

Abstract
The emergence of functional genomics and proteomics has added to the growing need for improved analysis methods that can detect and distinguish between protein variants resulting from allelic variation, mutation, or post-translational modification. Aptamers, single-stranded DNA or RNA molecules that fold into three-dimensional structures conducive to binding targets, have become an attractive alternative to antibodies for this type of analysis. Although aptamers have been developed for a wide range of target species, very few sequences have been identified that bind selectively to proteins with specific post-translational modifications. Using capillary electrophoresis-based selection, we have developed DNA aptamer sequences that selectively bind an N-glycosylated peptide fragment of vascular endothelial growth factor (VEGF). The selection method incorporates alternating positive- and counter-selection steps in free solution in order to obtain aptamers with both high affinity toward the glycosylated target and high selectivity versus a non-glycosylated variant. Affinity capillary electrophoresis and surface plasmon resonance binding assays indicate these sequences have low-µM dissociation constants and preferentially bind the glycosylated peptide with as much as 50-fold specificity. Such aptamers could serve as tools for rapid and simple monitoring of disease-linked functional changes in proteins, with potential applications in drug screening and disease diagnosis.
Introduction
In efforts to better understand the products resulting from more than 22,000 genes that comprise the human genome, it has become increasingly important to develop tools with the ability to identify individual proteins.1 In particular, there has been a continuing drive to develop high-quality affinity reagents such as antibodies for a wide variety of molecular and biomedical applications. An ever expanding library of post-translational modifications (PTMs) have also been identified on a significant portion of the proteome. PTMs add a layer of complexity to the already diverse set of biological molecules within the cell. Glycosylation, in particular, has been demonstrated to play a crucial role in determining the structure and function of proteins.2 In fact, it has been estimated that more than half of all human proteins are glycoproteins.3 Differentiating between proteins with unique PTMs such as glycosylation requires new approaches to develop reagents with both the necessary affinity and specificity.
Aptamers are single-stranded DNA or RNA oligonucleotides generated in vitro that fold into tertiary structures suitable for selective binding to target molecules. This class of molecules has received increasing attention as alternative binding ligands to antibodies.4, 5 Aptamers offer a number of distinct advantages as biorecognition molecules including chemical stability, facile and site-specific chemical modification, ease of handling, ability to introduce into cells for in-vivo applications, and relatively inexpensive synthesis.6, 7 In addition, the small size of aptamers (~20–40 bases in length) makes them readily applicable to high-resolution analytical methods.8 At the same time, aptamers have been shown to have dissociation constants to specific targets ranging generally from high-µM values for small-molecules to sub-nM dissociation constants for larger targets.
Despite the rapidly increasing number of identified aptamers for a wide variety of targets, only a few aptamers have been selected that target specific post-translational modifications. RNA aptamers that bind a K-Ras-derived farnesylated peptide have been reported, yielding approximately 10-fold selectivity versus an equivalent non-farnesylated peptide.9 More recently, Williams et al. developed a DNA aptamer toward an acetylated histone peptide.10 Impressively, this aptamer showed a 2400-fold specifity versus the unmodified histone analog, although the selectivity of this aptamer decreased significantly to 5-fold when used for atomic force microscopy imaging applications.11 Several groups have reported sequences that bind to glycoproteins, but only one report has specifically been aimed at differentiating glycosylation patterns. Li et al. reported an aptamer incorporating boronic acid-modified nucleotides that demonstrated high affinity for fibrinogen, a model protein with well-characterized glycan structures.12 The boronic-acid moiety was included because of its intrinsic ability to interact with diols and hydroxyl groups. The proposed aptamer yielded a 60-fold lower affinity toward deglycosylated fibrinogen.
While most aptamer selection to-date has followed some version of immobilizing target onto a solid support, a variation of this technique has been developed using capillary electrophoresis (CE) that allows oligonucleotide-target binding to occur in free solution.13 Termed CE-SELEX (systematic enrichment of ligands through exponential enrichment), this method isolates bound complexes from unbound oligonucleotides by differential migration in a capillary under an applied electric field.14 CE-SELEX has been used to identify aptamers for protein targets including human immunodeficiency virus reverse transcriptase,15 human immunoglobulin E,16 protein kinase C-δ,17 an egg-white lysozyme,18 and ricin.19 Aptamers have also been generated for targets smaller than the oligonucleotide itself, such as neuropeptide-Y,20 an acetylated histone peptide,10 and small molecules such as anthrax toxin.21 There are advantages inherent in using CE as an aptamer selection technique. First, because the selection occurs in free solution, the target molecule remains in its native, biologically-relevant conformation and fully accessible, thereby maximizing the number of potential binding sites. This also eliminates issues with nonspecific binding of oligonucleotides to the solid support itself. In addition, most reports have demonstrated that selective aptamers with moderate-to-high affinity (Kd values µM or below) can be generated in six or fewer rounds of selection, significantly lower than the number required for traditional SELEX.14
Our target for these studies is a peptide fragment of vascular endothelial growth factor (VEGF). VEGF is a central positive regulator of angiogenesis that binds tyrosine kinase receptors on cell surfaces and triggers blood vessel growth. The full-length 165-amino-acid protein is a disulfide-linked homodimer with a heparin-binding domain and is singly glycosylated in its native form at asparagine 75. VEGF has been implicated in the growth of tumor-associated vasculature.22 As such, VEGF has become an important target for pharmaceutical development – drugs targeting this protein are now available. We have chosen to target a 32-amino-acid peptide fragment of the protein which contains the natural glycosylation site of VEGF at asparagine 75, located in an exposed portion of the heparin-binding domain. The length of peptide for this work was optimized to simplify synthetic consideration while still being large enough to induce a significant mobility shift in CE.
