

Abstract
In our continuation of the structure-based design of anti-trypanosomatid drugs, parasite-selective adenosine analogues were identified as low micromolar inhibitors of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Crystal structures of Trypanosoma brucei, Trypanosoma cruzi, Leishmania mexicana, and human GAPDH’s provided details of how the adenosyl moiety of NAD+ interacts with the proteins, and this facilitated the understanding of the relative affinities of a series of adenosine analogues for the various GAPDH’s. From exploration of modifications of the naphthalenemethyl and benzamide substituents of a lead compound, N6-(1-naphthalenemethyl)-2′-deoxy-2′-(3-methoxybenzamido)adenosine (6e), N6-(substituted-naphthalenemethyl)-2′-deoxy-2′-(substituted-benzamido)adenosine analogues were investigated. N6-(1-Naphthalenemethyl)-2′-deoxy-2′-(3,5-dimethoxybenzamido)adenosine (6m), N6-[1-(3-hydroxy-naphthalene)methyl]-2′-deoxy-2′-(3,5-dimethoxybenzamido)adenosine (7m), N6-[1-(3-methoxy-naphthalene)methyl]-2′-deoxy-2′-(3,5-dimethoxybenzamido)adenosine (9m), N6-(2-naphthalene-methyl)-2′-deoxy-2′-(3-methoxybenzamido)adenosine (11e), and N6-(2-naphthalenemethyl)-2′-deoxy-2′-(3,5-dimethoxybenzamido)adenosine (11m) demonstrated a 2- to 3-fold improvement over 6e and a 7100- to 25000-fold improvement over the adenosine template. IC50’s of these compounds were in the range 2–12 μM for T. brucei, T. cruzi, and L. mexicana GAPDH’s, and these compounds did not inhibit mammalian GAPDH when tested at their solubility limit. To explore more thoroughly the structure–activity relationships of this class of compounds, a library of 240 N6-(substituted)-2′-deoxy-2′-(amido)adenosine analogues was generated using parallel solution-phase synthesis with N6 and C2′ substituents chosen on the basis of computational docking scores. This resulted in the identification of 40 additional compounds that inhibit parasite GAPDH’s in the low micromolar range. We also explored adenosine analogues containing 5′-amido substituents and found that 2′,5′-dideoxy-2′-(3,5-dimethoxy-benzamido)-5′-(diphenylacetamido)adenosine (49) displays an IC50 of 60–100 μM against the three parasite GAPDH’s.
Introduction
According to the World Health Organization, Trypanosoma brucei, the causative agent of African sleeping sickness, infected 300 000 to 500 000 people in 1999 and poses an escalating health threat to nearly 60 million people of 36 countries of sub-Saharan Africa.1,2 Introduced into the mammalian host via the blood-meal bite of the tsetse fly vector, T. brucei freely replicates in the bloodstream as it makes its way to the central nervous system (CNS). From there, this parasitic infection induces delirium, seizures, coma, and, if left untreated, death.3 Existing drug therapies, including suramin, pentamidine, melarsoprol, and difluoromethylornithine (DFMO), are inadequate because they suffer from one or more drawbacks such as ineffectiveness against more virulent strains, ineffectiveness against post-CNS invasion, toxicity, resistance, or parenteral administration.4,5 Other diseases caused by Trypanosomatidae, which share a similar state with respect to available drug treatment, include Chagas’ disease (Trypanosoma cruzi)6 and leishmaniasis (Leishmania spp.).7 Clearly, there exists a need for safer and more efficacious chemotherapeutic agents against trypanosomatids.
The focus of this work is on glycolysis in trypanosomes as a target for structure-based drug design. Rigorous studies of energy metabolism in T. brucei have established that, unlike the insect form, the bloodstream form lacks a functional TCA cycle and mitochondrial oxidative phosphorylation and depends solely on glycolysis with the excretion of pyruvate for energy production.8-10 As has been shown by Clarkson and Brohn, treatment of cultured parasites with combinations of glycerol and salicylhydroxamic acid causes inhibition of carbohydrate catabolism and severely impedes parasite proliferation.11 Computer modeling of T. brucei’s glycolytic flux suggests that, unlike in mammalian cells, glycosomal glyceraldehyde-3-phosphate dehydrogenase (GAPDH) exerts significant control over the glycolytic pathway. Competitive inhibitors of this enzyme, when present at concentrations 10- to 100-fold higher than their KI values, are predicted to reduce parasite glycolytic flux to zero.12-14 Energy metabolism in Leishmania spp. and in T. cruzi has been studied predominantly with the promastigote and epimastigote forms, respectively, because the intracellular forms are difficult to analyze because of interference from host cell metabolism. However, glycolysis is always active in these parasites and biochemical studies with the T. cruzi axenic amastigote intracellular form suggest that carbohydrate catabolism is its major source of energy.15 Thus, glycolysis inhibitors have the potential to serve as effective anti-T. cruzi and anti-Leishmania agents.
Guided by crystal structures of T. brucei, T. cruzi, and L. mexicana GAPDH’s16-18 in comparison with human GAPDH,16,19 the process of designing competitive and selective inhibitors is facilitated. X-ray structures co-bound with their natural substrates reveal the detailed binding mode of the adenosyl moiety of the NAD+ cosubstrate and have led to the successful design of adenosine analogues as selective and competitive inhibitors of trypanosomatid GAPDH’s.20-24 Although the active sites of the parasite and mammalian enzymes are highly conserved in the region that binds glyceraldehyde-3-phosphate, the adenosyl moiety is well removed from the catalytic cysteine and sits in a region that is far less conserved between parasite and mammalian enzymes. A detailed description of the targeted region has been described previously.21 The adenosine scaffold provides several opportunities not only for improving affinity but also for improving selectivity. Specifically, a hydrophobic cleft protruding from the ribosyl C2′ is a hallmark of the trypanosomatid GAPDH’s. This “selectivity cleft” is absent from the human enzyme because of a difference in protein backbone conformation. The area around the adenosyl N6 exhibits an additional surface for hydrophobic substituents in all enzymes, though the region appears somewhat larger in the trypanosomatid GAPDH’s. Both mentioned opportunities were exploited previously by our group, culminating in the design of a trypanosomatid-selective inhibitor N6-(1-naphthalenemethyl)-2′-deoxy-2′-(3-methoxybenzamido)adenosine (6e)24 with IC50 values that have been determined to be 25, 12, and 6 μM for T. brucei, T. cruzi, and L. mexicana GAPDH’s, respectively.
In this paper, the exploration of the C2′ and N6 regions is continued with a structure–activity relationship (SAR) series of benzamides and with modifications to the naphthalene ring, respectively. To further investigate the diversity of substituents that could be accommodated by these two regions, a 2.8 Å X-ray structure of an L. mexicana GAPDH/adenosine analogue complex (Figure 1)25 was used to generate a combinatorial library of 240 N6-(substituted)-2′-deoxy-2′-(amido)adenosine derivatives, via computational docking methodology. Additionally, the incorporation of 5′-amido substituents into adenosine analogues bearing either of the most potent N6 or C2′ substituents is explored.
Figure 1.

(Left) L. mexicana GAPDH/6m crystal structure. Circles denote the cleft recognized by the 2′-substituent, which provides for selectivity and affinity, while the N6 region provides for affinity. In human GAPDH (right), recognition of the C2′ substituent is precluded by steric occlusion.
Chemistry
There are three reported methods for the synthesis of N6-(substituted)-2′-deoxy-2′-(benzamido)adenosine analogues: (1) acylation of 2′-amino-2′-deoxy-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)adenosine20,26 followed by N1 alkylation with subsequent Dimroth rearrangement and removal of the Markiewicz protecting group;23,27 (2) acylation of 2′-amino-2′-deoxy-3′,5′-O-(1,1,3,3-tetra-isopropyldisiloxane-1,3-diyl)adenosine followed by diazotization-iodination of N6, C6 amination, and removal of the Markiewicz protecting group;24 (3) diazotization–chlorination of 2′-azido-2′-deoxy-3′,5′-O-(1,1,3,3-tetra-isopropyldisiloxane-1,3-diyl)adenosine, removal of the Markiewicz protecting group, amination of the C6 position, reduction of the C2′ azide, and polymer-assisted amidation.28 Route 1 suffers from poor yields of the alkylation and rearrangement steps and is limited in scope to the availability, either commercially or synthetically, of activated halide alkylating agents. Routes 2 and 3 both suffer from poor yields at the diazotization–halogenation step. The acylation step of route 3 is attractive because the desired end-product can simply be washed cleanly from the resin; however, the resin adds two additional steps to the synthesis because it also needs to be prepared.29,30 To more thoroughly explore N6,2′-disubstituted adenosine analogues, we needed to develop an economically robust route to a difunctionalized intermediate that would allow for the incorporation of a wide variety of substituents at both the N6 and C2′ positions.
Our route (Scheme 1) starts with the synthesis of 2′-deoxy-2′-aminoadenosine (1)20,26 and its quantitative conversion to 2′-deoxy-2′-aminoinosine (2) by preparative use of adenosine deaminase.31 Trifluoroacetylation of the C2′ amine and acetylation of the C3′ and C5′ hydroxyls to yield 2′-deoxy-2′-trifluoroacetamidoinosine (3) and 2′-deoxy-2′-trifluoroacetamido-3′,5′-O-(diacetyl)-inosine (4), respectively, is accomplished in greater than 95% yield for each step. By use of the methodology of Matsuda et al.,32 subsequent chlorination of the C6 position of 4 with phosphorus oxychloride to yield 9-[2′-deoxy-2′-trifluoroacetamido-3′,5′-O-(diacetyl)]-(1-β-d-ribofuranosyl)-6-chloropurine (5) is accomplished in better than 90% yield. Starting from the commercially available arabinose adenosine and transforming through nine steps, the intermediate 5 can be synthesized in 65% overall yield. Amination of the C6 position, in situ removal of the acetates, and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCl (EDCI) mediated acylation of the C2′ amino yield the desired disubstituted products (Scheme 1; 6a–6al, 7e–11e, 7f, 7m, 9m, 11m–34m, 7n, 9n, 11–14n, 19–34n, 12–14ac to 12–14al, and 19ac–34al). Typically, amination of 5 led to a variety of deacetylated products as determined by electrospray ionization mass spectrometry (ESI-MS), but this was of no adverse consequense.
Scheme 1a.

a (a) Adenosine deaminase in H2O at room temperature; (b) EtOC(O)CF3 in DMF at 60 °C; (c) Ac2O, Et3N, and DMAP in ACN at room temperature; (d) N,N-dimethylaniline, POCl3, and Et4NCl in ACN; (e) R1NH2 and Et3N in EtOH at 60 °C; (f) NH4OH in EtOH at 60 °C; (g) R2COOH and EDCI in CH2Cl2/DMF at room temperature.
Synthesis of N6-(1-naphthalenemethyl)-5′-deoxy-5′-(amido)adenosine analogues (Scheme 2) starts with the conversion of commercially available 6-chloropurine-9-riboside (37) to 6-chloropurine-9-[5′-deoxy-5′-(phthalimido)]riboside (38) via standard Mitsunobu conditions. Amination at C6 followed by in situ hydrazine-mediated cleavage of the phthalimide group yields the respective N6-(1-naphthalenemethyl)-5′-deoxy-5′-(phthalimido)adenosine (39) and N6-(1-naphthalenemethyl)-5′-deoxy-5′-aminoadenosine (40) intermediates. EDCI-mediated acylation or polymer-assisted acylation with the appropriately acylated safety-catch resin29,30 is chemoselective and gives the desired products N6-(1-naphthalenemethyl)-5′-deoxy-5′-(cyclohexylacetamido)adenosine (41) and N6-(1-naphthalenemethyl)-5′-deoxy-5′-(diphenylacetamido)adenosine (42) in high yield (Scheme 2). Despite the additional steps needed to prepare the resin, employing the safety-catch acylating agent has the added benefit of merely filtering off the solid support to obtain the desired product in nearly quantitative yield and in greater than 95% purity as determined by HPLC.
Scheme 2a.

