5 μg/mL in coating buffer (0.1 M NaHCO3, pH 8.6) was mixed with the reducing agent, DTT, at final concentration of 10 mM. The mixture was plated in a 96-well microtiter plate and incubated overnight at 4 °C. The plate was washed; nonreactive sites on the plastic blocked with 0.25% BSA in TBST buffer and the appropriate antibody titrated on the coated wells. The assay was developed with an AP-conjugated goat anti-mouse IgG-F(ab)2 (Pierce). The substrate used was PNPP (Sigma).
Sandwich CDI for human and bovine PrPSc
The CDI data described in this paper were generated with recFab HuM-P (24) labeled with Eu-chelate of N-(p-isothiocyanatobenzyl)-diethylenetriamine-N1,N2,N3,N3-tetraacetic acid (DTTA) at pH 8.5 for 16 h at RT as described (28). Additional data were generated with Eu-labeled mAb 3F4 (29, 30).
The principle, development, calibration, and calculation of PrPSc concentration from CDI data have been described (24, 28, 30, 31). Briefly, each sample was divided into two aliquots: (i) untreated (designated native, or N) and (ii) mixed to a final concentration of 4 M GdnHCl and heated for 5 min at 80°C (designated denatured, or D). Both samples were immediately diluted 20-fold with H2O containing protease inhibitors (5 mM PMSF; aprotinin and leupeptin, 4 μg/ml each), and aliquots were loaded on a 96-well black polystyrene plate (Packard, Meriden, CT) that was previously coated overnight with 5 μg/ml of mAb recFab D18 or mAb F4-31 in sodium phosphate buffer, pH 7.4 and blocked with TBS, pH 7.8, containing 0.25% BSA (w/v) and 0.1% Tween 20 (w/v) for 1 h at RT. The plates containing native and denatured aliquots of each sample were then incubated for 2 h at RT. The plates were washed 3 times with TBS, pH 7.8, containing 0.05% (v/v) Tween 20, incubated with Eu-labeled recFab P at RT for 2 h, then developed after seven washing steps in the enhancement solution provided by the Eu-label supplier (Wallac Inc.). The signal was counted on a Discovery dual-wavelength, time-resolved fluorometer (Packard). As a standard, we used denatured recHuPrP(23–231) stored at −80°C in 4 M GdnHCl and 50% (v/v) Stabilguard (SurModics, Eden Prairie, MN). The results were expressed as the (D–N) difference of the time-resolved fluorescence (TRF) measured in counts per minute (cpm). The concentration of PrPSc is directly proportional to (D–N) value and is calculated from a previously published formula (24, 28, 31).
Affinity measurements
Antibody affinity measurements for F4-31 and F20-29 were obtained using two independent methods, by ELISA and by surface plasmon resonance (SPR).
ELISA
The KD values in solution were measured by ELISA using the method described by Friguet et al. (32). Data analysis and calculation of KD used the following relationships as modified by Bobrovnik (33):
| (Eq. 1) |
| (Eq. 2) |
for which Ao equals the absorbency measured when no antigen was present (100% antibody binding to the plate); A, the absorbency measured when different concentrations of recPrP (Li) were present; Ka, the on-rate; and Kd, the off-rate. A minimum of five recPrP concentrations, ranging between 0.1 to 50 (× 10−9 M), was used in each analysis. A constant concentration of antibody (0.2 μg/mL) was used in these experiments. The antibody was mixed with the antigen in solution and incubated up to 18 h at room temperature. Incubation time in the ELISA was optimized (15 min) in order to not disturb the antibody/antigen complex at equilibrium. The KD value was determined graphically as the slope of the linear relationship detailed in Equation 1.
SPR
Surface plasmon resonance measurements were obtained with a Biacore (model 2000) instrument. Determination of the affinity constants of IgG in SPR is best accomplished by coupling the antibody to the chip. Due to the physicochemical properties of PrP, we were unable to regenerate the chip surface between sequential injections of antigen solution. Therefore, the single-cycle kinetic method (34) was used, and data analyzed by the Biacore control software method using the BiaEvaluation software fit algorithm kindly provided by Kevin Lindquist (GE Healthcare). Briefly, antibody (10 μg/mL) was coupled to a CM5 chip (Biacore) using 10 mM acetate buffer at optimum pH (pH 4.0, 4.5, 5.0, 5.5 from Biacore BR 100349, 100350, 100351, 100352, respectively), resulting in ~1000 relative units (RU) being immobilized on the CM5 chip using amine-coupling chemistry as described by the manufacturer.
For each determination, three sample cycles were performed. The first two sample cycles consisted of injections of running buffer [HBS-EP buffer (GE Healthcare), 0.05% Tween 20, 0.1 mg/mL BSA, pH 7.4] followed by injection of the antigen solutions. Five antigen solutions in running buffer ranging between 1–150 nM were injected sequentially from lowest to highest concentration with a 1-min stabilization between each injection. After the last injection, the antigen was allowed to dissociate for 30 min. Background (blank sample cycle data trace) was subtracted and data analyzed with the BiaEvaluation software to generate a calculated on-rate (Ka) and off-rate (Kd). KD values were calculated by dividing Kd/Ka.