We have developed an aptamer which selectively binds a glycosylated VEGF peptide with high affinity. By incorporating a novel on-column counter-selection versus a non-glycosylated variant, we are also able to achieve substantial selectivity. These results demonstrate that it is possible to differentiate between glycosylation states using aptamers evolved with CE-SELEX.
Experimental
Biochemicals, reagents, and buffers
A random DNA oligonucleotide library was obtained from Integrated DNA Technologies (IDT, Coralville, IA) and consisted of a 40-base random sequence flanked by two 20-base fixed sequences and fluorescently tagged with 6-carboxyfluorescein (6-FAM): 5'-(6-FAM)-AGC AGC ACA GAG GTC AGA TG-(N40)-CCT ATG CGT GCT ACC GTG AA-3'. Forward and reverse primers for PCR amplification were purchased from Invitrogen (Carlsbad, CA) and 5'-labeled with Alexa Fluor 488 and biotin, respectively. Sequences used for surface plasmon resonance (SPR) imaging were 5'-modified with a 12-carbon amine (IDT) in order to allow attachement to the SPR chip. All other oligonucleotides were synthesized by either IDT or GenoMechanix (Gainesville, FL) and labeled with Alexa Fluor 488. All oligonucleotides were HPLC purified.
Peptides were custom synthesized by Anaspec (Fremont, CA). The target peptide was a 32-residue fragment of VEGF that includes the heparin-binding domain of the native protein: PTEESNITMQIMRIKPHQGQHIGEMSFLQHNK. The glycosylated variant incorporated a β-N-acetylglucosaminyl asparagine derivative (EMD Chemicals, Gibbstown, NJ) that yielded an N-acetylglucosamine (GlcNAc) monosaccharide at the natural glycosylation site of the VEGF peptide (underlined in the above sequence). In addition, a nonhomologous control hexadecapeptide was acquired from Sussex Research (Ottowa, Canada) which includes a similar threonine-linked GlcNAc glycosylation (AETPALSESDSTEAFR).
All samples and buffers were prepared using deionized water (18.2 MΩ cm resistivity; Millipore). All other reagents were purchased from Sigma-Aldrich. The capillary electrophoresis separation buffer (TGK) consisted of 25 mM Tris, 192 mM glycine, and 5 mM K2HPO4, was adjusted to pH 7.8, and filtered through a 0.2-µm membrane filter (Whatman, Florham Park, NJ).
Capillary electrophoresis selection
All CE selections were performed on an instrument constructed in-house. Samples were irradiated by focusing the 488-nm line of an argon-ion laser (CVI Melles-Griot, Carlsbad, CA) onto a fused-silica capillary. Laser-induced fluorescence at 520 nm was collected at 90° by a 20X microscope objective, and detected with a Hamamatsu Photonics photomultiplier tube (Japan). Signals were amplified (PMT-4, Advanced Research Instruments, Golden, CO) prior to data acquisition and analysis by LabView Software (National Instruments, Austin, TX).
Experiments were conducted using zero-electroosmotic flow capillaries (MicroSolv, Eatontown, NJ). Prior to use, capillaries (linear polyacrylamide coated, 50-µm i.d., 360-µm o.d., total length = 50 cm, length to detector = 35 cm) were rinsed with separation buffer for 15 min. In between separations, the capillary was flushed again with buffer for at least 1 min.
For the initial selection, 10-nM glycosylated peptide was incubated with 10-µM DNA selection library (10 µL total volume) for 20 min at room temperature. Sample plugs (~3.5 nL) were reproducibly injected onto the capillary by 15-sec gravity injections (8.5-cm height difference between injection and outlet ends). Separations were performed by applying 20 kV across the capillary. With polyacrylamide-coated capillaries, unbound DNA should migrate more quickly than the larger DNA-peptide complexes. The early-eluting, unbound sequences were visualized by fluorescence and allowed to pass through the capillary. Application of potential was then halted, and the remaining sequences were collected by pushing the leftover solution from the capillary with a syringe into a separate vial containing 90 µL of separation buffer. A total of six collections were performed for each selection round in order to increase the number of individual sequences sampled.
Later selection rounds were performed in either an analogous positive format (incubating oligonucleotide pools with glycosylated peptide and collecting late-eluting bound sequences) or in a counter-selection mode (incubating oligonucleotide pools with non-glycosylated peptide and collecting early-eluting unbound sequences) (Fig. 1). On-column counter-selection rounds were included to improve the specificity of the developed aptamers for only the glycosylated variant. For rounds 2–5 of positive selection, a 1,000-fold molar excess of DNA from the previous round was incubated with the glycosylated peptide for 20 min at room temperature before separation. For the final selection round (round 6) a 10,000-fold excess of DNA was used to promote more competitive binding. Negative selections were conducted using a 25-fold molar excess of non-glycosylated peptide.
Fig. 1.