a (a) Phthalimide, Ph3P, and DIAD in THF at room temperature; (b) R1NH2 and Et3N in EtOH at 60 °C; (c) NH2NH2·H2O in EtOH at 80 °C; (d) method a, R2COOH and EDCI in CH2Cl2/DMF at room temperature, or method b, appropriately acylated safety-catch resin in DMF at room temperature.
Synthesis of 2′,5′-dideoxy-2′,5′-(bisamido)adenosine analogues also starts with 1 (Scheme 3). Trifluoroacetylation of the 2′ amine yields 2′-deoxy-2′-(trifluoroacetamido)adenosine (43). From use of standard Mitsunobu conditions, 43 is converted to 2′,5′-dideoxy-2′-(trifluoroacetamido)-5′-(phthalimido)adenosine (44), and subsequent in situ hydrazine-mediated cleavage of both the phthalimide and the trifluoroacetamide protecting group yields the desired 2′,5′-dideoxy-2′,5′-diamino-adenosine intermediate (45). Regioselective acylation of the 5′ amine is accomplished with the use of the appropriately acylated safety-catch resin at ambient temperature to give 2′,5′-dideoxy-2′-amino-5′-(cyclohexylacetamido)adenosine (46) and 2′,5′-dideoxy-2′-amino-5′-(diphenylacetamido)adenosine (47) in greater than 95% yield. EDCI-mediated acylation of the 2′ amine gives the final products 2′,5′-dideoxy-2′-(3,5-dimethoxy-benzamido)-5′-(cyclohexylacetamido)adenosine (48) and 2′,5′-dideoxy-2′-(3,5-dimethoxybenzamido)-5′-(diphenylacetamido)adenosine (49) (Scheme 3). Alternatively, acylation of 46 and 47 can be accomplished by employing the appropriately acetylated safety-catch resin at elevated temperatures.28,33,34
Scheme 3a.

a (a) EtOC(O)CF3 in DMF at 60 °C; (b) phthalimide, Ph3P, and DIAD in THF at room temperature; (c) NH2NH2·H2O in EtOH at 80 °C; (d) appropriately acylated safety-catch resin in DMF at room temperature; (e) method a, R2COOH and EDCI in CH2Cl2/DMF at room temperature, or method b, appropriately acylated safety-catch resin in DMF at 60 °C.
2-Naphthalenemethylamine hydrochloride (51), 4-methoxy-1-naphthalenemethylamine hydrochloride (55), and 3-methoxy-1-naphthalenemethylamine hydrochloride (57) were prepared by LiAlH4 reduction of 2-naphthonitrile (50),35 4-methoxy-1-naphthonitrile (54), and 3-methoxy-1-naphthonitrile (56),36 respectively. By use of the methodology of Deana et al.37,38 for the synthesis of 2-hydroxy-1-naphthalenemethylamine, 2-methoxy-1-naphthalenemethylamine (53) was prepared by treatment of 2-methoxynaphthalene (52) with 2-chloro-N-(hydroxymethyl)acetamide and hydrochloric acid. 3-Hydroxy-1-naphthalenemethylamine hydrobromide (58) was prepared by treatment of 57 with BBr3 (Scheme 4).
Scheme 4a.

a (a) LiAlH4 in THF at 0 °C; (b) ClCH2CONHCH2OH and H+ in EtOH at 80 °C; (c) [HCl] in EtOH at 80 °C; (d) BBr3 in CH2Cl2 at 40 °C.
Hydroxylated benzoic acids needed for the synthesis of compounds 6c, 6g, 6k, 6o, 6r, 6t, 6v, and 6x were acetylated under standard conditions.39 After they were coupled to the nucleoside, deprotection of the phenolic groups was accomplished in methanolic ammonia.
Results and Discussion
All inhibitors were screened against L. mexicana GAPDH (Tables 1-5). Additionally, all of the compounds were docked into the structure of L. mexicana GAPDH, which was derived from the X-ray structure of the complex with N6-(1-naphthalenemethyl)-2′-deoxy-2′-(3,5-dimethoxybenzamido)adenosine (6m) after removal of the inhibitor coordinates.25 Docking studies were performed with the program FLO in which molecular potential energy calculations with the QXP force field are accessible through a graphical interface.40 With this information, we attempted to rationalize the observed inhibition data.
Table 1.
Inhibition of L. mexicana GAPDH by N6-(1-Naphthalenemethyl)-2′-deoxy-2′-(benzamido)adenosine Analoguesa
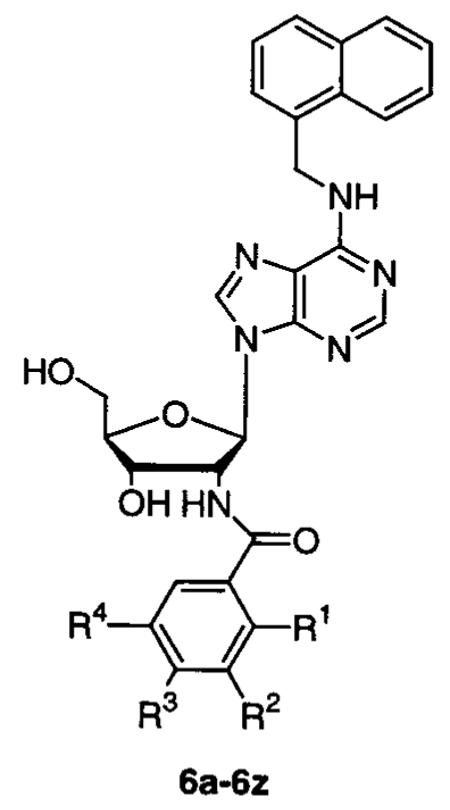
| compd | R1 | R2 | R3 | R4 | IC50 (μM) |
|---|---|---|---|---|---|
| 6a | OCH3 | H | H | H | inactiveb |
| 6b | Cl | H | H | H | inactive |
| 6c | OAc | H | H | H | inactive |
| 6d | OH | H | H | H | inactive |
| 6e | H | OCH3 | H | H | 6 |
| 6f | H | Cl | H | H | 6 |
| 6g | H | OAc | H | H | 50 |
| 6h | H | OH | H | H | 25 |
| 6i | H | H | OCH3 | H | 100 |
| 6j | H | H | Cl | H | 100 |
| 6k | H | H | OAc | H | 100 |
| 6l | H | H | OH | H | 80 |
| 6m | H | OCH3 | H | OCH3 | 2 |
| 6n | H | Cl | H | Cl | 12 |
| 6o | H | OAc | H | OAc | 10 |
| 6p | H | OH | H | OH | 5 |
| 6q | H | OCH3 | OCH3 | H | 4 |
| 6r | H | OAc | OAc | H | 25 |
| 6s | H | OH | OH | H | 18 |
| 6t | OAc | OCH3 | H | H | inactive |
| 6u | OH | OCH3 | H | H | inactive |
| 6v | H | OAc | OCH3 | H | 78 |
| 6w | H | OH | OCH3 | H | 37 |
| 6x | H | OCH3 | OAc | H | 10 |
| 6y | H | OCH3 | OH | H | 10 |
| 6z | H | N(CH3)2 | H | H | 25 |
For compounds with IC50’s ≤ 50 μM, inhibitor concentrations were varied, with at least five different concentrations used to determine the IC50 values. Statistical error limits on the IC50 values have been calculated and amount to 10% or less. For compounds with IC50’s > 50 μM, these values were extrapolated from at least three different concentrations.
Inactive = inactive at 50 μM.
Table 5.
Inhibition of L. mexicana GAPDH by 5′-Deoxy-5′-amido-, N6-(Substituted)-5′-deoxy-5′-amido-, and 2′,5′-Dideoxy-2′,5′-bisamidoadenosine Analoguesa
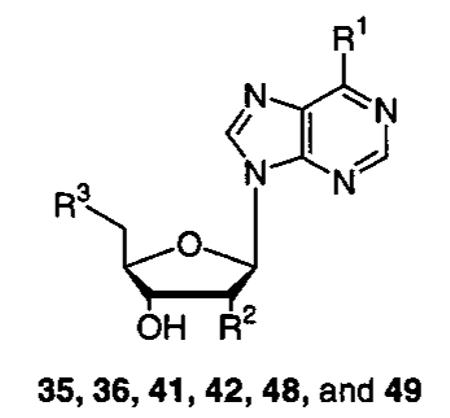
| Compound | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|
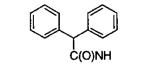
| 35 | NH2 | OH |
|
250 |
| 36 | NH2 | OH |
|
250 |
| 41 |
|
OH |
|
inactive |
| 42 |
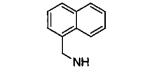
|
OH |
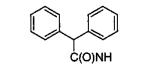
|
inactive |
| 48 | NH2 |
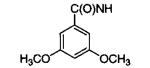
|
|
100 |
| 49 | NH2 |
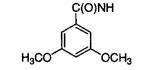
|
|
60 |
Inactive = inactive at 50 μM.
Initially we docked the inhibitor of the crystallographic complex, compound 6m, to verify whether the program could reproduce the experimentally observed binding mode. After docking, the root-mean-square (rms) deviation between the experimental coordinates and the docked model was only 0.70 Å for 42 atoms, confirming the validity of the docking protocol. This rms deviation is similar to the value of 0.76 Å found by the FLO authors for a diverse set of unrelated test cases. Subsequently, all other inhibitors were docked using the same protocol. The calculated binding energies (data not shown) comprise ligand and protein strain energies, van der Waals energy, electrostatic interaction energy (distance-dependent dielectric is 4R∈), and a term accounting for hydrophobic interaction. Because there is no explicit penalty term for desolvation of hydrophilic groups, the calculated interaction energies allow only for qualitative interpretation of ligand binding,41 as clearly demonstrated in a design study of potent angiotensin-converting enzyme (ACE) inhibitors.
Modifications of the C2′ Benzamide
To more thoroughly explore the conformational space of the C2′ selectivity cleft, an SAR of N6-(1-naphthalenemethyl)-2′-deoxy-2′-(benzamido)adenosine analogues was generated. This series of compounds incorporates methoxy, chloro, acetyl, and hydroxy groups at various positions on the benzamide ring.
Of the N6-naphthalenemethyl-2′-benzamido analogues, the most drastic decreases in binding affinity were noted with 2-substituted benzamides that had IC50’s greater than 100 μM (Table 1). In principle, two orientations of the benzamide fragments are possible: syn, which places the ortho substituent adjacent to the amide carbonyl oxygen and pointing into solvent; and anti, which places the ortho substituent opposite the carbonyl oxygen pointing into the selectivity cleft. Modeling experiments suggest the syn conformation is unlikely because the lone pairs of the two oxygen atoms point unfavorably toward each other. Work by Cignitti et al. would support this observation,42 and a Cambridge Structural Database (CSD)43 search for o-methoxy- and o-chlorobenzamides reveals that the 26 available crystal structures all exhibit the anti conformation. In contrast, the anti conformation allows the methoxy and hydroxy substituents to make a 2.7 Å hydrogen bond with the amide nitrogen. Docking of the anti conformation of the ortho-substituted compounds by FLO reveals that the benzamido is pushed out of the selectivity cleft by 1.3–1.8 Å compared to that of 6m because of steric constraints. The end result is that a key hydrogen bond between the amide nitrogen and the carboxylate of Asp-38 is significantly weakened, and the benzamide ring buries less hydrophobic surface in the cleft. This most likely explains the poor activity of these compounds.
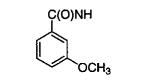
With the exceptions of 6e and 6f, for which affinities had been previously determined,24 the meta- and para-substituted benzamides (Table 1) displayed IC50’s of 25–100 μM against L. mexicana GAPDH. Essentially, these compounds all had the same coordinates as 6m and FLO preferentially oriented the meta substituents to point into the selectivity cleft. Compounds 6m, 6o, and 6p, with a 3,5-disubstituted benzamido substituent, exhibited a 3- to 5-fold increase in affinity for L. mexicana GAPDH when compared to their 3-substituted analogues 6e, 6g, and 6h, while 6n exhibited a 2-fold decrease in activity. The marginal differences in the inhibitor potency suggests that the 3-monosubstituted benzamides can adopt either the anti or syn conformation. Compound 6z was synthesized in an attempt to more completely fill the “selectivity cleft” and to explore alternative conformations of the methoxy group of the lead inhibitor 6e. Modeling suggested that replacing methoxy with dimethylamino might accomplish that goal, but the IC50 of 6z was 4 times worse than 6e.