Histoblots
Frozen coronal sections (10 μm) from Tg(BoPrP+/+)Prnp0/0/FVB4092 and Tg(HuPrP,M129)Prnp0/0/FVB440 mice as well as from normal human and bovine cerebrum were transferred onto nitrocellulose membranes for overnight reduction with 10% 2-mercaptoethanol (Sigma-Aldrich). After reduction, immunohistochemistry was performed by incubating membranes for 2 h with F20-29 and F4-31 (1:1000) antibodies, followed by a 1-h incubation with anti-mouse, AP-conjugated secondary antibody (Promega, Madison, WI). The signal was then developed using NBT/BCIP (Roche) substrate.
Immunofluorescence microscopy
Brains were dissected from normal or Sc263K-infected hamsters and frozen on dry ice. Serial cryosections (5-μm thick) were cut and fixed on slides in 3.7% formaldehyde/zinc fixative (Electron Microscopy Sciences, PA). Slides subject to antigen retrieval were submerged in 1:10 dilution of citric acid–based antigen unmasking solution (Vector, CA), microwaved on high power for 5 min, allowed to cool to RT and processed with the remaining slides by incubation for 20 min in PBS with 0.1% TritonX-100, then blocked for 1 h in PBS containing 1% IgG-free BSA (Jackson, PA). Samples were subjected to PK digestion (250 μg/ml; Roche, NJ) in PBS at 37 °C for 1 h; the reaction was stopped by addition of 50 mM PMSF followed by repeated dilution in wash buffer. The following primary antibodies were diluted in PBS-BSA with 0.1% Tween 20 and applied to slides overnight at 4 °C in a humidified chamber: anti-PrP mAb F4-31 (1:100), rabbit anti-glial fibrillary acid protein (1:500; Novus, CO), and rabbit anti-pan neurofilaments (1:200; BIOMOL, PA). The following secondary antibodies were diluted in PBST-BSA with 4',6-diamidino-2-phenylindole and applied to slides for 2 h: donkey anti-mouse CY2 (1:800) and donkey anti-rabbit-CY5 (1:500; Jackson). Cover slips were applied with Hydromount (National Diagnostics, GA) and sections imaged using a Leica SP5 confocal microscope equipped with AOTF/AOBS and blue Argon (488 nm), HeNe (633 nm) and 405-nm lasers (Leica, Germany).
RESULTS
Antibody production
High serum titers were observed following immunization of Prnp0/0/FVB mice (35) with either recHuPrP(90–231) or recBoPrP(102–241). Western blot analyses using normal brain homogenates from mice, Syrian hamsters, humans, cattle, sheep, and deer suggested that the Prnp0/0/FVB mice were producing antibodies reactive with PrPC (data not shown). Following multiple cell-fusion experiments, hundreds of putative hybridomas were obtained in the direct binding ELISA (a response of 3–5× above background was considered positive). However, only a few were positive in both the direct-binding ELISA and the dsDELFIA (Table I).
Table I.
Properties of the mAbs.†
| mAb | ds DELFIA | Western blot | Epitope | Ratio O/R | |||||
|---|---|---|---|---|---|---|---|---|---|
| Bo | Hu | Ov | Mo | SHa | MD | ||||
| F4-31 | + | + | − | + | + | + | + | N | 3.9 |
| F10-26 | − | + | + | W | W | W | W | BoPrP(100–111) GWGQGGTHXQWN |
1.2 |
| F20-29 | − | − | + | − | − | − | − | N | 3.8 |
| F20-49 | + | − | + | − | − | + | − | HuPrP(108–115) MKHMAGA |
1.3 |
| F20-89a | + | − | + | + | − | − | + | HuPrP(97–100) SQWN |
1.2 |
| F20-108a | + | + | + | + | + | + | + | N | 4.6 |
| F20-130a | + | + | + | + | + | + | + | N | 4.4 |
| F20-80 | − | + | + | + | + | + | + | N | 3.0 |
In these assays, + indicates positive signal, − indicates no signal, W indicates weak response. For Western blots, antibodies probed PrPC in 10% brain homogenates prepared from uninfected cattle (Bo), humans (Hu), sheep (Ov), mouse (Mo), Syrian hamsters (SHa), and mule deer (MD). N, nonlinear (conformational or discontinuous) epitope. Ratio O/R shows binding to oxidized PrP over reduced PrP, using a 1/10 dilution of the antibody cell culture supernatant.
Eight mAbs (F4-31, F10-26, F20-29, F20-49, F20-80, F20-89a, F20-108a and F20-130a) were selected for further study. Western blots using brain homogenates from normal, control animals demonstrated a diverse set of binding patterns (Table I). Two mAbs, F4-31 and F10-26, were isolated from Prnp0/0/FVB mice immunized with recBoPrP(102–241) while the remaining 6 mAbs were isolated from Prnp0/0/FVBmice immunized with recHuPrP(90–231). Isotype analysis indicated that these 8 antibodies are of subclass IgG1 with kappa light chains. Three mAbs (F20-108a, F20-130a, and F20-80) showed binding to PrPC across all species tested (cattle, human, sheep, mouse, hamster, and mule deer). F4-31 bound to all but human PrPC, while F20-29 bound only to human PrPC.