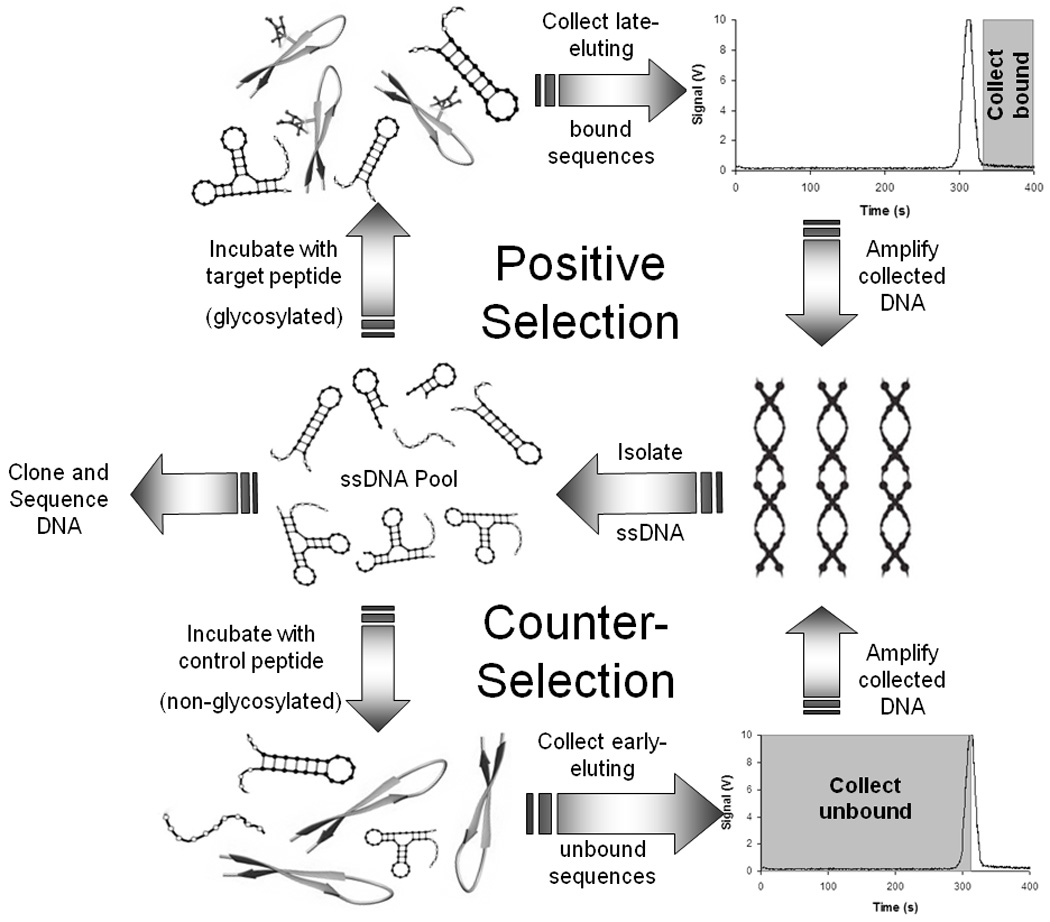
After each selection round (positive or counter), oligonucleotides were amplified using PCR. PCR mixtures contained 50 µL of Taq 2X Master Mix (New England Biolabs, Ipswich, MA), 2.55 µL each of 20 µM forward and reverse primers, and 6 µL of the collection solution in a total of 100 µL. For each round, 16 PCR reaction vials and two control vials without collected DNA were placed in a thermal cycler and heated for 5 min at 94°C to allow time for DNA template and primer denaturation, followed by cycles of 30 sec at 94°C, 30 sec at 53°C, and 20 sec at 72°C. Positive selection rounds were amplified by 23 amplification cycles, while negative selections were followed by 19 rounds of PCR amplification. Limiting amplification rounds in the higher concentration samples of heterogeneous sequences such as those found in the counter-selection pools has been demonstrated to better amplify DNA pools while reducing undesired products.23 A final extension was performed for 5 min at 72°C. PCR amplification of the DNA template was verified by gel electrophoresis on a 2% agarose gel stained with ethidium bromide. Double-stranded DNA at ~80 base pairs was observed after each round of selection, with no bands evident in negative control lanes.
In order to isolate the desired ssDNA from the dsDNA PCR product, the amplified sequences were added to a column loaded with 300 µL of streptavidin-agarose (Thermo Fisher Scientific, Rockford, IL) and 500 µL of DNA binding buffer (10-mM Tris, 50-mM NaCl, and 1 mM EDTA, pH 7.5). The column was incubated at room temperature with occasional vortexing for 30 min and then rinsed with 10 1-mL aliquots of buffer. The ssDNA was eluted from the column using 2 × 200 µL of 0.15 M NaOH at 37°C. The resulting solution was immediately neutralized with acetic acid and concentrated by ethanol precipitation. Oligonucleotide concentrations for successive rounds of selection were determined by absorbance spectroscopy with a Shimadzu BioSpec-nano (Columbia, MD).
Aliquots of the ssDNA pool after 6 rounds of positive selection and 2 rounds of counter-selection were PCR amplified with alternative conditions in preparation for cloning (unlabeled primers, 6 cycles with a final 10-min extention at 72°C). The PCR products were purified with a MinElute PCR Purification Kit (Qiagen, Valencia, CA) and ligated into the pGEM-T Vector System (Promega, Madison, WI). The ligation product was then transformed into DH5α chemically competent E. coli cells (Invitrogen, Carlsbad, CA), and 28 colonies were chosen randomly for sequencing (Sequetech, Mountain View, CA).
Determination of dissociation constants (Kd)
Affinity capillary electrophoresis was used as the basis for evaluating aptamer binding and selectivity, and specifically for assessing Kd values for candidate aptamer sequences. Binding was assessed by incubating potential aptamers with target peptide, and estimating the percent of oligonucleotide bound as being equal to the percent decrease of the free-DNA peak height. To determine dissociation constants, low concentrations (50 nM or 100 nM) of oligonucleotide were mixed with increasing concentrations (up to 50 µM) of the glycosylated peptide, non-glycosylated peptide, or control peptide. The decreasing peak height of the free aptamer peak served as the basis for Kd calculations by plotting peak height versus peptide concentration as described previously20 and fitting to a 1:1 binding model using Igor Pro 6.01 software (WaveMetrics, Portland, OR).