Modifications of the N6-Naphthalenemethyl
Computational modeling experiments suggested that a hydroxyl group incorporated into the 3-position of the naphthyl ring or a methoxy group in the 3- or 4-position of the naphthyl ring might be well tolerated or might enhance binding affinity of the lead compound 6e (Table 2). The hydrogen bond provided by N6-(3-hydroxy-1-naphthalenemethyl)-2′-deoxy-2′-(3-methoxybenzamido)-adenosine (7e) to the backbone carbonyl of Ala-90 was well tolerated, and the compound displayed an IC50 of 6 μM. The 3-hydroxynaphthalenemethyl substituent was also incorporated into compounds bearing other C2′ benzamides that were well tolerated by L. mexicana GAPDH (3-chlorobenzamide, 3,5-dimethoxybenzamide, and 3,5-dichlorobenzamide), but there were only marginal improvements or, in the case of 7n, a marginal decrease in affinities. Comparable results were also obtained with the incorporation of the methoxy group into the 3- and 4-positions of the naphthalene ring, and docking experiments with FLO place all of these compounds such that they essentially superimpose with 6m. Integration of the methoxy group into the 2-position of the naphthyl ring, yielding 8e, was predicted to drastically decrease binding affinity because the methoxy would clash with either the adenosyl N6 or the residues Ala-90 and Gln-91 of the protein backbone. In fact, FLO displaced the entire molecule partially out of the binding site. The 2-methoxynaphthalenemethyl analogue (8e) was synthesized as proof of principle, and it was inactive against L. mexicana GAPDH when tested up to its solubility limit of 50 μM.
Table 2.
Inhibition of L. mexicana GAPDH by N6-Naphthalenemethyl-2′-deoxy-2′-(3-methoxybenzamido)-adenosine Analogues
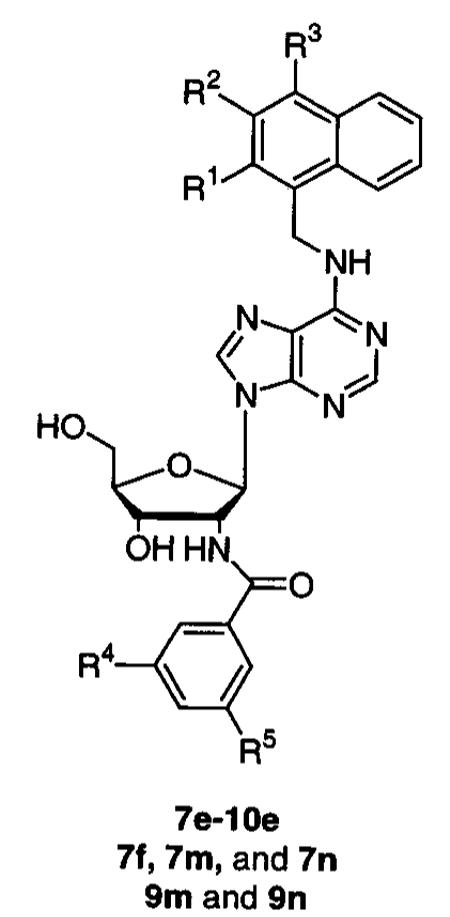
| compd | R1 | R2 | R3 | R4 | R5 | IC50 (μM) |
|---|---|---|---|---|---|---|
| 7e | H | OH | H | OCH3 | H | 6 |
| 7f | H | OH | H | Cl | H | 4 |
| 7m | H | OH | H | OCH3 | OCH3 | 2 |
| 7n | H | OH | H | Cl | Cl | 25 |
| 8e | OCH3 | H | H | OCH3 | H | inactivea |
| 9e | H | OCH3 | H | OCH3 | H | 3 |
| 9m | H | OCH3 | H | OCH3 | OCH3 | 2 |
| 9n | H | OCH3 | H | Cl | Cl | 25 |
| 10e | H | H | OCH3 | OCH3 | H | 6 |
Inactive = inactive at 50 μM.
An alternative connectivity of the naphthalene ring was explored with the incorporation of 2-naphthalenemethyl and 2-naphthalene substituents. Compounds 11e, 11m, and 11n were well tolerated, especially 11e and 11m with IC50’s of 2 μM. FLO docked these compounds with their benzamido adenosine fragments in the same position as in 6m, and the analysis showed that the β-naphthyl ring makes favorable contacts with the guanadinium of Trp-92. Docking of 18m (IC50 = 60 μM), which has a β-naphthyl directly attached to N6, elucidated poor packing of the naphthalene ring with the aliphatic portion of Arg-92 and the side chains of Met-39 and Leu-113, which caused a small rotation of the purine ring and a pivoting of the C1′ of the ribose.
Modifications of the N6 Position
Incorporation of N6 and C2′ amido substituents into one adenosine scaffold reveals that there is a degree of “cross-talk” between these two regions. For example, N6-benzyl-adenosine and N6-(1-naphthalenemethyl)adenosine have IC50’s of 4 and 0.15 mM, respectively.23 Despite the 27-fold difference in binding affinity between these two compounds, incorporation of the C2′-(3-methoxy)benzamide into both of them resulted in compounds showing only a 3-fold affinity difference; the IC50 of N6-benzyl-2′-deoxy-2′-(3-methoxybenzamido)adenosine for L. mexicana GAPDH is 16 μM,23 and the IC50 of N6-(1-naphthalenemethyl)-2′-deoxy-2′-(3-methoxybenzamido)adenosine for L. mexicana GAPDH is 6 μM. A variety of N6-monosubstituted adenosine analogues have been reported that exhibited IC50’s against L. mexicana GAPDH in the range 4–0.15 mM (N6-(2-methylbenzyl)-adenosine, IC50 = 0.7 mM; N6-(3-methylbenzyl)adenosine, IC50 = 0.7 mM; N6-(1,2,3,4-tetrahydro-1-naphthyl)-adenosine, IC50 = 0.36 mM; N6-[(2-(hydroxymethyl)-phenylthio)benzyl]adenosine, IC50 = 0.34 mM; N6-(diphenylmethyl)adenosine, IC50 = 0.24 mM).23 We wished to see if these too would exhibit the same increase in binding affinity as N6-benzyladenosine and N6-(1-naphthalenemethyl)adenosine when a well-tolerated C2′ benzamide, 3,5-dimethoxybenzamido, was incorporated into the molecule.
A combination of benzyl (12m), 2-methylbenzyl (13m), or 3-methylbenzyl (14m) with C2′-(3,5-dimethoxybenzamide) resulted in activities in the 20–25 μM range. The diphenylmethyl (15m), tetrahydronaphthyl (16m), and 2-(2-(hydroxymethyl)phenylthio)benzyl (17m) N6 analogues exhibited IC50’s in the range 55–75 μM. All of these N6 substituents that exhibited some modest binding affinity toward parasite GAPDH as monosubstituted adenosine analogues did exhibit some degree of “cross-talk” when incorporated into analogues bearing the C2′ benzamide group, yielding tighter binding derivatives, but none better than what had previously been obtained with the incorporation of the naphthalene ring, 6m.
N6-(Substituted)-2′-deoxy-2′-(amido)adenosine Combinatorial Library
According to the methodology and criteria of Aronov et al.,44 UC_Select,45 in conjunction with the Daylight version of the Available Chemicals Directory (ACD), was used to select primary amines and carboxylic acids that were devoid of undesirable pharmaceutical and chemical properties (i.e., substituents without phosphates or sulfates). Sybyl’s CONCORD module (version 6.5, 1999; Tripos Associates, St. Louis, MO) was used to generate the three-dimensional coordinates of substituent libraries that were then fused to the adenosyl template. When the coordinates of 6m, from the L. mexicana GAPDH complex, were used, the amine test library (consisting of approximately 460 amines) was fused to the C6 position of 6m after the removal of the naphthalenemethyl substituent. Likewise, the carboxylic acid test library (consisting of approximately 1400 acids) was fused to the C2′ position of 6m after the removal of the dimethoxybenzamide. DOCK 4.0146 was used as previously described44 to evaluate the potential binding energies of the candidate adenosine derivatives to GAPDH, and then a scoring algorithm that takes into account differential solvation of the adenosine substituents was applied.47 The best scoring molecules were visually inspected using Sybyl and Insight II (version 98.0, 1998; Molecular Simulations, Inc., San Diego, CA) software packages.
Our initial hope in venturing into the exploration of the virtual library described above was to discover substituents that were structurally different from naphthylenemethyl and benzoyl and could be accommodated in the N6 and C2′ regions of parasite GAPDH’s. This hope was not realized because many of the top DOCK-ranked amines had benzyl and naphthalenemethyl substituents, or substituents that were structurally similar to groups that had already been used to probe the N6 and C2′ areas. The majority of top DOCK-ranked carboxylic acids were benzoic acid derivatives. Still, we attempted to select a diverse group of ligands while, at the same time, including top DOCK-ranked substituents known to enhance adenosine affinity (Table 4; 3-methylbenzyl-, 2-methylbenzyl-, naphthylenemethyl-, and benzylamine; 4-phenylbenzoic acid,22 3,5-dimethoxybenzoic acid, and 3,5-dichlorobenzoic acid). This led to a selection of 20 amines and 12 carboxylic acids (Table 4) that were used to generate a 240-compound library of N6-(substituted)-2′-deoxy-2′-(amido)adenosine analogues via parallel solution-phase synthesis based on the common intermediate 5. The solvation-corrected DOCK scores for the 1400 carboxylic acids ranged from 10.22 to 87.13 kcal/mol, while those for the 460 amines ranged from 14.17 to 1814.83 kcal/mol. The 12 acids and 20 amines selected for parallel synthesis were chosen from the top 200 scoring compounds of their respective virtual libraries for which the difference in force field scores between the best and the worst compounds was no more than 10 kcal/mol.
Table 4.
Primary Amines and Carboxylic Acids Selected from Docked ACD Answer Seta
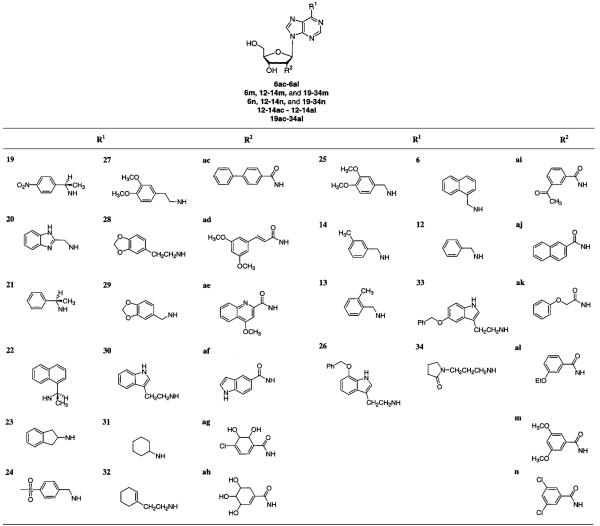
|
Substituents are presented in qualitative order of decreasing DOCK scores. For example, reading down the column, 19 > 20 > 14 > 26
N6-(Substituted)-2′-deoxy-2′-aminoadenosine intermediates were purified by preparative HPLC, and the final acylated library was spot-checked by ESI-MS and HPLC (as described in the Experimental Section) to ensure that the desired final products were present in reasonable yield. The crude library was screened against L. mexicana GAPDH at an approximate concentration of 30 μM to yield a number of low-micromolar inhibitors (Chart 1), but none were superior to 6m. Compounds 6m, 6af, 28m, 29m, and 30m were isolated from their respective crude reaction, characterized, and assayed against L. mexicana GAPDH to give IC50’s of 2, 20, 20, 8, and 20 μM, respectively.
Chart 1.