Antibody specificity
To ascertain the patterns of immunoreactivity of the 8 mAbs with PrPSc, brain homogenates containing BSE, sCJD, sheep scrapie, mouse scrapie or Syrian hamster scrapie prions were digested with proteinase K (PK) for 1 h at 37 °C. The undigested and digested samples were subjected to Western blotting using the 8 mAbs (Fig. 1). Antibody binding to infected brain homogenates from mule deer with CWD was not tested. Our data show binding behavior similar to that observed with uninfected brain homogenates, demonstrating that mAbs recognizing PrPC also bound to the denatured, PK-resistant PrPSc isoform (rPrPSc), also called PrP 27-30 (Fig. 1). The strongest binding to BSE-infected bovine brain was observed with mAb F4-31, which also was able to bind PrPSc in scrapie-infected sheep, RML-infected mouse, and Sc237-infected hamster brains (Fig. 1). The F20-29 mAb bound only to human PrPC in uninfected brain (Table I) and to denatured human PrP 27-30 in sCJD brain homogenates (Fig. 1). The F20-89a mAb did not bind to PrPC in uninfected mouse or hamster brain (Table I), but weakly recognized PrP 27-30 in infected mouse and hamster brain, suggesting that the affinity for these PrP antigens may be low (Fig. 1).
Figure 1.

Western blots show the binding of each of the 8 mAbs to PrP in infected brain homogenates from different species. Samples were prepared from (paired lanes 1) the obex of a cow with BSE, (paired lanes 2) human with sCJD, (paired lanes 3) sheep with scrapie, (paired lanes 4) mouse infected with RML prions, and (paired lanes 5) Syrian hamster infected with Sc237 prions; samples were either undigested (−) or digested with 20 μg/ml PK (+) for 1 h at 37 °C. Brain homogenates from PrP-knockout mice are shown as a control (C). Western blots were developed with 1 μg/mL of the mAb indicated and peroxidase-labeled anti-mouse secondary antibody, followed by addition of a chemiluminescent substrate. The Mr of the protein fragments are shown in kDa.
To quantify the avidity of F4-31 for PrPs from various mammals, antibody binding to recPrPs was analyzed using the dsDELFIA (Fig. 2). The most avid binding was seen with the F4-31 antibody with recombinant polypeptides: recBoPrP(102–241), recMoPrP(89–231) and recSHaPrP(90–231). Approximately 10-fold weaker binding was observed to recOvPrP(23–231) polypeptide and the lowest activity was observed using recHuPrP(90–231). Measurements of the KD values for F4-31 mAb binding to recBoPrP(102–241) in solution by ELISA and for immobilized antigen by SPR gave values of 630 pM and 255 pM, respectively (Fig. 3 and Table II). Each value represents the average of three independent measurements in triplicate. The dissociation constants for F20-29 mAb binding to recHuPrP(90–231) by ELISA and SPR were 276 pM and 463 pM, respectively. While the dissociation constants determined by ELISA and SPR were in good agreement for F4-31 mAb binding to recBoPrP(102–241) and F20-29 mAb binding to recHuPrP(90–231), the KD value for F4-31 mAb binding to recMoPrP(89–231) obtained by ELISA was nearly 40-fold higher than that found by SPR (Table II).
Figure 2.
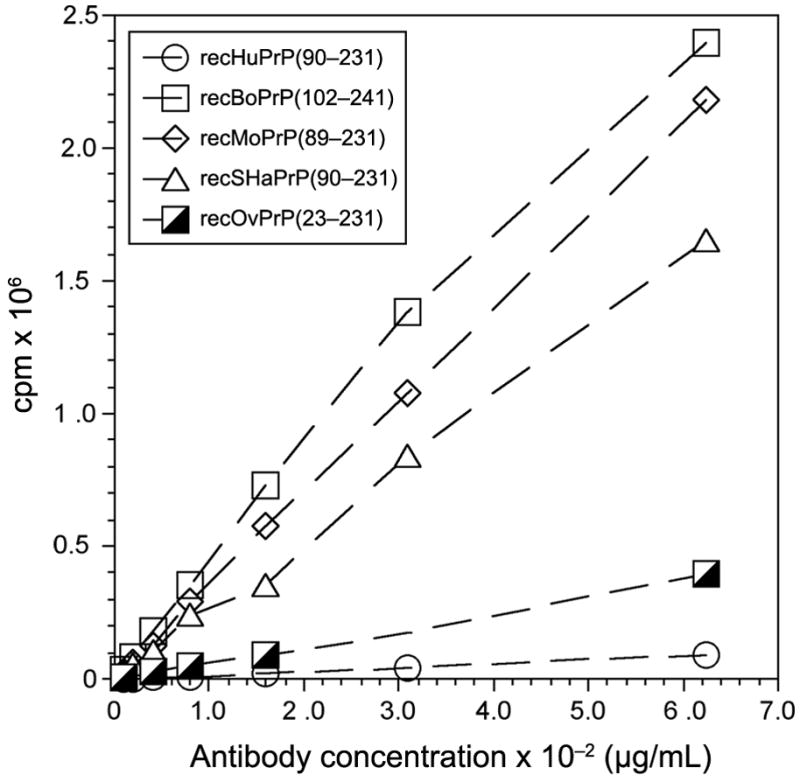
Binding of mAb F4-31 to recPrP in a dsDELFIA. Antigens: recHuPrP(90–231), open circle; recBoPrP(102–241), open squares; recMoPrP(89–231), open diamond; recSHaPrP(90–231), open triangles; and recOvPrP(23–231), partially filled squares. Binding was measured as the fluorescence signal in counts per minute (cpm). For each recPrP antigen, five different concentrations of mAb F4-31 were tested.
Figure 3.