Surface plasmon resonance imaging (SPRI)
As a complement to affinity capillary electrophoresis, surface plasmon resonance imaging was used to confirm binding of potential aptamers to the target peptide. Single-stranded DNA microarrays were created as described by Chen et al.24 A monolayer of poly(L-glutamic acid) (pGlu) was electrostatically adsorbed onto a chemically modified gold thin film prior to attaching amine-modified oligonucleotides. Sequences were individually spotted onto an SPR chip and allowed to incubate for at least four hours prior to analysis.
SPRI measurements of differential reflectivity (Δ %R) at 830 nm were obtained with a GWC Technologies SPRimager II (Madison, WI). Briefly, polarized light strikes a prism/gold film/flow cell assembly at an angle near the SPR angle. Reflected light is then collected with a CCD camera.
Results and Discussion
Selection of DNA aptamers for glycosylated VEGF peptide
CE-SELEX for small target molecules is particularly challenging, in that the size-to-charge ratio of the oligonucleotide does not change dramatically upon binding the target. While larger targets such as proteins will generally yield separate peaks for bound and unbound DNA, this is not always the case with smaller targets like peptides. Here, the 80-nucleotide ssDNA (~25 kDa) is significantly larger than the glycosylated peptide target (3.9 kDa). In addition, at the buffer pH of 7.8, the peptide charge is only ~ +1 (pI = 9.9). Thus, a very small migration shift is expected upon binding. Williams et al. were able to resolve peaks for bound vs. unbound DNA in their CE-based selection of aptamers for an even smaller histone peptide (MW ~ 1600 g/ mol), but this was likely due to the substantial +6 positive charge of the peptide under their separation conditions.10 Mendonsa and Bowser, however, did not observe resolved peaks in their development of aptamers for neuropeptide Y, a peptide more similar in both size and charge to the glycosylated VEGF peptide of interest in the present work.20 Instead, formation of the DNA-peptide complex was observed by monitoring a decrease in the fluorescence of the unbound DNA peak.
One concern in utilizing CE-SELEX is that the small sample volume limits the actual number of unique sequences being tested for binding. Here, each separation injects only 2 ×1010 DNA sequences onto the capillary. In order to partially mitigate this issue, six CE collections were performed and pooled together for each round, increasing the number of DNA sequences to 1 × 1011. This number is still at least two orders of magnitude lower than that used for most aptamer selection methods, and lowering the number of sequences in the starting DNA pool almost certainly decreases the abundance of potential high-affinity aptamers. Increasing the amount of injected DNA, however, would increase peak widths and therefore would make it even more challenging to isolate bound sequences from free DNA.
Our selection strategy incorporated rounds of both positive selection for the glycosylated peptide target and counter-selection against the non-glycosylated isoform. Initially, a pool of sequences with affinity toward the desired target was generated through three successive rounds of positive selection. For each round, a 1,000-fold molar excess of DNA was utilized to provide sufficient competition to enrich the DNA pools with molecules that bound to the glycosylated variant. With polyacrylamide-coated capillaries, unbound DNA should migrate more quickly than the larger DNA-peptide complexes. Therefore, the free DNA signaled by a large fluorescence peak in the electropherogram was allowed to pass through the capillary (accounting for the distance between the detection window and the capillary exit), and the remaining solution containing bound DNA-peptide was collected (see Fig. 1).
Following the third round of positive selection, an on-column counter-selection was implemented to remove sequences with affinity for the non-glycosylated VEGF peptide. For these experiments, the early-eluting unbound sequences were collected. This negative selection was performed using a 25-fold molar excess of the non-glycosylated peptide in order to most efficiently bind sequences with even moderate affinity, thereby removing them from later selection pools. The counter-selection was followed by another positive round of selection, a final counter-selection, and two concluding rounds of positive selection. The final selection was performed with 10,000-fold molar excess of DNA in order to promote more competitive binding and retain only sequences with the highest affinity toward the glycosylated peptide.