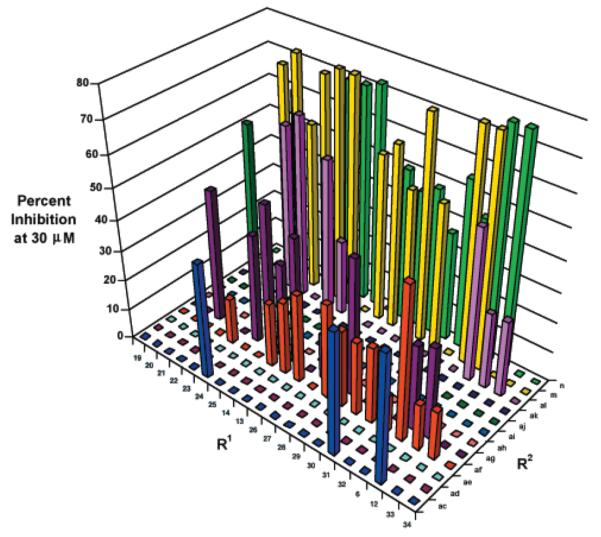
Screening of N6-(Substituted)-2′-deoxy-2′-(amido)Adenosine Combinatorial Library against L. mexicana GAPDH
The relative experimental potencies of the library compounds did not correlate well with their solvation-corrected DOCK scores, but it should be noted that the scores for these 240 compounds were all among the very best scores (within 10 kcal/mol of each other) compared to the much larger range of energies for all compounds analyzed computationally, as noted above. Indeed, 25% of the 240 compounds display an IC50 that was within 4- to 15-fold of our best inhibitor, 6m. Thus, the process of using DOCK provided approximately 40 new, reasonably potent GAPDH inhibitors. It is presumed that many of the poor-scoring compounds would be poor GAPDH inhibitors and thus were not synthesized. To put the DOCK program to a rigorous test, a large number of compounds with poor DOCK scores would need to be synthesized. We refrained from doing this because we believe that we have thoroughly explored 2′,N6-disubstituted adenosines as parasite GAPDH inhibitors.
Modifications of the C5′ of N6-(1-Naphthalenemethyl)adenosine and 2′-Deoxy-2′-(3,5-dimethoxy-benzamido)adenosine (Table 5)
Both 5′deoxy-(5′-cyclohexylacetamido)adenosine (35)23 and 5′deoxy-(5′-diphenylacetamido)adenosine (36)22 inhibit L. mexicana GAPDH with IC50’s of 250 μM. We wished to investigate if the integration of these C5′ substituents with either the N6-naphthalenemethyl or C2′-(3,5-dimethoxybenzamido) substituents would increase binding affinity. Surprisingly, N6-(1-naphthalenemethyl)-5′deoxy-(5′-cyclohexylacetamido)adenosine (41) and N6-(1-naphthalenemethyl)-5′deoxy-(5′-diphenylacetamido)adenosine (42) did not inhibit GAPDH when tested up to their solubility limit of 50 μM. 2′,5′-Dideoxy-2′-(3,5-dimethoxybenzamido)-5′-(cyclohexylacetamido)adenosine (48) and 2′,5′-dideoxy-2′-(3,5-dimethoxybenzamido)-5′-(diphenylacetamido)adenosine (49) did inhibit L. mexicana GAPDH with IC50’s of 100 and 60 μM, respectively. Docking experiments positioned all four C5′-substituted compounds such that their adenosyl moieties and their respective N6 or C2′ substituents were superimposed with the coordinates of 6m. It is unclear why compounds 41 and 42 did not benefit from the additive effect that 48 and 49 did. Solubility limitations hindered a careful examination of the competitive and selective nature of 49.
GAPDH Cross Species Evaluation and Tryp-anosomatid Growth Inhibition Studies (Table 6)
Table 6.
Effect of Adenosine Analogues on Trypanosomatide GAPDH and on Growth of Cultured Parasites
| comp |
L. mexicana IC (μM) |
T. brucei |
T. cruzi |
rabbit muscle IC (μM) |
3T3 fibroblasts ED (μM) |
||
|---|---|---|---|---|---|---|---|
| IC50 (μM) |
ED (μM) |
IC (μM) |
ED (μM) |
||||
| 6e | 6 | 25 | 18, 16 | 12 | 10, 12 | >50 | 40, 50 |
| 6m | 2 | 7 | 17, 17 | 7 | 8, 13 | >50 | 40, 37 |
| 6n | 12 | 100 | 5, 4 | 75 | 7, 7 | >50 | 35, 33 |
| 6o | 10 | 14 | >50, >50 | 12 | >50, >50 | >50 | >50, >50 |
| 6p | 5 | 26 | 50, 35 | 14 | >50, >50 | >50 | >50, >50 |
| 6q | 4 | 47 | 36, 40 | 7 | 22, 28 | >50 | >55, 44 |
| 7e | 6 | 25 | 35, 34 | 12 | >50, >50 | >50 | >50, >50 |
| 7f | 4 | 25 | 16, 20 | 12 | 30, 48 | >50 | >50, >50 |
| 7m | 2 | 8 | 38, 38 | 10 | >50 | >50 | >50, >50 |
| 7n | 25 | 55 | 12, 20 | 40 | 41, 38 | >50 | 43, 50 |
| 9e | 2 | 12 | 31, 32 | 12 | 20, 33 | >50 | >50, >50 |
| 9m | 2 | 5 | 35, 40 | 7 | 35, 30 | >50 | 50, >50 |
| 9n | 25 | 45 | 16, 16 | 40 | 16, 16 | >50 | 32, 38 |
| 10e | 5 | 26 | 10, 12 | 50 | 12, 12 | >50 | 50, 50 |
| 11e | 2 | 24 | 19, 36 | 10 | 10, 17 | >50 | >50, >50 |
| 11m | 2 | 10 | 37, 35 | 5 | 25, 21 | >50 | 50, >50 |
| 11n | 25 | 30 | 16, 17 | 30 | 15, 13 | >50 | 34, 38 |
| pentamidine | a | a | 0.006, 0.004 | a | a | >50 | a |
| benznidazole | a | a | a | a | 0.5, 0.5 | >50 | >50, >50 |
Not determined.
Only the more potent compounds (6e, 6m–6q, 7e, 7f, 7m, 7n, 9e, 9m, 9n, 10e, 11e, 11m, and 11n) were screened against T. brucei and T. cruzi GAPDH (Table 5) because these two enzymes are more difficult to overexpress and purify, and only small quantities were available. In general, the activity of the adenosine analogues decreases in the order L. mexicana, T. cruzi, and T. brucei (Table 6). This was also true for 2′,5′-dideoxy-2′-(3,5-dimethoxybenzamido)-5′-(diphenylacetamido)adenosine, 49, which displayed IC50’s of 60, 82, and 100 μM for the respective parasite GAPDH’s. Although the homology between the adenosyl binding regions is highly conserved across trypanosomatids, the replacement of Ser-40 in the selectivity cleft of L. mexicana with an asparagine in T. cruzi and T. brucei may account for the small differences in affinity. All of these compounds were tested against rabbit muscle GAPDH (mammalian control) and were found to be completely selective for the parasite enzymes up to their solubility limit. Compound 6m was shown to be competitive with NAD+ (data not shown), which is consistent with the crystallographic data.
The above-mentioned inhibitors were tested for the ability to block the growth of bloodstream T. brucei, mammalian stage T. cruzi grown in murine 3T3 fibroblasts, and murine 3T3 fibroblasts (Table 6). All cell culture assays were performed twice with very similar results. Suprisingly, ED50’s for the cultured parasites were within a 2- to 4-fold range of the IC50’s measured against GADPH in vitro, with the exception of 6n. On the basis of the aforementioned computational studies performed by Baker et al.,12-14 a 10- to 100-fold difference between IC50’s and EC50’s might be expected. It is possible that these analogues accumulate in parasites to a higher concentration than present extracellularly. Alternatively, it is possible these compounds act on a target other than or in addition to GAPDH. The ED50’s of the compounds against mammalian cells were consistently higher than the ED50’s against parasites. This is consistent with the selectivity of the adenosine analogues for parasitic GAPDH over mammalian.
Conclusions
New robust routes for the synthesis of N6-(substituted)-2′-deoxy-2′-(amido)adenosine and N6-(substituted)-5′-deoxy-5′-(amido)adenosine analogues and a route to a new class of derivative, 2′,5′-dideoxy-2′,5′-(bisamido)-adenosine, have been developed. The synthetic route from the common intermediate 9-[2′-deoxy-2′-trifluoro-acetamido-3′,5′-O-(diacetyl)]-(1-β-d-ribofuranosyl)-6-chloropurine (5) allows a library of hundreds of N6-(substituted)-2′-deoxy-2′-(amido)andenosines to be readily synthesized via parallel solution-phase synthesis. Utilizing structure-based design methodology, we synthesized a number of inhibitors that are 7000- to 25000-fold more active (across trypanosmatid species) than adenosine. Additionally, the application of rational design and combinatorial chemistry led to a number of low-micromolar inhibitors of L. mexicana GAPDH. The most potent GAPDH inhibitors block the growth of bloodstream T. brucei and the intracellular stage T. cruzi in the low-micromolar range.
Experimental Section
General Synthetic Procedures
CH2Cl2 was freshly distilled over CaH2 under argon. 6-Chloropurine-9-riboside and anhydrous DMF were purchased from Aldrich Chemical Co. Ltd. 4-Sulfonamidobutyryl-AM resin (loading, 0.4–1.2 mmol/g; polymer matrix, copoly(styrene–1% DVB); 200–400 mesh) was purchased from Novabiochem. 1H NMR spectra were recorded on a Bruker AF 300 MHz or AM 500 MHz spectrometer in DMSO-d6. Electrospray ionization mass spectra (EISMS) were obtained on a Bruker Esquire LC ion trap or Kratos Profile HV4 mass spectrometer. Reactions were monitored by TLC on precoated silica gel 60 F254 glass plates (EM Science) with CH2Cl2/EtOH (9:1) or CHCl3/MeOH (80:20) as eluent and visualized at 254 nm. Medium-pressure flash column chromatography was carried out on silica gel 60 (230–400 mesh; EM Science) with CHCl3/MeOH (98:2 to 80:20) eluent. Preparative HPLC was performed on a C18 column (Vydac 218TP1010) using one of the following elution gradients: (method A) linear gradient of 10 mM triethylammonium acetate (aq, pH = 7.0) from 0 to 100% ACN over 60 min at 4.0 mL/min; (method B) linear gradient of 5% HCOOH (aq) from 0 to 100% MeOH over 60 min at 4.0 mL/min; (method C) isocratic 10 mM triethylammonium acetate (aq, pH = 7.0) of 0% over 15 min, then a linear gradient of 10 mM triethyl-ammonium acetate (aq, pH = 7.0) from 0 to 25% ACN over 45 min at 4.0 mL/min; (method D) same as method B except a flow rate of 7.0 mL/min on a C18 column (Vydac 218TP1022) was used. The purity of all compounds was verified by HPLC with methods A and B on a C18 column (Vydac 218TP52) at 1.0 mL/min.
Synthesis of 2′-Amino-2′-deoxyadenosine (1)
Compound 1 was synthesized via 2′-azido-2′-deoxy-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)adenosine as per the reliable methodologies of Robins et al.26 and Van Calenberg et al.20 Because minor modifications to the procedure were made that resulted in slightly improved yields, we have included the details of the preparation in Supporting Information.
Synthesis of 2′-Amino-2′-deoxyinosine (2)
Starting with 1 (4.0 g, 15.0 mmol), the procedure of Kotra et al..31 was used to give 4.0 g of 2 (99.0%). 1H NMR: δ 3.54 and 3.63 (2 × m, 2H, C5′-Ha,b), 3.95 (m, 2H, C3′-H and C4′-H), 4.18 (m, 1H, C2′-H), 5.64 (d, 1H, C1′-H), 8.10 (d, 1H, C2-H), 8.27 (s, 1H, C8-H), 12.51 (s, 1H, N1-H). ESI-MS: m/z 268.1 (M + H+)+.