Measurement of the KD values of mAbs in solution by ELISA. (A) F4-31 with different concentrations of recBoPrP(102–241) and recMoPrP(89–230) and (B) F20-29 using different concentrations of recHuPrP(90–231).
Table II.
KD values in pM for mAbs F4-31 and F20-29.
| mAb | ELISA | SPR | ||||
|---|---|---|---|---|---|---|
| recHuPrP | recMoPrP | recBoPrP | recHuPrP | recMoPrP | recBoPrP | |
| F4-31 | — | 1700± 200 | 630 ± 4.0 | — | 45.1 ± 5.5 | 255.3 ± 47.4 |
| F20-29 | 276 ± 5.0 | — | — | 463.3 ± 52.6 | — | — |
Differential antibody binding
Immunoprecipitation experiments as well as immunostaining of frozen brain sections, denoted histoblots, showed that the F4-31 and F20-29 mAbs bound to native PrPC but not to native PrPSc (Fig. 4A and B, Fig. S1). The F4-31 and F20-29 mAbs (Fig. 4A and B, lanes 1) immunoprecipitated native PrPC in uninfected brain homogenates prepared from an FVB mouse and a normal, human control, respectively. The F4-31 and F20-29 mAbs (Fig. 4A and B, lanes 2) immunoprecipitated native PrPin prion-infected brain homogenates prepared from an RML prion-infected mouse and a sCJD case, respectively. Neither F4-31 nor F20-29 (Fig. 4A and B, lanes 3) immunoprecipitated native PrPSc in prion-infected brain homogenates prepared from an RML prion-infected mouse and a sCJD case, respectively. Prior to immunoprecipitation, the samples in lanes 3 were digested with 20 μg/ml of PK for 1 h at 37 °C to remove PrPC and the N-terminus of the PrPSc to form PrP 27-30. Staining of PrP 27-30 was confirmed by immunohistochemistry (data not shown).
Figure 4.

F4-31 and F20-29 mAbs recognize to native PrPC and denatured PrPSc, but not native PrPSc. (A, B) Immunoprecipitation shows F4-31 binding to MoPrP (A) and F20-29 binding to HuPrP (B) in uninfected, control brain homogenates (lanes 1) and undigested, infected brain homogenates (lanes 2). F4-31 and F20-29 mAbs did not bind to native rPrPSc in PK-digested, infected brain homogenates (lanes 3). Apparent molecular masses based on the migration of protein standards are shown in kDa. (C, D) dsELISA data showing binding of F4-31 to PrP in RML-infected mouse brain homogenates (C) and of F20-29 to PrP in human sCJD brain homogenates (D). Samples were untreated (circles), digested with PK only (squares), denatured with GdnHCl only (diamonds), or digested with PK and then denatured with 4 M GdnHCl (triangles). Protease-digested samples were incubated with 20 μg/ml of PK for 1 h at 37 °C. Denatured samples were incubated with GdnHCl for 15 min at room temperature, after PK digestion. HuM-P was used as the detection antibody.
In ELISA studies, F4-31 bound PrP in undigested, nondenatured brain homogenates of RML-infected mice (Fig. 4C, circles) but the signal disappeared when brain samples were digested with PK (squares). When the RML-infected brain homogenates were denatured with 4 M GdnHCl after limited digestion with PK (Fig. 4C, triangles) or without digestion (diamonds), the F4-31 mAb gave similar ELISA signals. Similar to the immunoprecipitation and histoblot findings, the ELISA results showed that the F4-31 mAb binds to native PrPC, denatured PrPC, and denatured PrPSc, but not to native PrPSc, in RML-infected mouse brain homogenates. Comparable findings were obtained using the anti-HuPrP–specific F20-29 mAb with sCJD brain homogenates (Fig. 4D). When the sCJD brain homogenates were denatured with 4 M GdnHCl after limited digestion with PK (Fig. 4D, triangles) or without digestion (diamonds), the F20-29 mAb gave similar ELISA signals. Moreover, F20-29 bound human PrPC in undigested, nondenatured sCJD brain homogenate (Fig. 4C, circles) but the signal disappeared when brain samples were digested with PK (squares).
Disruption of the disulfide bond interferes with antibody binding
PrP contains a single intramolecular disulfide bond (36). In recBoPrP(102–241), this disulfide bond is located between Cys190 and Cys225, linking α-helices B and C. We investigated the effect of reducing the disulfide in recBoPrP(102–241) on antibody binding. The PrP antigen was absorbed onto microtiter plates in the absence or presence of 10 mM DTT. The plates were then blocked and immediately used to determine antibody binding by ELISA (Fig. 5). While binding of F20-89a and F20-49 to oxidized and reduced recBoPrP(102–241) were similar, the F20-29 and F4-31 mAbs were unable to bind the reduced recHuPrP(90–231) and recBoPrP(102–241), respectively (Fig. 5A and B). The ratios of antibody binding to oxidized and reduced PrP (O/R) were determined for the 8 mAbs (Table I).
Figure 5.

Binding of mAb F20-29 to reduced (squares) and nonreduced (circles) recPrP(90–231) (A) and mAbs F4-31 (B), F20-89a (C), and F20-49 (D) to reduced (squares) and nonreduced (circles) recBoPrP(102–241), as measured by ELISA. Reduced samples were incubated overnight with 10 mM DTT at 4 °C. (E) Western blot of reduced (odd-numbered lanes) and nonreduced (even-numbered lanes) recBoPrP(102–241), probed with recFab HuM-P (lanes 3–4), mAb F4-31 (lanes 5–6); and F10-26 (lanes 7–8). PrP was silver stained as a control (lanes 1–2). Apparent molecular masses based on the migration of protein standards are shown in kDa.