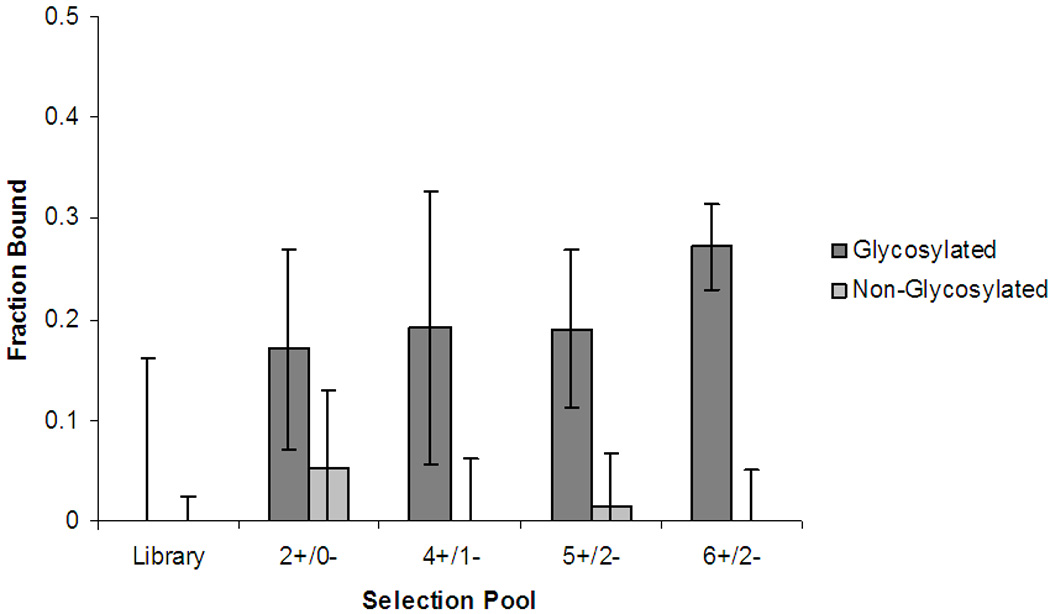
Binding affinity toward both the glycosylated and non-glycosylated peptide was monitored throughout the selection process by affinity CE. Fig. 2 shows how the fraction of DNA bound in the presence of 50-fold molar excess of peptide progressed with increasing rounds of selection. The fraction of bound sequences was calculated by:
where Io is the height of the free DNA fluorescence peak in the absence of peptide and I is the free-DNA peak height at a given peptide concentration. The affinity of the ssDNA pools for the glycopeptide increased significantly after only two rounds of selection (2+/0−). While the random DNA library yielded virtually no binding to the peptide, nearly 20% of the 2+/0− pool showed affinity toward the glycosylated target. The affinity of the pools for the non-glycosylated peptides also increased, although not as substantially (5% bound), suggesting that many sequences at this stage in selection bound both variants. After incorporating counter-selection rounds, the affinity of the DNA pools for the non-glycosylated peptide decreased markedly. After five rounds of positive selection and 2 rounds of counter-selection (5+/2−), the binding affinity of the pools appeared to plateau. The final, more stringent round of positive selection (6+/2−) yielded a selection pool with both higher affinity and excellent specificity for the glycosylated peptide.
Fig. 2.
Following 6 rounds of positive selection and 2 rounds of counter-selection, the ssDNA pool was amplified, ligated into the pGEM-T vector system, and transformed into chemically competent E. coli cells. Twenty-eight colonies were randomly chosen and sequenced (Table 1). The resulting sequences were examined for sequence homology using ClustalW. No common motifs or conserved nucleotides were apparent in the cloned sequences. These results are consistent with those observed by others using CE-SELEX.10
Table 1.
Nucleotide sequences of aptamers after 6 rounds of positive selection and 2 rounds of counter-selection cloned from randomly selected colonies.
| Clone | Sequence |
|---|---|
| 1 | TCAATCATGAGCATGTATAATTTGGTCCACATTAAACGGG |
| 2 | GGGGTAGTGAAGGGTAAAGTAAGAGGAATGCCCAGTACGT |
| 3 | GGGACAGGGGGTCAGGTGGAGCAAGGATTTGCTAGCGTAC |
| 4 | CCATGCAAATCCGAGTTGGGCGTCAACTCCGGAAGGGGTC |
| 5 | GGCATGGTGTGTATCAGGTGATGCGGTAATACTACTCGAA |
| 6 | GGGTGTGGACGTCTATCTGCCGTATGGGTCACAAGTTTGC |
| 7 | GTCATAGGAGCATATGCTGGCATCTTGGAGCGGTCAGGTC |
| 9 | CAGGTGCACTTGGCGTGACTATAGTAGACGGTCGCCGGCC |
| 10 | GGTGGAAGGCACGGACACAGCCACAAACATACGCGGTGAC |
| 11 | GNGCTGAATGTACATTAGGGAGCATGATACAGCATGCCGC |
| 13 | GGTAGTGTCAGCTGCGGCGGTGAGCCGGGAGAGGCTCCCC |
| 14 | GAGGTCACGACCGGTTCAGTAATATGGAGATATAAACACA |
| 15 | GTCCGAGTGGCATCATTGAAGCTAGGGAAGATGGGCGGGG |
| 16 | CCACTTCTTTATCTAGGAAGACTTTTGAACTACGTTACAA |
| 17 | GGTAGTGTACGACGTGCAGGGCGATGGGGGGCAGGGGAGT |
| 18 | GCATCATCTCAAATGTTGACGTGTCATGTGCGTATTAGAG |
| 19 | GACCACGCGAGGTGACATTTGGCATTATAAGCGTAAGTAC |
| 20 | TGGTAAGTGCACAGATATGTAACGTGAAGCTCGTCGCTGC |
| 21 | GCATCATATGTAAGTGTACTGTGGTACGGAAACGTACGTG |
| 22 | ACGTGGTAGGCGCGTGCGTAAAGGCGAGATCCGAAGGCGG |
| 25 | GCGGCAAAATGACGACGAGGAGGGGAGCAGGGTCGGGCCG |
| 26 | GAGGGCTAGGAGTTTATGTAAGGAGATACGGCGGTGTGGC |
| 27 | GTGAGCCGGTGTAGGAGCGGCAGGAACGTCCCGCACAGTA |
| 28 | GCACCCTAATGTGCAAGCTAATGCGGAATGGGGTCGGTTT |
Characterization of binding affinity and selectivity of candidate aptamers
A random-number generator was used to select 12 clones for synthesis and further characterization. Each sequence was evaluated for relative binding affinity for both the glycosylated and non-glycosylated VEGF peptide variants by affinity CE. Most of the sequences demonstrated a significant percentage of binding to the glycosylated target under these conditions, and two sequences (clones 26 and 28) yielded binding significantly higher than that of the overall selection pool (Table 2). Four of the cloned sequences showed little-to-no binding of the intended target, suggesting that the low number of selection rounds did not completely remove non-binding sequences. Encouragingly, the incorporation of only two rounds of on-column counter-selection into the CE-SELEX protocol led to reasonable selectivity in many of the evaluated clones. Of the 6 clones showing more than 15% binding to the glycopeptide at these conditions, all of them demonstrated substantially less binding upon incubation with the non-glycosylated variant (Table 2).