Synthesis of 2′-Deoxy-2′-trifluoroacetamidoinosine (3)
Ethyl trifluoroacetate (5.36 mL, 45.0 mmol) was added to a solution of 2 (4.0 g, 15.0 mmol) in anhydrous DMF (50 mL), and the reaction was heated at 60 °C under argon for 3 h. The reaction was evaporated via rotary evaporation at 50 °C and then further dried in vacuo for 24 h. The resulting amorphous solid was of sufficient purity to use in the subsequent reaction. For characterization, a small sample was purified via flash chromatography. 1H NMR: δ 3.57 and 3.68 (2 × m, 2H, C5′-Ha,b), 4.03 (m, 1H, C4′-H), 4.34 (m, 1H, C3′-H), 5.02 (m, 1H, C2′-H), 5.19 (br s, 1H, C5′-OH), 5.92 (br s, 1H, C3′-OH), 6.18 (d, 1H, C1′-H), 8.10 and 8.27 (2 × s, 2H, C2-H and C8-H), 9.65 (d, 1H, C2′–NH), 12.54 (s, 1H, N1-H). ESI-MS: m/z 364.1 (M + H+)+.
Synthesis of 2′-Deoxy-2′-trifluoroacetamido-3′,5′-O-(diacetyl)inosine (4)
To a mixture of 3 (4.5 g, ~12.4 mmol) in anhydrous ACN (250 mL) was added sequentially Ac2O (11.7 mL, 124 mmol), anhydrous Et3N (13.8 mL, 99.2 mmol), and DMAP (128 mg, 1.0 mmol) at ambient temperature under argon. After the mixture was stirred for 1 h at ambient temperature, MeOH (15.0 mL) was added and then the reaction was evaporated via rotary evaporation at 40 °C. The resulting residue was partitioned between EtOAc (250 mL) and H2O (250 mL), washed with H2O (2 × 250 mL), washed with saturated NaCl (aq, 2 × 250 mL), dried over MgSO4, filtered, and then evaporated. The crude material was purified via flash chromatography to yield 6.2 g of 4 (93% based on 2 as the starting material). 1H NMR: δ 2.04 (s, 3H, C5′-OAc), 2.08 (s, 3H, C3′-OAc), 4.23 (m, 1H, C5′-Ha), 4.38 (m, 2H, C4′-H and C5′-Hb), 5.37 (m, 1H, C3′-H), 5.44 (m, 1H, C2′-H), 6.23 (d, 1H, C1′-H), 8.10 and 8.34 (2 × s, 1H, C2-H and C8-H), 10.10 (br s, 1H, C2′-NH), 12.50 (s, 1H, N1-H). ESI-MS: m/z 448.1 (M + H+)+.
Synthesis of 9-[2′-Deoxy-2′-trifluoroacetamido-3′,5′-O-(diacetyl)]-(1-β-d-ribofuranosyl)-6-chloropurine (5)
With minor modifications, the methodology of Matsuda et al.32 was employed. To a rapidly stirring mixture of 4 (6.0 g, 13.4 mmol), Et4NCl (8.9 g, 53.7 mmol), and dry N,N-dimethylaniline (1.7 mL, 13.4 mmol) in anhydrous ACN (30 mL) was added POCl3 (3.1 mL, 33.6 mmol) at ambient temperature under argon. The reaction was placed in a preheated oil bath and refluxed at 100 °C for 10 min. The reaction was then evaporated via rotary evaporation at 40 °C, and the resulting green foam was dissolved in CH2Cl2 (100 mL) and then stirred vigorously over crushed ice for 15 min. The layers were separated, and the aqueous phase was extracted with CH2Cl2 (2 × 100 mL). The combined organic phase was then washed with H2O (2 × 100 mL), washed with 5% NaHCO3 (aq, pH ~7, 2 × 100 mL), dried over MgSO4, filtered, and evaporated to dryness. The resulting yellow foam was then purified via flash chromatography to give 5.7 g of 5 as a white foam (92.0%). 1H NMR: δ 2.15 (s, 3H, C5′-OAc), 2.20 (s, 3H, C3′-OAc), 4.28 and 4.41 (2 × m, 2H, C5′-Ha,b), 4.44 (m, 1H, C4′-H), 5.50 (m, 2H, C2′-H and C3′-H), 6.42 (d, 1H, C1′-H), 8.87 and 8.95 (2 × s, 1H, C2-H and C8-H), 10.09 (d, 1H, C2′-NH). ESI-MS: m/z 448.1 (M + H+)+. ESI-MS: m/z 466.1 (M + H+)+.
General Procedure for the Preparation of N6-(Substituted)-2′-Deoxy-2′-aminoadenosine Analogues (6–18)
To a solution of 5 (10.0 mg, 2.2 × 10−2 mmol) in EtOH (500 μL), in a 4.0 mL screw cap vial with Teflon septum, was added the appropriate amine or amine hydrochloride (6.4 × 10−2 mmol) and then Et3N (9.0 μL, 6.4 × 10−2 mmol for free amines; 18.0 μL, 12.8 × 10−2 mmol for amine hydrochlorides). The reaction was heated at 60 °C for 18 h and then cooled to ambient temperature. Concentrated NH4OH (aq, 500 μL) was then added, and the reaction was heated at 80 °C for 24 h. Without further workup, the reaction was purified via preparative HPLC using method A. The desired fraction was concentrated and then lyophilized to obtain the desired products in 85–95% yield.
N6-(1-Naphthalenemethyl)-2′-deoxy-2′-aminoadenosine (6)
1H NMR: δ 3.53 and 3.64 (2 × m, 2H, C5′-Ha,b), 3.98 (m, 3H, C2′-H, C3′-H, and C4′-H), 5.19 (br s, 3H, N6-CH2 and C5′-OH), 5.50 (br s, 1H, C3′-OH), 5.68 (d, 1H, C1′-H), 7.42 (m, 2H, naphthyl-H), 7.55 (m, 2H, naphthyl-H), 7.81 (d, 1H, naphthyl-H), 7.94 (d, 1H, naphthyl-H), 8.15 and 8.36 (2 × s, 2H, C2-H and C8-H), 8.19 (d, 1H, naphthyl-H), 8.50 (br s, 1H, N6-H). ESI-MS (MeOH): m/z 407.2 (M + H)+.
General Procedure for the Preparation of N6-(Substituted)-2′-deoxy-2′-(amido)adenosine Analogues
EDCI (3.7 × 10−2 mmol) was added to a solution of the appropriate carboxylic acid (3.7 × 10−2 mmol) in anhydrous CH2Cl2 (100 μL), and the solution was stirred under argon for 30 min. A total of 100 μL of this solution was added to a solution of N6-(substituted)-2′-deoxy-2′-aminoadenosine (1.2 × 10−2 mmol) in anhydrous DMF (100 μL), and the mixture was stirred for 30 min. Without further workup, the reaction mixture was purified via preparative HPLC using method B to obtain the desired products in 85–95% yields after removal of solvents. For reactions involving acetylated hydroxybenzoic acids, the solutions were evaporated to dryness. The resulting solid was dissolved in MeOH (250 μL) and cooled to 0 °C, and then NH3-(g) was bubbled into the solution. The reaction mixture was stirred at ambient temperature for 15 min and then, without further workup, was purified by preparative HPLC using method B.
N6-(1-Naphthalenemethyl)-2′-deoxy-2′-(3,5-dimethoxybenzamido)adenosine (6m)
1H NMR: δ 3.61 and 3.72 (2 × m, 2H, C5′-Ha,b), 3.75 (s, 6H, OCH3), 4.10 (m, 1H, C4′-H), 4.33 (m, 1H, C3′-H), 5.16 (br s, 2H, N6-CH2), 5.34 (m, 1H, C2′-H), 5.60 (br s, 1H, C5′-OH), 5.79 (br s, 1H, C3′-OH), 6.23 (d, 1H, C1′-H), 6.63 (s, 1H, Ph-H), 6.98 (s, 2H, Ph-H), 7.39 (m, 2H, naphthyl-H), 7.52 (m, 2H, naphthyl-H), 7.80 (d, 1H, naphthyl-H), 7.94 (d, 1H, naphthyl-H), 8.21 and 8.31 (2 × s, 2H, C2-H and C8-H), 8.22 (d, 1H, naphthyl-H), 8.42 (d, 1H, C2′-NH), 8.55 (br s, 1H, N6-H). ESI-MS: (MeOH) m/z 571.2 (M + H)+.
Combinatorial Library
The 240 N6-(substituted)-2′-deoxy-2′-(amido)adenosine derivatives were synthesized individually via solution-phase parallel synthesis. Starting with 5 (50 mg for each amine reaction, 0.11 μmol), the above procedure for the synthesis of N6-substituted-2′-deoxy-2′-aminoadenosine analogues was followed using the set of 20 amines shown in Table 4. These intermediates were purified by preparative HPLC using method D and characterized by NMR and ESI-MS (see Supporting Information) to yield the desired intermediates in 85–95% yield. Acylation of the 2′-aminoadenosine derivatives (approximately 4.4 μmol each) was accomplished as described above, and the reaction progress was monitored by TLC. Two members from each carboxylic acid vector were chosen (such that each amine vector was represented at least once) from the 20 × 12 matrix and were analyzed by ESI-MS and HPLC. For carboxylic acid vectors ac, ad, ai, aj, ak, al, m, and n (Table 4), acylation reactions gave 80–95% conversion to the desired products while ae and af gave approximately 50% and ag and ah gave approximately 25%. Upon completion of the reaction, the solutions were diluted with MeOH and then evaporated to dryness in a Speed-Vac. The crude material was dissolved in DMSO and used for in vitro enzyme inhibition analysis.
6-Chloropurine-5′-deoxy-(5′-phthalamido)riboside (38)
To a solution of 6-chloropurine-9-riboside (37, 1.0 g, 3.5 mmol), phthalimide (615 mg, 4.2 mmol), and triphenylphosphine (1.8 g, 7.0 mmol) in anhydrous THF (10 mL) was added DIAD (1.3 mL, 7.0 mmol) at ambient temperature under argon. After the mixture was stirred for 2 h, MeOH (10 mL) was added to the reaction mixture that was then stirred for an additional 15 min. Without further workup, the reaction was absorbed onto silica gel and purified via flash chromatography to give 1.2 g of 38 (86.0%). 1H NMR: δ 3.98 (m, 2H, C5′-H), 4.18 (m, 1H, C4′-H), 4.25 (m, 1H, C3′-H), 4.80 (m, 1H, C2′-H), 5.41 (d, 1H, OH), 5.62 (d, 1H, OH), 6.01 (s, 1H, C1′-H), 7.82 (s, 4H, phthalimide-H), 8.59 and 8.96 (2 × s, 2H, C2-H and C8-H). ESI-MS: m/z 416.1 (M + H+)+.
N6-(1-Naphthalenemethyl)-5′deoxy-5′-aminoadenosine (40)
To a solution of 38 (100 mg, 2.4 × 10−1 mmol) and Et3N (101 μL, 7.2 × 10−1 mmol) in EtOH (5 mL) was added 1-naphthalenemethylamine (113 μL, 7.2 × 10−1 mmol). After the solution was refluxed for 18 h, conversion to 39 was complete, as determined by TLC and ESI-MS, and the sample was used without further workup. Hydrazine hydrate (50% aq, 82 μL, 2.4 mmol) was then added to the crude reaction mixture and was refluxed for 18 h. Without further workup the reaction was purified via preparative HPLC method A to give 91 mg of 40 (93.6%). 1H NMR: δ 2.79 and 2.87 (2 × m, 2H, C5′-Ha,b), 3.92 (m, 1H, C4′-H), 4.15 (m, 1H, C3′-H), 4.69 (m, 1H, C2′-H), 5.17 (br s, 3H, N6-CH2), 5.87 (d, 1H, C1′-H), 7.41 (m, 2H, naphthyl-H), 7.55 (m, 2H, naphthyl-H), 7.80 (d, 1H, naphthyl-H), 7.94 (d, 1H, naphthyl-H), 8.17 and 8.41 (2 × s, 2H, C2-H and C8-H), 8.22 (d, 1H, naphthyl-H), 8.44 (br s, 1H, N6-H). ESI-MS (MeOH): m/z 407.2 (M + H)+.