These observations were confirmed by electrophoretic analysis. Silver staining revealed that reduced recBoPrP(102 241) migrated more slowly on SDS-PAGE than the nonreduced polypeptide (Fig. 5E, lanes 1 and 2). Antibodies F4-31, F10-26 and recFab HuM-P were used in Western blots. The recHuM-P Fab recognized both reduced and nonreduced recBoPrP (Fig. 5E, lanes 3 and 4), which was expected because its linear epitope is remote from the disulfide bond and therefore should not be influenced by its reduction. The F4-31 mAb bound nonreduced recBoPrP(102–241), but not reduced BoPrP (Fig. 5E, lanes 5 and 6). In contrast, the F10-26 mAb detected both oxidized and reduced BoPrP (Fig. 5E, lanes 7–8), reflecting its O/R binding ratio of 1.2 (Table I). The inability of the F20-29 and F4-31 mAbs to bind reduced HuPrP and BoPrP, respectively, argues that these two antibodies bind to discontinuous epitopes that are conformation-dependent.
Further evidence that the F20-29 and F4-31 mAbs bind to conformation-dependent epitopes comes from histoblot analyses (37). Coronal sections were taken from the brains of uninfected cattle, humans, FVB mice, Tg(BoPrP+/+)4092 and Tg(HuPrP)440 mice. These frozen brain sections were then developed with either F4-31 or F20-29 mAbs. The F4-31 mAb bound to sections from cattle, FVB mice, Tg(BoPrP+/+)4092 mice, whereas F20-29 bound only to sections from humans and Tg(HuPrP)440 mice. Exposure of the brain sections to 10% β-mercaptoethanol in order to reduce the PrP disulfide abolished the antibody binding (Figs. 6–7, Fig. S1).
Figure 6.

Histoblots developed with F4-31 and F20-29 mAb. Coronal sections of the hippocampus of FVB, Tg(BoPrP+/+)4092, and Tg(HuPrP)440 mice were used. Reduction with 10% 2-mercaptoethanol abolished the signal. Am, amygdala; CC, corpus callosum; Fi, fimbria; Hp, hippocampus; Hy, hypothalamus; Ic, internal capsule; Pu, putamen; Th, thalamus.
Figure 7.

F4-31 detects PrPC in the both the gray and white matter in bovine cerebrum (A) but not in human tissue (B). In comparison, F20-29 detects only PrPC in the gray matter of the human cerebrum (D) but not in bovine tissue (C). Reduction of the disulfide in PrP using 10% β-mercaptoethanol abolished the antibody signal (E–H). The bar in F represents 0.5 cm and applies to all panels.
F4-31 as a capture antibody in ELISA
To test the ability of mAb F4-31 to function as a capture antibody, we compared its fluorescence signals to those of the capture antibody HuM-D18 recFab using brain homogenates from BSE-infected cattle, scrapie-infected sheep, CWD-infected deer, Sc237-infected Syrian hamster, RML-infected mouse, and sCJD-infected human brain (Fig. 8A).Using the conformation-dependent immunoassay (CDI) format (28), we measured the differential antibody binding to native (N) PrP versus chemically denatured (D) PrP. The CDI employs antibodies that recognize an epitope available on PrPC but buried in native PrPSc; chemical denaturation of PrPSc reveals the epitope, thereby allowing detection of PrPSc even in the presence of PrPC. Formatted as a sandwich immunoassay, we asked if the CDI might be improved by substituting the F4-31 mAb for HuM-D18 recFab in the detection of BSE and CWD prions (24).
Figure 8.
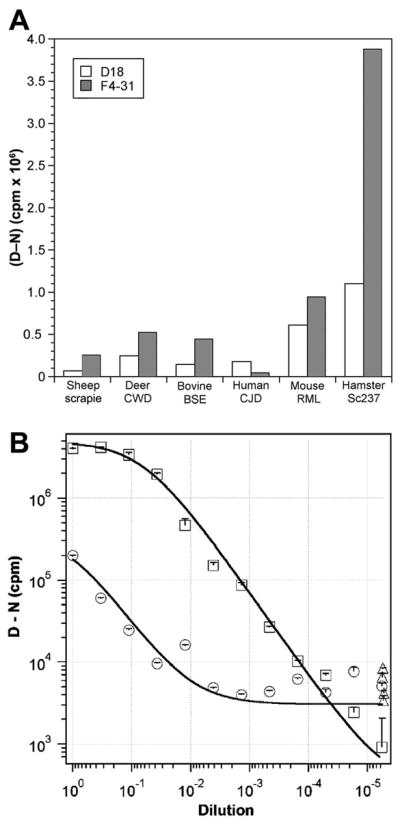
Improved detection of BSE prions. (A) Detection of PrPSc in infected brain homogenates of different species, as indicated, using F4-31 mAb (shaded bars) or HuM-D18 recFab (open bars) as the capture antibody in the CDI. Results are presented as the (D–N) difference in fluorescence signals, measured in counts per minute (cpm), from denatured (D) and nondenatured (N) samples. (B) Measurement of BoPrPSc in BSE-infected bovine brain samples diluted into normal bovine brain homogenate, using either HuM-D18 recFab (circles) or F4-31 mAb (squares) as the capture antibody in the CDI. In both experiments, europium-conjugated HuM-P recFab was used as the detection antibody. The (D–N) difference is directly proportional to the concentration of PrPSc in the sample. Data points and bars represent the averages and standard deviations, respectively, from three independent measurements.