Table 2.
Binding characterization of cloned sequences. Each clone (either 50 nM or 100 nM) was incubated with a 50-fold molar excess of either glycosylated or non-glycosylated peptide. Binding was evaluated through affinity capillary electrophoresis.
| Colony | Glycosylated | Non-glycosylated |
|---|---|---|
| 1 | 0.21 ± 0.22 | 0.00 ± 0.26 * |
| 3 | 0.00 ± 0.01 * | 0.15 ± 0.11 |
| 9 | 0.28 ± 0.02 | 0.00 ± 0.11 * |
| 11 | 0.18 ± 0.03 | 0.00 ± 0.19 * |
| 13 | 0.35 ± 0.14 | 0.00 ± 0.11 * |
| 14 | 0.09 ± 0.03 | 0.04 ± 0.06 |
| 16 | 0.00 ± 0.24 * | 0.01 ± 0.07 |
| 17 | 0.15 ± 0.15 | 0.20 ± 0.13 |
| 18 | 0.00 ± 0.01 * | 0.13 ± 0.18 |
| 20 | 0.04 ± 0.12 | 0.28 ± 0.12 |
| 26 | 0.41 ± 0.28 | 0.12 ± 0.05 |
| 28 | 0.59 ± 0.23 | 0.20 ± 0.10 |
Data are presented as fraction of DNA bound (± SD).
Values marked with * represent slightly negative numbers rounded to zero percent fraction bound.
To validate our assessment of relative binding, we used surface plasmon resonance as a complementary and more sensitive approach. SPRI imaging allowed for multiple sequences to be spotted onto a single chip and evaluated for binding simultaneously. For this analysis, four sequences were chosen (clones 11, 16, 20, and 28) which showed differential binding to the glycosylated peptide by affinity CE. The array was exposed to a solution of 1-µM glycosylated VEGF peptide in TGK buffer (pH = 7.8) for approximately twenty min, and the change in reflectivity was continuously monitored. All four sequences demonstrated a rapid rise in reflectivity in the presence of the peptide target, quickly reaching a plateau (Fig. 3). This pattern suggests significant DNA-peptide binding with rapid kinetics. Of the four spotted sequences, clone 28 demonstrated the highest binding affinity for the glycosylated peptide as the association curve achieved the highest plateau of Δ%R. Clones 11, 20, and 16 followed in order of decreasing plateau height with clone 16 being the sequence with the lowest affinity for the glycosylated peptide. Three of four sequences show typical SPRI binding curves, while one (clone 11) depicts some dissociation of peptide during the SPR run. This might be reflective of non-specific binding that is gradually removed by constantly flowing solution over the DNA array.
Fig. 3.
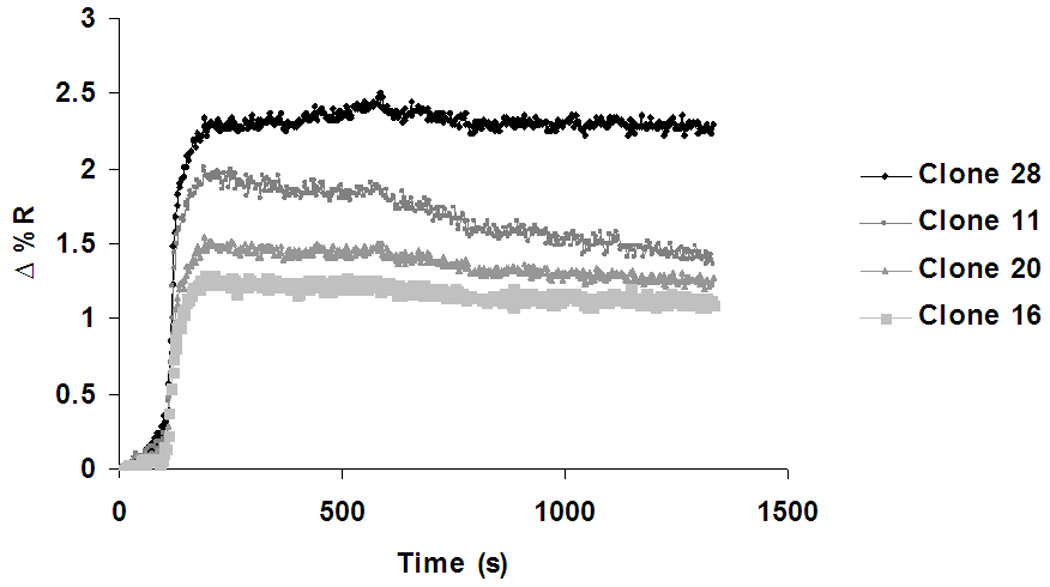
This pattern of relative binding closely matches that observed for the same four sequences using affinity CE (Table 2). Of the four sequences, clone 28 demonstrated the highest affinity for the glycosylated peptide with a fraction bound of 0.59, followed in order by clones 11, 20, and 16. Interestingly, while affinity CE showed little indication of binding between clone 16 and the target peptide, SPRI data suggest that some binding does indeed occur.