General Procedure for the Preparation of N6-(1-Naphthalenemethyl)-5′-deoxy-5′-(amido)adenosine Analogues. Method A
EDCI (3.7 × 10−2 mmol) was added to a solution of the appropriate carboxylic acid (3.7 × 10−2 mmol) in anhydrous CH2Cl2 (100 μL), and the mixture was stirred under argon for 30 min. A total of 100 μL of this solution was added to a solution of 40 (5.0 mg, 1.2 × 10−2 mmol) in anhydrous DMF (100 μL), and the reaction mixture was stirred for 30 min. Without further workup, the reaction mixture was purified via preparative HPLC using method B to obtain the desired products 41 and 42 in 75 and 90% yields, respectively.
Method B
A mixture of the appropriately acylated safety-catch resin (6.0 × 10−2 mmol) and 40 (5.0 mg, 1.2 × 10−2 mmol) was stirred under argon in anhydrous DMF (500 μL) for 18 h at ambient temperature. The product was filtered from the resin, and the resin was washed with DMF (500 μL), CH2Cl2/MeOH (1:1, 2 × 500 μL), and CH2Cl2 (2 × 500 μL). The filtrate and washings were combined and then evaporated to dryness via rotary evaporation at 40 °C. The desired products were obtained in >95% yield and purity as determined by HPLC. 1H NMR and ESI-MS of samples prepared via methods A and B were identical.
N6-(1-Naphthalenemethyl)-5′deoxy-(5′-cyclohexylacetamido)adenosine (41)
1H NMR: δ 0.85–1.65 (band, 11H, cyclohexyl-H), 1.99 (d, 2H, C5′-NHC(O)CH2), 3.35 and 3.45 (2 × m, 2H, C5′-Ha,b), 3.95 (m, 1H, C4′-H), 4.01 (m, 1H, C3′-H), 4.69 (m, 1H, C2′-H), 5.18 (br s, 2H, N6-CH2), 5.28 (br s, 1H, OH), 5.49 (br s, 1H, OH), 5.85 (d, 1H, C1′-H), 7.41 (m, 2H, naphthyl-H), 7.55 (m, 2H, naphthyl-H), 7.80 (d, 1H, naphthyl-H), 7.94 (d, 1H, naphthyl-H), 8.12 (m, 1H, C5′-NH), 8.20 and 8.35 (2 × s, 2H, C2-H and C8-H), 8.22 (d, 1H, naphthyl-H), 8.44 (br s, 1H, N6-H). ESI-MS: m/z 531.3 (M + H+)+.
N6-(1-Naphthalenemethyl)-5′deoxy-(5′-diphenylacetamido)adenosine (42)
1H NMR: δ 3.42 and 3.53 (2 × m, 2H, C5′-Ha,b), 3.95 (m, 1H, C4′-H), 4.01 (m, 1H, C3′-H), 4.64 (m, 1H, C2′-H), 4.98 (s, 1H, C5′-NHC(O)CH), 5.18 (br s, 1H, N6-CH), 5.28 (br s, 1H, OH), 5.46 (br s, 1H, OH), 5.85 (d, 1H, C1′-H), 7.17–7.32 (band, 10H, Ph-H), 7.41 (m, 2H, naphthyl- H), 7.55 (m, 2H, naphthyl-H), 7.80 (d, 1H, naphthyl-H), 7.94 (d, 1H, naphthyl-H), 8.20 and 8.35 (2 × s, 2H, C2-H and C8-H), 8.21 (d, 1H, naphthyl-H), 8.44 (br s, 1H, N6-H), 8.57 (m, 1H, C5′-NH). ESI-MS: m/z 601.3 (M + H+)+.
2′-Deoxy-2′-trifluoroacetamidoadenosine (43)
Ethyl trifluoroacetate (135 μL, 1.1 mmol) was added to a solution of 1 (100 mg, 3.7 × 10−1 mmol) in anhydrous DMF (1.25 mL), and the reaction mixture was heated at 60 °C under argon for 3 h. The reaction mixture was evaporated via rotary evaporation at 50 °C and then further dried in vacuo for 24 h. The resulting amorphous solid was purified via flash chromatography to yield 130 mg of 43 (95.5%). 1H NMR: δ 3.57 and 3.68 (2 × m, 2H, C5′-Ha,b), 4.05 (m, 1H, C4′-H), 4.35 (m, 1H, C3′-H), 5.12 (m, 1H, C2′-H), 5.57 (m, 1H, C5′-OH), 5.84 (d, 1H, C3′-OH), 6.21 (d, 1H, C1′-H), 7.41 (br s, 2H, N6-H), 8.13 and 8.24 (2 × s, 2H, C2-H and C8-H), 9.60 (br s, 1H, C2′-NH). ESI-MS: m/z 363.1 (M + H+)+.
2′,5′-Dideoxy-2′-(trifluoroacetamido)-5′-phthalimido-adenosine (44)
To a solution of 43 (100 mg, 2.8 × 10−1 mmol), phthalimide (49.4 mg, 3.4 × 10−1 mmol), and triphenylphosphine (144 mg, 5.5 × 10−1 mmol) in anhydrous THF (1.0 mL) was added DIAD (80.5 μL, 5.5 × 10−1 mmol) at ambient temperature under argon. After the solution was stirred for 2 h, MeOH was added to the reaction mixture (1.0 mL), which was then stirred for an additional 15 min. Without further workup, the reaction was absorbed onto silica gel and purified via flash chromatography to give 111 mg of 44 (81.5%). 1H NMR: δ 3.85 and 4.02 (2 × dd, 2H, C5′-Ha,b), 4.22 (m, 1H, C4′-H), 4.49 (m, 1H, C3′-H), 5.28 (t, 1H, C2′-H), 5.91 (br s, 1H, C3′-OH), 6.12 (d, 1H, C1′-H), 7.31 (br s, 2H, N6-H), 7.84 (s, 4H, phthalimide-H), 7.91 and 8.34 (2 × s, 2H, C2-H and C8-H), 9.60 (d, 1H, C2′-NH). ESI-MS: m/z 492.1 (M + H+)+.
2′,5′-Dideoxy-2′,5′-(bisamino)adenosine (45)
A solution of 44 (100 mg, 2.0 × 10−1 mmol) and hydrazine hydrate (50% aq, 94 μL, 2.8 mmol) in EtOH (5.0 mL) was refluxed for 18 h. Without any further workup, the crude reaction mixture was purified via preparative HPLC method C to yield 48 mg of 45 (88.9%). 1H NMR: δ 2.88 (m, 2H, C5′-H), 3.95 (m, 1H, C4′-H), 4.02 (m, 2H, C2′-H and C3′-H), 5.65 (d, 1H, C1′-H), 7.23 (br s, 2H, N6-H), 8.12 and 8.32 (2 × s, 2H, C2-H and C8-H). ESI-MS: m/z 266.2 (M + H+)+.
General Procedure for the Preparation of 2′,5′-Dideoxy-2′-amino-5′-(amido)adenosine Analogues
See Method B of General Procedure for the Preparation of N6-(1-Naphthalenemethyl)-5′-deoxy-5′-(Amido)adenosine Analogues.
2′,5′-Dideoxy-2′-amino-5′-(cyclohexylacetamido)adenosine (46)
1H NMR: δ 0.87–1.65 (band, 11H, cyclohexyl-H), 2.04 (d, 2H, C5′-NHC(O)CH2), 3.38 and 3.51 (2 × m, 2H, C5′-Ha,b), 3.87 (m, 1H, C4′-H), 3.96 (m, 2H, C2′-H and C3′-H), 5.61 (d, 1H, C1′-H), 7.33 (br s, 2H, N6-H), 8.14 and 8.29 (2 × s, 3H, C2-H, C8-H, and C5′-NH). ESI-MS: m/z 390.2 (M + H+)+.
2′,5′-Dideoxy-2′-amino-5′-(diphenylacetamido)adenosine (47)
1H NMR: δ 3.38 and 3.51 (2 × m, 2H, C5′-Ha,b), 3.82 (m, 2H, C3′-H and C4′-H), 3.89 (m, 1H, C2′-H), 5.02 (s, 1H, N6-CH), 5.61 (d, 1H, C1′-H), 7.18−7.32 (band, 12H, N6-H and Ph-H), 8.16 and 8.25 (2 × s, 2H, C2-H and C8-H), 8.75 (m, 1H, C5′-NH). ESI-MS: m/z 460.2 (M + H+)+.
General Procedure for the Preparation of 2′,5′-Dideoxy-2′-amido-5′-(amido)adenosine Analogues
See Method A of General Procedure for the Preparation of N6-(1-Naphthalenemethyl)-5′-deoxy-5′-(amido)adenosine Analogues. The desired products 48 and 49 were obtained in 86 and 92% yields, respectively.
Method B
As per the methodology of Link et al.,33 a mixture of the activated 3,5-dimethoxybenzylsulfonamide safety-catch resin (6.0 × 10−2 mmol) and 46 or 47 (1.2 × 10−2 mmol) was stirred under argon in anhydrous DMF (500 μL) for 6 h at 60 °C. The product was filtered from the resin, and the resin was washed with DMF (500 μL), CH2Cl2/MeOH (1:1, 2 × 500 μL), and CH2Cl2 (2 × 500 μL). The filtrate and washings were combined and then evaporated to dryness via rotary evaporation at 40 °C to give 48 and 49 in >90% yield and in >95% purity as determined by HPLC. 1H NMR and ESI-MS of samples prepared via methods A and B were identical.
2′,5′-Dideoxy-2′-(3,5-dimethoxybenzamido)-5′-(cyclohexylacetamido)adenosine (48)
1H NMR: δ 0.89–1.65 (band, 11H, cyclohexyl-H), 2.04 (d, 2H, C5′-NHC(O)CH2), 3.58 (m, 2H, C5′-H), 3.78 (s, 6H, OCH3), 4.10 (m, 1H, C4′-H), 4.19 (m, 1H, C3′-H), 5.34 (m, 1H, C2′-H), 5.92 (br s, 1H, OH), 6.17 (d, 1H, C1′-H), 6.64 (s, 1H, Ph-H), 6.98 (s, 2H, Ph-H), 7.39 (br s, 2H, N6-H), 8.19 and 8.25 (2 × s, 2H, C2-H and C8-H), 8.39 (m, 1H, C5′-NH), 8.42 (d, 1H, C2′-NH). ESI-MS: m/z 554.3 (M + H+)+.
2′,5′-Dideoxy-2′-(3″,5″-dimethoxybenzamido)-5′-(di-phenylacetamido)adenosine (49)
1H NMR: δ 3.58 (m, 2H, C5′-H), 3.75 (s, 6H, OCH3), 4.07 (m, 1H, C4′-H), 4.23 (m, 2H, C3′-H), 5.03 (s, 1H, C5′-NHC(O)CH), 5.37 (m, 1H, C2′-H), 5.92 (br s, 1H, OH), 6.14 (d, 1H, C1′-H), 6.62 (s, 1H, Ph-H), 6.96 (s, 2H, Ph-H), 7.19–7.33 (band, 12H, N6-H and Ph-H), 8.09 and 8.26 (2 × s, 2H, C2-H and C8-H), 8.43 (d, 1H, C2′-NH), 8.73 (m, 1H, C5′-NH). ESI-MS: m/z 624.3 (M + H+)+.
Preparation of Acetylated Hydroxybenzoic Acids
The method of White et al. was employed.39
Preparation of Cyclohexylmethyl- and Diphenyl-methylsulfonamidobutyryl-AM resin
These resins were prepared from sulfonamidobutyryl-AM resin as per the methodology of Kenner et al.29 and Backes et al.30
General Procedure for the Synthesis of Naphthalene-methylamines from Naphthylnitriles
LiAlH4 (2.7 mmol) was added to naphthylnitrile (2.7 mmol) in anhydrous THF (10.0 mL) at 0 °C, and the mixture was stirred for 1 h. The reaction was quenched with MeOH and then washed with H2O (2 × 10 mL). The organic layer was then extracted with 1 N HCl (2 × 10 mL), and the combined extracts were left to stand for 24 h at 4 °C to yield crystals of the desired amine hydrochloride in 60–80% yield.