In the CDI format, mAb F4-31 showed higher fluorescence signals for all species tested except human PrP, to which it does not bind. With Sc237-infected hamster brain, we observed a 4-fold increase in signal from F4-31 compared to D18. Additionally, F4-31 demonstrated a ~3-fold increase in signal with infected cattle and sheep brain homogenates and a 2-fold increase in signal with infected deer and mouse samples (Fig. 8A).
Next, we compared the sensitivity of the CDI for detection of BoPrPSc in BSE-infected bovine brain using D18 or F4-31 as capture antibodies (Fig. 8B). BoPrPSc in BSE-infected bovine brain was serially diluted into uninfected brain homogenate and either HuM-D18 recFab (circles) or F4-31 mAb (squares) was used as the capture antibody. Europium-conjugated HuMP recFab was used as the detection antibody. The (D–N) difference was directly proportional to the concentration of PrPSc in the sample. Data points and bars represent the averages and standard deviations, respectively, from three independent measurements. Compared to HuM-D18 recFab, the F4-31 mAb was found to increase the sensitivity ~500-fold for the detection of BSE prions.
Antibody epitope mapping
To identify the epitopes of the F4-31 mAb, we evaluated antibody binding to a series of peptides with overlapping residues by ELISA. For BoPrP, we synthesized 12-residue peptides with a 7-amino-acid overlap, spanning BoPrP(95–241) (Supplemental Table I). Binding of recFab HuMP to these peptides served as a positive control. Binding of F4-31 was not observed to any of these peptides, whereas strong binding was observed to the intact recBoPrP(102–241) polypeptide (Supplemental Table I). However, binding of F4-31 to longer bovine peptides [BoPrP sequences 102–190, 186–212, 110–242, 205–237, 222–241, 186–212, 206–237, and 222–241] was negative (data not shown). In contrast, mAb F10-26, which failed as a capture antibody in the dsDELFIA and CDI (Table I), demonstrated strong binding by ELISA to the bovine peptides (Supplemental Table I). Together, these data suggest binding at a linear epitope within BoPrP(107–116), corresponding to the epitope motif HSQWNKPSKP, similar to the epitope for HuM-P (24). This finding explains the failure of mAb F10-26 to capture BoPrP in the dsDELFIA and CDI when the HuM-P recFab was used as the detection (sandwich) antibody. In these assays, the detection and capture antibodies must bind to different epitopes on PrP in order to give positive responses. In competitive binding experiments (data not shown), pre-incubation of recBoPrP(102–241) with mAb F10-26 inhibited subsequent binding of recFab HuM-P, again suggesting that these antibodies share an identical or overlapping epitope.
We performed similar experiments to determine the epitopes of F20-89a on HuPrP. We synthesized 10-residue peptides, overlapping by 7 amino acids, spanning HuPrP(88–226) (Supplemental Table II). Binding of antibody 3F4 was used as a positive control. Of the mAbs generated using the recHuPrP immunogen, only F20-49 (data not shown) and F20-89a (Supplemental Table II) bound specific peptides. These data suggest an epitope motif of HuPrP(108–115) (MKHMAGA) for F20-49 and HuPrP(97–100) (SQWN) for F20-89a.
When mAbs F20-29, F20-108a, F20-130a, and F20-80 were tested on the individual peptides, no binding was observed, which suggests a nonlinear epitope for these mAbs (Table I).
DISCUSSION
Once PrP 27-30 was discovered (38, 39), a substantial effort led to the production of polyclonal (40, 41) and later mAbs (42). Western blotting and immunohistochemistry open new avenues of research including the demonstration that amyloid plaques in prion disease contain PrP (40) in accord with earlier studies showing PrP 27-30 polymerized into rod-shaped structures that were indistinguishable ultrastructurally and tinctorially from amyloid (43). Localization of PrPSc in brain sections was made possible by development of the histoblotting technique (37) that was used here to show that reduction of the disulfide bond of PrPSc in situ abolished immunostaining with the conformation-dependent F20-29 and F4-31 mAbs (Figs. 6–7 and Fig. S1).
While anti-PrP antibodies have been crucial in advancing prion research, bioassays in rodents and cultured cells continue to be essential (25, 44, 45). Using Tg mice expressing luciferase under control of the glial fibrillary acidic protein promoter, it has been possible to correlate the increase in bioluminescence with the production of PrPSc and prion infectivity long before clinical signs of neurological dysfunction appear (46). A non-immunoassay–based procedure has been developed utilizing the amyloid-seeding properties of PrPSc and is capable of detecting both protease-resistant and -sensitive forms of PrPSc (47).
mAbs were generated by immunizing mice with the recombinant polypeptides recHuPrP(90–231) and recBoPrP(102–241), sequences that correspond to the PK-resistant cores of human and bovine prions, respectively. We employed a tiered screening strategy that identified early in the screening process which mAbs are highly active as capture reagents. Initial fusion experiments resulted in several hundred positive signals. However, only 6 candidates were identified that consistently gave high signals when used as a capture reagent. These antibodies were subsequently cloned, resulting in mAbs F4-31, F20-29, F20-49, F20-89a, F20-108a, and F20-130a. Two additional mAbs were selected, F10-26 and F20-80, which gave strong signals in the ELISA but failed as capture antibodies in the dsDELFIA. Western immunoblotting using brain homogenates from prion-infected cattle, humans, sheep, mice and Syrian hamsters demonstrated that all 8 mAbs bound native PrPC and denatured PrPSc but not native PrPSc (Fig. 4). Each mAb showed different abilities to recognize PrP of the five species examined. mAb F20-29 recognized only HuPrP whereas antibody F4-31 bound PrP from all species tested except for human.