Due to its combination of high target affinity (as suggested by both CE and SPRI data) and high selectivity, clone 28 was characterized more carefully as the best aptamer candidate for the glycosylated VEGF peptide. Fig. 4a shows the binding curves resulting from the incubation of clone 28 with the glycosylated VEGF peptide, the non-glycosylated VEGF peptide, and the non-homologous hexadecapeptide similarly modified with GlcNAc. Binding curves were constructed by measuring the fraction of 50-nM aptamer bound in the presence of target peptides ranging in concentration from 0–50 µM (Fig. 4a). Analysis of the affinity plots reveals that clone 28 binds the glycosylated target with a Kd of 2.5 (± 0.4) µM. The affinity of the initial DNA library for the same peptide, meanwhile, was substantially lower, with a Kd of ~22 mM (data not shown). These data demonstrate a dramatic 8800-fold increase in binding affinity after only 6 rounds of positive selection for the target. The specificity of the selected aptamer sequence is also impressive. Kd values for the non-glycosylated VEGF peptide and the glycosylated hexadecapeptide were 130 (± 120) µM and 170 (± 4) µM, respectively. With only two rounds of counter-selection, the generated aptamer has a 52-fold selectivity for its intended target over the non-glycosylated variant, and a 68-fold selectivity versus the control glycosylated hexadecapeptide. These results suggest that we have successfully generated an aptamer with specificity toward both the glycosylation and the sequence itself. In the presence of only one of these factors, affinity decreases by a factor of more than 50 (Table 3).
Fig. 4.
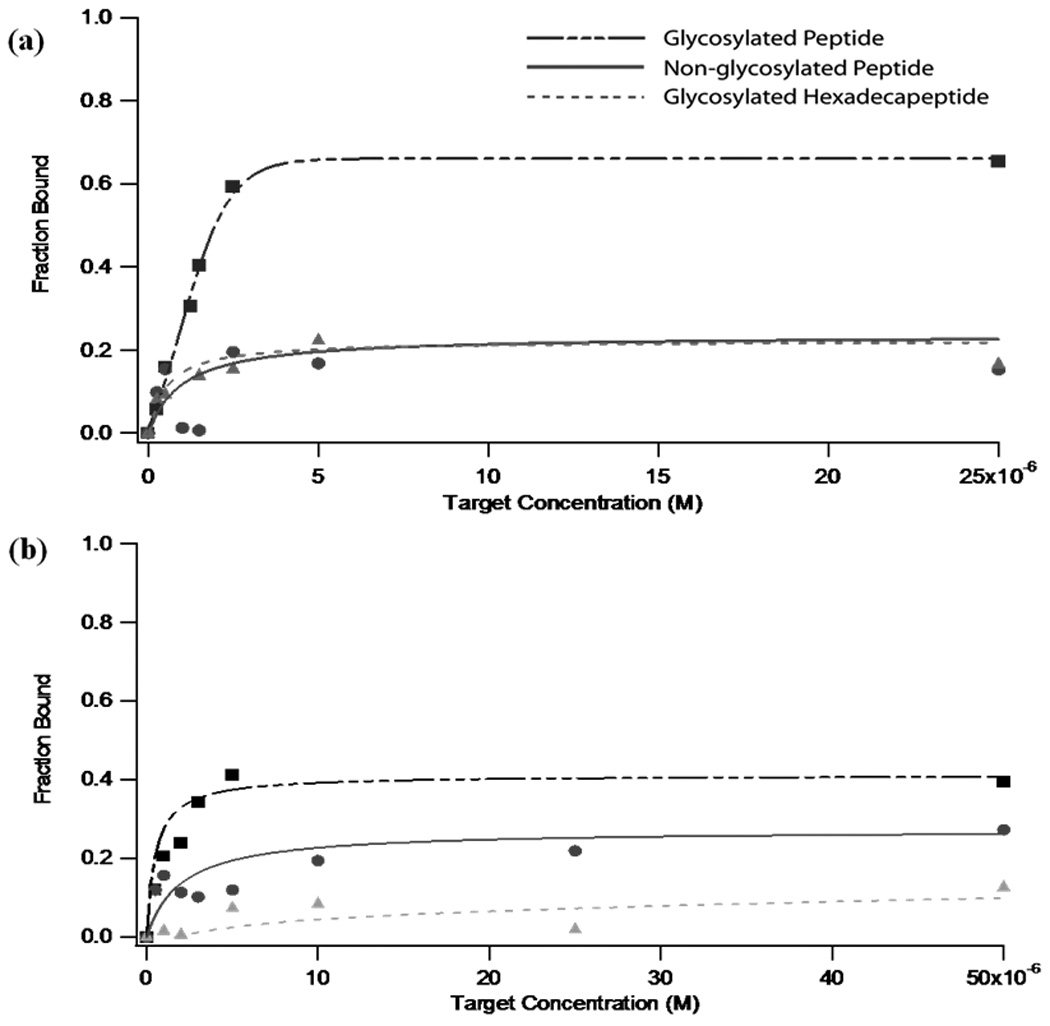
Table 3.
Dissociation constants and fold specificity of selected aptamers. Kd values were obtained through affinity capillary electrophoresis. Fold specificity demonstrates preferential binding of aptamer towards glycosylated peptide.
| Clone | Glycosylated Peptide |
Non- Glycosylated Peptide |
Fold Specificity |
Glycosylated Hexadecapeptide |
Fold Specificity |
|---|---|---|---|---|---|
| 28 | 2.5 ± 0.4 µM | 130 ± 120 µM | 52 | 170 ± 4 µM | 68 |
| 26 | 11 ± 11 µM | 91 ± 59 µM | 8 | 380 ± 270 µM | 34 |
| Selection Library |
22 ± 11 mM | --- | --- | ||
Clone 26, the candidate sequence with the second-highest fraction bound observed (Table 2), was similarly characterized using 100-nM aptamer in order to attain adequate sensitivity. Binding curves demonstrate that this oligonucleotide binds the glycosylated target peptide with a Kd of approximately 11 µM and has lower affinity to both the non-glycosylated variant and the glycosylated hexadecapeptide control (Fig. 4b). A comparison of the dissociation constants and specificity (Table 3) shows that while both sequences have reasonably high affinity for the intended target, clone 28 yields significantly greater fold-specificity versus both of the control peptides.