2-Naphthalenemethylamine Hydrochloride (49)
1H NMR: δ 4.18 (br s, 1H, C2–CH2), 7.55 (m, 2H), 7.62 (d, 1H), 7.89–7.99 (band, 4H), 8.46 (br s, 3H, NH3). ESI-MS: m/z 158.1 (M + H+)+.
1-(Aminomethyl)-4-methoxynaphthalene Hydrochloride (53)
1H NMR: δ 3.98 (s, 3H, OCH3), 4.41 (br s, 2H, C1–CH2), 7.01 (d, 1H), 7.57 (m, 2H), 7.65 (t, 1H), 8.09 (d, 1H), 8.23 (d, 1H), 8.41 (br s, 3H, NH3). ESI-MS: m/z 188.1 (M + H+)+.
1-(Aminomethyl)-3-methoxynaphthalene Hydrochloride (55)
3-Methoxy-1-naphthonitrile was synthesized as described by DeCosta et al.36 1H NMR: δ 3.89 (s, 3H, OCH3), 4.49 (d, 2H, C1-CH2), 7.32 (s, 1H), 7.38 (s, 1H), 7.45 (t, 1H), 7.53 (t, 1H), 7.89 (d, 1H), 8.02 (d, 1H), 8.42 (br s, 3H, NH3). ESI-MS: m/z 188.1 (M + H+)+.
1-(Aminomethyl)-2-methoxynaphthalene Hydrochloride (51)
To a solution of 2-methoxynaphthalene (500 mg, 3.2 mmol) in EtOH (2.5 mL) was added concentrated HCl (125 μL) and 2-chloro-N-(hydroxymethyl)acetamide (395 mg, 3.2 mmol). The reaction mixture was refluxed for 30 min and then was cooled to ambient temperature. Additional concentrated HCl (1.25 mL) was added, and the reaction mixture was refluxed for 1.5 h and then was cooled to ambient temperature. The desired product crystallized upon cooling to give 375 mg of 34 (53.0%). 1H NMR: δ 4.06 (s, 3H, OCH3), 4.38 (d, 2H, C1–CH2), 7.34–7.60 (band, 3H), 7.92 (d, 2H), 8.06 (d, 1H), 8.11 (d, 1H), 8.38 (br s, 3H, NH3). ESI-MS: m/z 188.1 (M + H+)+.
3-Hydroxy-1-naphthalenemethylamine Hydrobromide (56)
To a solution of 41 (200 mg, 0.89 mmol) in CH2Cl2 (2.0 mL) was added BBr3 (422 μL, 4.5 mmol) at ambient temperature. The reaction was refluxed for 1 h, cooled to ambient temperature, and then evaporated to dryness via rotary evaporation at 40 °C. The resulting solid was partitioned between CH2Cl2 (25 mL) and H2O (25 mL). The organic layer was washed with H2O (3 × 25 mL), and the aqueous layers were combined. The aqueous layer was reduced in volume to approximately 25 mL and was left to stand at 4 °C for 24 h to yield crystals of 56 (71%). 1H NMR: δ 4.46 (d, 2H, C1–CH2), 7.18 (s, 1H), 7.20 (s, 1H), 7.37 (t, 1H), 7.46 (t, 1H), 7.75 (d, 1H), 7.98 (d, 1H), 8.28 (br s, 3H, NH3), 9.97 (s, 1H, OH). ESI-MS: m/z 174.1 (M + H+)+.
Inhibition Studies
Assays were performed at 23 °C in a final volume of 0.50 mL of a 0.10 M triethanolamine/HCl (pH = 7.5) assay buffer while the absorption at 340 nm was monitored using a quartz cuvette. Activity for GAPDH was measured in the direction of NADH formation. Fresh glyceraldehyde-3-phosphate was prepared from its diethylacetal [di-(cyclohexylammonium) salt] as described by the manufacturer (Sigma). All compounds tested were dissolved in DMSO-d6 (the combinatorial library was dissolved in DMSO), and the final concentration of DMSO-d6 in the assay was kept at 5%. The reaction was initiated by addition of enzyme, and control reactions were run in the absence of inhibitors but in the presence of 5% DMSO-d6. Remaining activity was calculated as a percent of control using the initial velocities measured from 0 to 2 min. For compounds with IC50’s less than or equal to 50 μM, inhibitor concentrations in the cuvette were varied, with at least five different concentrations used to determine the IC50 values. Statistical error limits on the IC50 values have been calculated and amount to 10% or less. For compounds with IC50’s greater than 50 μM, these values were extrapolated from at least three different concentrations. Final concentration of the components in the assay buffer was as follows: 5.0 mM MgSO4, 1.0 mM EDTA, 12.8 mM KH2PO4, 1.0 mM DTT, 1.0 mM NaN3, 0.45 mM NAD+ (Sigma), and 1.87 mM glyceraldehyde-3-phosphate. Trypanosome and Leishmania GAPDH were obtained as described previously.24
Parasite Cultures and Drug Screening Assays
T. brucei (bloodstream form strain 427 from K. Stuart, Seattle Biomedical Research Institute, Seattle, WA) was cultured in HMI-9 medium containing 10% fetal bovine serum, penicillin, and streptomycin at 37 °C with 5% CO2.48 Drug sensitivity of the T. brucei strain was determined in 96-well microtiter plates in triplicate with an initial inoculum of 5 × 103 trypomastigotes per well. Drugs were added in serial dilutions for a final volume of 200 μL/well. Parasite growth was quantified at 48 h by the addition of Alamar Blue (Alamar Biosciences, Sacramento, CA).49 Alamar Blue quantification corresponded with microscopic counts determined with a hemacytometer. The Tulahuen strain of T. cruzi was provided by S. Reed (Infectious Diseases Research Institute, Seattle, WA). This strain was stably transfected with the E. coli β-galactosidase gene (lacZ) so that intracellular growth could be monitored with a colorimetric assay. The mammalian stages were grown in coculture with mouse 3T3 fibroblasts in RPMI 1640 containing 10% fetal bovine serum, penicllin, and streptomycin at 37 °C with 5% CO2. Drug sensitivity studies with mammalian stage T. cruzi were carried out in triplicate using the β-galactosidase assay as described.50
Supplementary Material
Table 3.
Inhibition of L. mexicana GAPDH by N6-(Subsutituted)-2′-deoxy-2′-(benzamido)adenosine Analogues
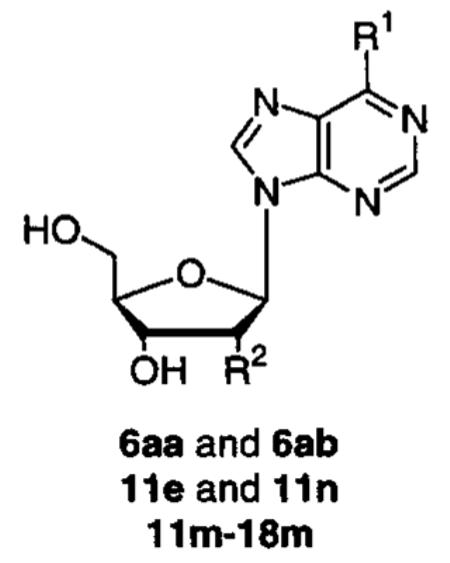
| Compound | R1 | R2 | IC50 (μM) |
|---|---|---|---|
| 6aa |
|
|
inactive |
| 6ab |
|
|
inactive |
| 11e |
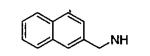
|
|
2 |
| 11m |
|
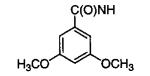
|
2 |
| 11n |
|
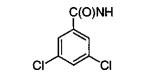
|
25 |
| 12m |
|
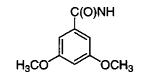
|
20 |
| 13m |
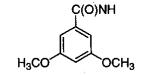
|
|
25 |
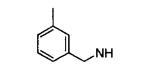
| 14m |
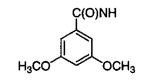
|
|
25 |
| 15m |
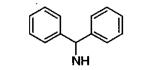
|
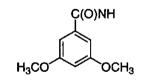
|
55 |
| 16m |
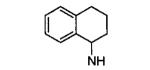
|
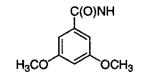
|
75 |
| 17m |
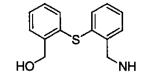
|
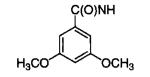
|
75 |
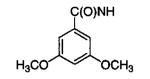
| 18m |
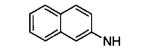
|
|
60 |
Acknowledgment
This work was supported by Grant AI44119 from the National Institutes of Health to Michael H. Gelb. Jerome C. Bressi is the recipient of the University of Washington Shain Fellowship. We thank Dr. Paul A. Michels for providing the trypanosmatid GAPDH enzymes.
Footnotes
Supporting Information Available: Synthetic and analytical data for 1 and analytical data for N6-(substituted)-2′-deoxy-2′-aminoadenosine and N6-(substituted)-2′-deoxy-2′-(amido)adenosine analogues. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1). Web site: http://www.who.org.