When we disrupted the single disulfide bond in PrP, binding of mAbs F20-29a, F4-31, F20-108a, F20-130a, and F20-80 was virtually eliminated (Figs. 5–7 and S1; Table I). In contrast, binding of the other 3 antibodies was not reduced by removal of the disulfide bond. Differences in the O/R ratios argue linear or discontinuous epitopes for the antibodies, the latter of which are conformation dependent (Table I; Suppl. Tables I and II). F10-26, F20-49, and F20-89a had O/R ratios near 1.0 and bound to short, overlapping peptides, which indicate linear epitopes (Table I). The five other antibodies had O/R ratios of ≥3.0 and did not bind any overlapping peptides, arguing for nonlinear, conformation-dependent epitopes. Reduction of the disulfide bridge disrupted the conformational or discontinuous epitopes, but did not affect the linear epitopes, for binding. The atomic structures of the epitopes for F4-31, F20-29, F20-108a, F20-130a, and F20-80 mAbs remain to be determined.
F4-31 was selected for further study because of its strong binding to BoPrP, its broad cross-species reactivity, and its ability to function as a capture antibody. F4-31 binds to native PrPC and Gdn-denatured PrPSc, but not to native PrPSc (Fig. 4A and C). F4-31 gave higher signals in the CDI compared to HuM-D18 recFab for all PrP sequences tested except for HuPrP (Fig. 8A), demonstrating a substantial improvement for prion detection. Experiments in which BSE-infected brain homogenates were diluted into normal bovine brain homogenate demonstrated a >500-fold increase in sensitivity when F4-31 was used as the capture reagent in the CDI (Fig. 8B). It seems likely that the improved performance we observed in the CDI using F4-31 results from its high affinity to BoPrP and its conformational epitope, the atomic structure of which remains to be elucidated. Nevertheless, our data indicate that the F4-31 mAb can be employed as an excellent capture mAb for the highly sensitive detection of PrP.
The studies described here report a novel set of conformation-dependent mAbs, the activities of which can be abolished by reducing the single disulfide bond in PrP. The extremely low backgrounds after reduction of PrP in various testing formats suggest that these conformation-dependent mAbs are likely to find application in a wide variety of immunoassays.
Supplementary Material
Abbreviations
- Bo
bovine
- BSE
bovine spongiform encephalopathy
- CDI
conformation-dependent immunoassay
- CJD
Creutzfeldt-Jakob disease
- CWD
chronic wasting disease
- D
denatured samples
- Hu
human
- Mo
mouse
- N
native samples
- O/R
ratio of antibody binding to oxidized and reduced PrP
- Ov
ovine
- PK
proteinase K
- Prnp0/0
PrP-ablated
- PrP
prion protein
- PrP 27-30
protease-resistant core of PrPSc
- PrPC
normal, cellular PrP isoform
- PrPSc
disease-causing isoform
- rec
recombinant
- RML
Rocky Mountain Laboratory
- SHa
Syrian hamster
Footnotes
This work was supported by a USDA Cooperative Agreement (58-5325-3-246), by grants from the National Institutes of Health (AG02132, AG10770, and AG021601), and by a gift from the G. Harold and Leila Y. Mathers Charitable Foundation.
The online version of this article contains supplemental material.