Conclusions
Here, we demonstrate the successful development of an aptamer able to distinguish between peptide variants differing only by a single glycosylation. The CE selection scheme proved efficient at generating high-affinity candidate aptamers, requiring only six rounds of positive selection. Using a novel on-column CE-SELEX counter-selection strategy, we were able to attain more than 50-fold specificity versus the non-glycosylated variant. The generated aptamers are also selective against a non-homologous peptide with an equivalent glycosylation, proving that the sequences are not simply specific for the sugar moiety itself, but rather the combination of the peptide sequence and the glycosylation. The ability to rapidly evolve aptamers with selectivity for particular PTMs, such as that demonstrated, offers tremendous potential for distinguishing between different active forms of the same protein.
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (R15GM081854) and an IBM Research Grant from Santa Clara University. C.M.R. thanks the Doelger Scholars Program, and M.J.H. gratefully acknowledges the Gerald and Sally DeNardo Science Scholars Program and a Provost Research Scholarship, all through Santa Clara University. The authors thank Daniel Daugherty for contributions to selection development; Christian Paquet and Brian Jameson for efforts in optimizing separation conditions; Dr. Arup Sen for developmental discussions; Dr. Michael Bowser and Meng Jing for guidance with CE-SELEX; and Dr. Robert Corn, Ting Nico Hu, and Yulin Chen for substantial SPR assistance.
References
- 1.Blow N. Nature. 2007;447:741–744. doi: 10.1038/447741a. [DOI] [PubMed] [Google Scholar]
- 2.Fukuda M. In: Molecular Glycobiology. Fukuda M, Hindsgaul O, editors. New York: Oxford University Press; 1994. pp. 1–52. [Google Scholar]
- 3.Apweiler R, Hermjakob H, Sharon N. Bba-Gen Subjects. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 4.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 5.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 6.Jayasena SD. Clinical Chemistry. 1999;45:1628–1650. [PubMed] [Google Scholar]
- 7.Klussmann S. The Aptamer Handbook: Functional Oligonucleotides and their Applications. Wiley-VCH: Weinheim; 2006. [Google Scholar]
- 8.German I, Buchanan DD, Kennedy RT. Anal. Chem. 1998;70:4540–4545. doi: 10.1021/ac980638h. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert BA, Sha M, Wathen ST, Rando RR. Bioorgan. Med. Chem. 1997;5:1115–1122. doi: 10.1016/s0968-0896(97)00047-3. [DOI] [PubMed] [Google Scholar]
- 10.Williams BAR, Lin L, Lindsay SM, Chaput JC. J. Am. Chem. Soc. 2009;131:6330–6331. doi: 10.1021/ja900916p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin LY, Fu Q, Williams BAR, Azzaz AM, Shogren-Knaak MA, Chaput JC, Lindsay S. Biophys. J. 2009;97:1804–1807. doi: 10.1016/j.bpj.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li MY, Lin N, Huang Z, Du LP, Altier C, Fang H, Wang BH. J. Am. Chem. Soc. 2008;130:12636–12638. doi: 10.1021/ja801510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendonsa SD, Bowser MT. J. Am. Chem. Soc. 2004;126:20–21. doi: 10.1021/ja037832s. [DOI] [PubMed] [Google Scholar]
- 14.Mosing RK, Bowser MT. In: Handbook of Capillary and Microchip Electrophoresis and Associated Microtechniques. 3rd. Landers JP, editor. Boca Raton, FL: CRC Press; 2008. pp. 825–839. [Google Scholar]
- 15.Mosing RK, Mendonsa SD, Bowser MT. Anal. Chem. 2005;77:6107–6112. doi: 10.1021/ac050836q. [DOI] [PubMed] [Google Scholar]
- 16.Mendonsa SD, Bowser MT. Anal. Chem. 2004;76:5387–5392. doi: 10.1021/ac049857v. [DOI] [PubMed] [Google Scholar]
- 17.Mallikaratchy P, Stahelin RV, Cao Z, Cho W, Tan W. Chem. Commun. 2006:3229–3231. doi: 10.1039/b604778e. [DOI] [PubMed] [Google Scholar]
- 18.Tran DT, Janssen KPF, Pollet J, Lammertyn E, Anne J, Van Schepdael A, Lammertyn J. Molecules. 2010;15:1127–1140. doi: 10.3390/molecules15031127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang J-J, Xie J-W. Gaodeng Xuexiao Huaxue Xuebao. 2006;27:1840–1843. [Google Scholar]
- 20.Mendonsa SD, Bowser MT. J. Am. Chem. Soc. 2005;127:9382–9383. doi: 10.1021/ja052406n. [DOI] [PubMed] [Google Scholar]
- 21.Cella LN, Sanchez P, Zhong W, Myung NV, Chen W, Mulchandani A. Anal. Chem. 2010;82:2042–2047. doi: 10.1021/ac902791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 23.Musheev MU, Krylov SN. Anal Chim Acta. 2006;564:91–96. doi: 10.1016/j.aca.2005.09.069. [DOI] [PubMed] [Google Scholar]
- 24.Chen YL, Nguyen A, Niu LF, Corn RM. Langmuir. 2009;25:5054–5060. doi: 10.1021/la804021t. [DOI] [PMC free article] [PubMed] [Google Scholar]