- (2).Smith DH, Pepin J, Stich AH. Human African trypanosomiasis: An Emerging Public Health Crisis. Br. Med. Bull. 1998;54:341–355. doi: 10.1093/oxfordjournals.bmb.a011692. [DOI] [PubMed] [Google Scholar]
- (3).Despommier DD, Gwadz RW, Hotez PJ. Parasitic Diseases. 3rd ed Springer-Verlag; New York: 1995. [Google Scholar]
- (4).Croft SL. The Current Status of Antiparasite Chemotherapy. Parasitology. 1997;114:S3–S15. [PubMed] [Google Scholar]
- (5).Tropical Disease Research: Progress 1997–1998. World Health Organization; Geneva: 1999. [Google Scholar]
- (6).Urbina JA. Chemotherapy of Chagas’ Disease: The How and the Why. J. Mol. Med. (Berlin) 1999;77:332–338. doi: 10.1007/s001090050359. [DOI] [PubMed] [Google Scholar]
- (7).Pepin J, Milord F. The Treatment of Human African trypanosomiasis. Adv, Parasitol. 1994;33:1–47. doi: 10.1016/s0065-308x(08)60410-8. [DOI] [PubMed] [Google Scholar]
- (8).Opperdoes FR. Biochemical Peculiarities of Trypanosomes, African and South American. Br. Med. Bull. 1985;41:130–136. doi: 10.1093/oxfordjournals.bmb.a072039. [DOI] [PubMed] [Google Scholar]
- (9).Opperdoes FR. Compartmentation of Carbohydrate Metabolism in Trypanosomes. Annu. Rev. Microbiol. 1987;41:127–151. doi: 10.1146/annurev.mi.41.100187.001015. [DOI] [PubMed] [Google Scholar]
- (10).Clayton C, Hausler T, Blattner J. Protein Trafficking in Kinetoplastid Protozoa. Microbiol. Rev. 1995;59:325–344. doi: 10.1128/mr.59.3.325-344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Clarkson AB, Jr., Brohn FH. Trypanosomiasis: An Approach to Chemotherapy by the Inhibition of Carbohydrate Catabolism. Science. 1976;194:204–206. doi: 10.1126/science.986688. [DOI] [PubMed] [Google Scholar]
- (12).Bakker BM, Michels PA, Opperdoes FR, Westerhoff HV. Glycolysis in Bloodstream Form Trypanosoma brucei Can Be Understood in Terms of the Kinetics of the Glycolytic Enzymes. J. Biol. Chem. 1997;272:3207–3215. doi: 10.1074/jbc.272.6.3207. [DOI] [PubMed] [Google Scholar]
- (13).Bakker BM, Michels PA, Opperdoes FR, Westerhoff HV. What Controls Glycolysis in Bloodstream Form Trypanosoma brucei? J. Biol. Chem. 1999;274:14551–14559. doi: 10.1074/jbc.274.21.14551. [DOI] [PubMed] [Google Scholar]
- (14).Bakker BM, Mensonides FI, Teusink B, van Hoek PA, Michels P, Westerhoff HV. From the Cover: Compartmentation Protects Trypanosomes from the Dangerous Design of Glycolysis. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2087–2092. doi: 10.1073/pnas.030539197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Engel JC, de Cazzulo B. M. Franke, Stoppani AO, Cannata JJ, Cazzulo JJ. Aerobic Glucose Fermentation by Trypanosoma cruzi Axenic Culture Amastigote-like Forms During Growth and Differentiation to Epimastigotes. Mol. Biochem. Parasitol. 1987;26:1–10. doi: 10.1016/0166-6851(87)90123-x. [DOI] [PubMed] [Google Scholar]
- (16).Vellieux FM, Hajdu J, Verlinde CLMJ, Groendijk H, Read RJ, Greenhough TJ, Campbell JW, Kalk KH, Littlechild JA, Watson HC, Hol WGJ. Structure of Glycosomal Glyceraldehyde-3-phosphate Dehydrogenase from Trypanosoma brucei Determined from Laue Data. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2355–2359. doi: 10.1073/pnas.90.6.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Souza DH, Garratt RC, Araujo AP, Guimaraes BG, Jesus WD, Michels PA, Hannaert V, Oliva G. Trypanosoma cruzi Glycosomal Glyceraldehyde-3-phosphate Dehydrogenase: Structure, Catalytic Mechanism and Targeted Inhibitor Design. FEBS Lett. 1998;424:131–135. doi: 10.1016/s0014-5793(98)00154-9. [DOI] [PubMed] [Google Scholar]
- (18).Kim H, Feil IK, Verlinde CLMJ, Petra PH, Hol WGJ. Crystal Structure of Glycosomal Glyceraldehyde-3-phosphate Dehydrogenase from Leishmania mexicana: Implications for Structure-Based Drug Design and a New Position for the Inorganic Phosphate Binding Site. Biochemistry. 1995;34:14975–14986. doi: 10.1021/bi00046a004. [DOI] [PubMed] [Google Scholar]
- (19).Mercer WD, Winn SI, Watson HC. Twinning in Crystals of Human Skeletal Muscle d-Glyceraldehyde-3-phosphate Dehydrogenase. J. Mol. Biol. 1976;104:277–283. doi: 10.1016/0022-2836(76)90013-9. [DOI] [PubMed] [Google Scholar]
- (20).Van Calenbergh S, Van Den Eeckhout E, Herdewijn P, De Bruyn A, Verlinde CLMJ, Hol WGJ, Callens M, Van Aerschot A, Rozenski J. Synthesis and Conformational Analysis of 2′-Deoxy-2′-(3-methoxybenzamido)adenosine. A Rational-Designed Inhibitor of Trypanosomal Glyceraldehyde Phosphate Dehydrogenase (GAPDH) Helv. Chim. Acta. 1994;77:631–644. [Google Scholar]
- (21).Verlinde CLMJ, Callens M, Van Calenbergh S, Van Aerschot A, Herdewijn P, Hannaert V, Michels PA, Opperdoes FR, Hol WGJ. Selective Inhibition of Trypanosomal Glyceraldehyde-3-phosphate Dehydrogenase by Protein Structure-Based Design: Toward New Drugs for the Treatment of Sleeping Sickness. J. Med. Chem. 1994;37:3605–3613. doi: 10.1021/jm00047a017. [DOI] [PubMed] [Google Scholar]
- (22).Van Calenbergh S, Verlinde CLMJ, Soenens J, De Bruyn A, Callens M, Blaton NM, Peeters OM, Rozenski J, Hol WGJ, Herdewijn P. Synthesis and Structure–Activity Relationships of Analogues of 2′-Deoxy-2′-(3-methoxybenzamido)adenosine. A Selective Inhibitor of Trypanosomal Glycosomal Glyceraldehyde-3-phosphate Dehydrogenase. J. Med. Chem. 1995;38:3838–3849. doi: 10.1021/jm00019a014. [DOI] [PubMed] [Google Scholar]
- (23).Aronov AM, Verlinde CLMJ, Hol WGJ, Gelb MH. Selective Tight Binding Inhibitors of Trypanosomal Glyceraldehyde-3-phosphate Dehydrogenase via Structure-Based Drug Design. J. Med. Chem. 1998;41:4790–4799. doi: 10.1021/jm9802620. [DOI] [PubMed] [Google Scholar]
- (24).Aronov AM, Suresh S, Buckner FS, Van Voorhis WC, Verlinde CLMJ, Opperdoes FR, Hol WGJ, Gelb MH. Structure-Based Design of Submicromolar, Biologically Active Inhibitors of Trypanosomatid Glyceraldehyde-3-phosphate Dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4273–4278. doi: 10.1073/pnas.96.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Suresh S, Bressi JC, Kennedy KJ, Verlinde CLMJ, Gelb MH, Hol WJG. Manuscript in preparation.
- (26).Robins MJ, Wilson JS, Hansske F. Nucleic Acid Related Compounds. 42. A General Procedure for the Efficient Deoxygenation of Secondary Alcohols. Regiospecific and Stereoselective Conversion of Ribonucleosides to 2′-Deoxynucleosides. J. Am. Chem. Soc. 1983;105:4059–4065. [Google Scholar]
- (27).Robins MJ, Trip EM. Nucleic Acid Related Compounds. 6. Sugar-Modified N6-(3-Methyl-2-butenyl)adenosine Derivatives, N6-Benzyl Analogues, and Cytokinin-Related Nucleosides Containing Sulfur or Formycin. Biochemistry. 1973;12:2179–2187. doi: 10.1021/bi00736a001. [DOI] [PubMed] [Google Scholar]
- (28).Golisade A, Van Calenbergh S, Link A. 2′-Amino-2′-deoxy-N6-(1-naphthylmethyl)adenosine as Novel Scaffold for a Polymer-Assisted Amidation Protocol. Tetrahedron. 2000;56:3167–3172. [Google Scholar]
- (29).Kenner GW, McDermott JR, Sheppard RC. Safety Catch Principle in Solid Phase Peptide Synthesis. J. Chem. Soc. D. 1971:636–637. [Google Scholar]
- (30).Backes BJ, Virgilio AA, Ellman JA. Activation Method To Prepare a Highly Reactive Acylsulfonamide “Safety-Catch” Linker for Solid-Phase Synthesis. J. Am. Chem. Soc. 1996;118:3055–3056. [Google Scholar]
- (31).Kotra LP, Manouilov KK, Cretton-Scott E, Sommadossi J-P, Boudinot FD, Schinazi RF, Chu CK. Synthesis, Biotransformation, and Pharmacokinetic Studies of 9-(β-d-Arabinofuranosyl)-6-azidopurine: A Prodrug for Ara-A Designed To Utilize the Azide Reduction Pathway. J. Med. Chem. 1996;39:5202–5207. doi: 10.1021/jm960339p. [DOI] [PubMed] [Google Scholar]
- (32).Matsuda A, Shinozaki M, Yamaguchi T, Homma H, Nomoto R, Miyasaka T, Watanabe Y, Abiru T. Nucleosides and Nucleotides. 103. 2-Alkynyladenosines: A Novel Class of Selective Adenosine A2 Receptor Agonists with Potent Antihypertensive Effects. J. Med. Chem. 1992;35:241–52. doi: 10.1021/jm00080a007. [DOI] [PubMed] [Google Scholar]
- (33).Link A, Van Calenbergh S, Herdewijn P. Practical Method for the Parallel Synthesis of 2′-Amido-2′-deoxyadenosines. Tetrahedron Lett. 1998;39:5175–5176. [Google Scholar]
- (34).Golisade A, Bressi JC, Calenbergh S, Gelb MH, Link A. Polymer-Assisted Solution-Phase Synthesis of 2′-Amido-2′-deoxyadenosine Derivatives Targeted at the NAD+-Binding Sites of Parasite Enzymes. J. Comb. Chem. 2000;2:537–544. doi: 10.1021/cc0000343. [DOI] [PubMed] [Google Scholar]
- (35).Kumar HMS, Reddy BVS, Reddy PT, Yadav JS. Efficient One-Pot Preparation of Nitriles from Aldehydes Using N-Methyl-pyrrolidone. Synthesis. 1999:586–587. [Google Scholar]
- (36).DeCosta DP, Pincock JA. Photochemistry of substituted 1-naphthylmethyl esters of phenylacetic and 3-phenylpropanoic acid: radical pairs, ion pairs, and Marcus electron transfer. J. Am. Chem. Soc. 1993;115:2180–90. [Google Scholar]
- (37).Stokker GE, Deana AA, DeSolms SJ, Schultz EM, Smith RL, Cragoe EJ, Jr., Baer JE, Ludden CT, Russo HF, et al. 2-(Aminomethyl)phenols, a New Class of Saluretic Agents. 1. Effects of Nuclear Substitution. J. Med. Chem. 1980;23:1414–1427. doi: 10.1021/jm00186a024. [DOI] [PubMed] [Google Scholar]
- (38).Deana AA, Stokker GE, Schultz EM, Smith RL, Cragoe EJ, Jr., Russo HF, Watson LS. 2-(Aminomethyl)phenols, a New Class of Saluretic Agents. 5. Fused-Ring Analogues. J. Med. Chem. 1983;26:580–585. doi: 10.1021/jm00358a023. [DOI] [PubMed] [Google Scholar]
- (39).White BB, Barabash E. Chem. Abstr. 1952 No. 2,581,565. [Google Scholar]
- (40).McMartin C, Bohacek RS. QXP: Powerful, Rapid Computer Algorithms for Structure-Based Drug Design. J. Comput. Aided Mol. Des. 1997;11:333–344. doi: 10.1023/a:1007907728892. [DOI] [PubMed] [Google Scholar]
- (41).Bohacek R, De Lombaert S, McMartin C, Priestle J, Gruetter M. Three-Dimensional Models of ACE and NEP Inhibitors and Their Use in the Design of Potent Dual ACE/NEP Inhibitors. J. Am. Chem. Soc. 1996;118:8231–8249. [Google Scholar]
- (42).Cignitti M, Ramusino MC, Farina A, Rajevic M. Conformational Properties of o-Alkoxybenzamides in Different Solvents. J. Mol. Struct. 1996;384:9–16. [Google Scholar]
- (43).Allen FH, Kennard O. 3D Search and Research Using the Cambridge Structural Database. Chem. Des. Autom. News. 1993;8:31–37. [Google Scholar]
- (44).Aronov AM, Munagala NR, de Montellano P. R. Ortiz, Kuntz ID, Wang CC. Rational Design of Selective Submicromolar Inhibitors of Tritrichomonas foetus Hypoxanthine-Guanine-Xanthine Phosphoribosyltransferase. Biochemistry. 2000;39:4684–4691. doi: 10.1021/bi992555g. [DOI] [PubMed] [Google Scholar]
- (45).Skillman AG, Kuntz ID. Unpublished results.
- (46).Ewing TJA, Kuntz ID. Critical Evaluation of Search Algorithms for Automated Molecular Docking and Database Screening. J. Comput. Chem. 1997;18:1175–1189. [Google Scholar]
- (47).Zou X, Sun Y, Kuntz ID. Inclusion of Solvation in Ligand Binding Free Energy Calculations Using the Generalized Born Model. J. Am. Chem. Soc. 1999;121:8033–8043. [Google Scholar]
- (48).Hirumi H. Continuous Cultivation of Trypanosoma brucei Blood Stream Forms in a Medium Containing a Low Concentration of Serum Protein without Feeder Cell Layers. J. Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- (49).Raz B, Iten M, Buhler Y. Grether, Kaminsky R, Brun R. The Alamar Blue Assay To Determine Drug Sensitivity of African Trypanosomes (T.b. rhodesiense and T.b. gambiense) In vitro. Acta Trop. 1997;68:139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- (50).Buckner FS, Verlinde CLMJ, La Flamme AC, Van Voorhis WC. Efficient Technique for Screening Drugs for Activity against Trypanosoma cruzi Using Parasites Expressing beta-Galactosidase. Antimicrob. Agents Chemother. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.