References
- 1.DeArmond SJ, Mobley WC, DeMott DL, Barry RA, Beckstead JH, Prusiner SB. Changes in the localization of brain prion proteins during scrapie infection. Neurology. 1987;37:1271–1280. doi: 10.1212/wnl.37.8.1271. [DOI] [PubMed] [Google Scholar]
- 2.Bruce ME, McBride PA, Farquhar CF. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett. 1989;102:1–6. doi: 10.1016/0304-3940(89)90298-x. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Scrapie prions. Annu Rev Microbiol. 1989;43:345–374. doi: 10.1146/annurev.mi.43.100189.002021. [DOI] [PubMed] [Google Scholar]
- 4.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 5.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Safar J, Roller PP, Gajdusek DC, Gibbs CJJ. Thermal-stability and conformational transitions of scrapie amyloid (prion) protein correlate with infectivity. Protein Sci. 1993;2:2206–2216. doi: 10.1002/pro.5560021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-231) Nature. 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 8.Hope J, Reekie LJD, Hunter N, Multhaup G, Beyreuther K, White H, Scott AC, Stack MJ, Dawson M, Wells GAH. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature. 1988;336:390–392. doi: 10.1038/336390a0. [DOI] [PubMed] [Google Scholar]
- 9.Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 10.Philipp WJ, Groth D, Giles K, Vodrazka P, Schimmel H, Feyssaguet M, Toomik R, Schacher P, Osman A, Lachmann I, Wear A, Arsac JN, Prusiner SB. Transgenic mouse brains for evaluation and quality control of BSE tests. Biol Chem. 2007;388:349–354. doi: 10.1515/BC.2007.040. [DOI] [PubMed] [Google Scholar]
- 11.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of "new variant" CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 12.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 13.Scott MR, Will R, Ironside J, Nguyen HOB, Tremblay P, DeArmond SJ, Prusiner SB. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci USA. 1999;96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 15.Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203:733–739. doi: 10.1002/path.1580. [DOI] [PubMed] [Google Scholar]
- 16.Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schulz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, Ehrensperger F, Hornemann S, Glockshuber R, Riek R, Billeter M, Wuthrick K, Oesch B. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature. 1997;389:74–77. doi: 10.1038/36337. [DOI] [PubMed] [Google Scholar]
- 17.Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou WQ, Estey LA, Lamontagne J, Lehto MT, Kondejewski LH, Francoeur GP, Papadopoulos M, Haghighat A, Spatz SJ, Head M, Will R, Ironside J, O'Rourke K, Tonelli Q, Ledebur HC, Chakrabartty A, Cashman NR. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med. 2003;9:893–899. doi: 10.1038/nm883. [DOI] [PubMed] [Google Scholar]
- 18.Moroncini G, Kanu N, Solforosi L, Abalos G, Telling GC, Head M, Ironside J, Brockes JP, Burton DR, Williamson RA. Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc. Proc Natl Acad Sci USA. 2004;101:10404–10409. doi: 10.1073/pnas.0403522101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moroncini G, Mangieri M, Morbin M, Mazzoleni G, Ghetti B, Gabrielli A, Williamson RA, Giaccone G, Tagliavini F. Pathologic prion protein is specifically recognized in situ by a novel PrP conformational antibody. Neurobiol Dis. 2006;23:717–724. doi: 10.1016/j.nbd.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Mehlhorn I, Groth D, Stockel J, Moffat B, Reilly D, Yansura D, Willett WS, Baldwin M, Fletterick R, Cohen FE, Vandlen R, Henner D, Prusiner SB. High-level expression and characterization of a purified 142-residue polypeptide of the prion protein. Biochemistry. 1996;35:5528–5537. doi: 10.1021/bi952965e. [DOI] [PubMed] [Google Scholar]
- 21.Legname G, I, Baskakov V, Nguyen HOB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 22.Prusiner SB, editor. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2004. [Google Scholar]
- 23.Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor: 1988. [Google Scholar]
- 24.Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, Serban H, Groth D, Burton DR, Prusiner SB, Williamson RA. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol. 2002;20:1147–1150. doi: 10.1038/nbt748. [DOI] [PubMed] [Google Scholar]
- 25.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol. 1982;11:353–358. doi: 10.1002/ana.410110406. [DOI] [PubMed] [Google Scholar]
- 26.Carlson GA, Kingsbury DT, Goodman PA, Coleman S, Marshall ST, DeArmond S, Westaway D, Prusiner SB. Linkage of prion protein and scrapie incubation time genes. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 27.Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. Mapping the prion protein using recombinant antibodies. J Virol. 1998;72:9413–9418. doi: 10.1128/jvi.72.11.9413-9418.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 29.Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller BL, DeArmond SJ, Prusiner SB. Diagnosis of human prion disease. Proc Natl Acad Sci USA. 2005;102:3501–3506. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB, Safar JG. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol. 2004;78:2088–2099. doi: 10.1128/JVI.78.4.2088-2099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 33.Bobrovnik SA. Determination of antibody affinity by ELISA. Theory. J Biochem Biophys Methods. 2003;57:213–236. doi: 10.1016/s0165-022x(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 34.Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG. Analyzing a kinetic titration series using affinity biosensors. Anal Biochem. 2006;349:136–147. doi: 10.1016/j.ab.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 35.Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci USA. 1993;90:10608–10612. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem. 1988;176:21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- 37.Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brains. Proc Natl Acad Sci USA. 1992;89:7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 39.Prusiner SB, Bolton DC, Groth DF, Bowman KA, Cochran SP, McKinley MP. Further purification and characterization of scrapie prions. Biochemistry. 1982;21:6942–6950. doi: 10.1021/bi00269a050. [DOI] [PubMed] [Google Scholar]
- 40.Bendheim PE, Barry RA, DeArmond SJ, Stites DP, Prusiner SB. Antibodies to a scrapie prion protein. Nature. 1984;310:418–421. doi: 10.1038/310418a0. [DOI] [PubMed] [Google Scholar]
- 41.Diringer H, Rahn HC, Bade L. Antibodies to protein of scrapie-associated fibrils. Lancet. 1984;324:345. doi: 10.1016/s0140-6736(84)92708-9. [DOI] [PubMed] [Google Scholar]
- 42.Barry RA, Prusiner SB. Monoclonal antibodies to the cellular and scrapie prion proteins. J Infect Dis. 1986;154:518–521. doi: 10.1093/infdis/154.3.518. [DOI] [PubMed] [Google Scholar]
- 43.Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 44.Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. J Virol. 2000;74:4377–4386. doi: 10.1128/jvi.74.9.4377-4386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klohn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci USA. 2003;100:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamguney G, Francis KP, Giles K, Lemus A, DeArmond SJ, Prusiner SB. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci USA. 2009;106:15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D, Prusiner SB. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci USA. 2007;104:20914–20919. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.