

Abstract
Alternative splicing of mRNA precursors provides an important means of genetic control and is a crucial step in the expression of most genes. Alternative splicing markedly affects human development, and its misregulation underlies many human diseases. Although the mechanisms of alternative splicing have been studied extensively, until the past few years we had not begun to realize fully the diversity and complexity of alternative splicing regulation by an intricate protein–RNA network. Great progress has been made by studying individual transcripts and through genome-wide approaches, which together provide a better picture of the mechanistic regulation of alternative pre-mRNA splicing.
Alternative splicing is a crucial mechanism for gene regulation and for generating proteomic diversity. Recent estimates indicate that the expression of nearly 95% of human multi-exon genes involves alternative splicing1,2. In metazoans, alternative splicing plays an important part in generating different protein products that function in diverse cellular processes, including cell growth, differentiation and death.
Splicing is carried out by the spliceosome, a massive structure in which five small nuclear ribonucleoprotein particles (snRNPs) and a large number of auxiliary proteins cooperate to accurately recognize the splice sites and catalyse the two steps of the splicing reaction1,2 (BOX 1). Spliceosome assembly (BOX 1) begins with the recognition of the 5′ splice site by the snRNP U1 and the binding of splicing factor 1 (SF1) to the branch point3 and of the U2 auxiliary factor (U2AF) heterodimer to the polypyrimidine tract and 3′ terminal AG4,5. This assembly is ATP independent and results in the formation of the E complex, which is converted into the ATP-dependent, pre-spliceosomal A complex after the replacement of SF1 by the U2 snRNP at the branch point. Further recruitment of the U4/U6–U5 tri-snRNP complex leads to the formation of the B complex, which is converted into to the catalytically active C complex after extensive conformational changes and remodelling.
Box 1. Splicing and spliceosome assembly.
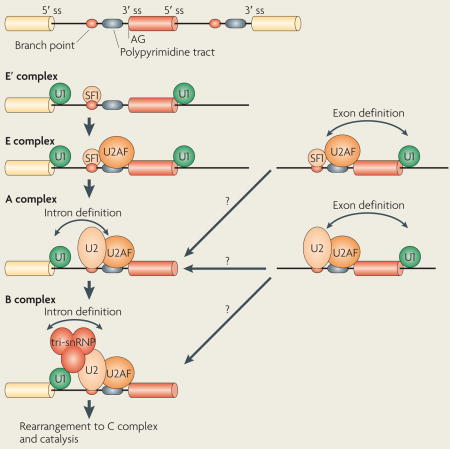
Pre-mRNA splicing is a process in which intervening sequences (introns) are removed from an mRNA precursor. Splicing consists of two transesterification steps, each involving a nucleophilic attack on terminal phosphodiester bonds of the intron. In the first step this is carried out by the 2′ hydroxyl of the branch point (usually adenosine) and in the second step by the 3′ hydroxyl of the upstream (5′) exon1,2. This process is carried out in the spliceosome, a dynamic molecular machine the assembly of which involves sequential binding and release of small nuclear ribonucleoprotein particles (snRNPs) and numerous protein factors as well as the formation and disruption of RNA–RNA, protein–RNA and protein–protein interactions.
The basic mechanics of spliceosome assembly are well known. Briefly, the process begins with the base pairing of U1 snRNA to the 5′ splice site (ss) and the binding of splicing factor 1 (SF1) to the branch point3 in an ATP-independent manner to form the E′ complex (see the figure; double-headed arrows indicate an interaction). The E′ complex can be converted into the E complex by the recruitment of U2 auxiliary factor (U2AF) heterodimer (comprising U2AF65 and U2AF35) to the polypyrimidine tract and 3′ terminal AG158. The ATP-independent E complex is converted into the ATP-dependent pre-spliceosome A complex by the replacement of SF1 by U2 snRNP at the branch point. Further recruitment of the U4/U6–U5 tri-snRNP leads to the formation of the B complex, which contains all spliceosomal subunits that carry out pre-mRNA splicing. This is followed by extensive conformational changes and remodelling, including the loss of U1 and U4 snRNPs, ultimately resulting in the formation of the C complex, which is the catalytically active spliceosome.
The decision as to which exon is removed and which exon is included involves RNA sequence elements and protein regulators. Depending on the position and function of the cis-regulatory elements, they are divided into four categories: exonic splicing enhancers (ESEs), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs) and intronic splicing silencers (ISSs). ESEs are usually bound by members of the SR (Ser–Arg) protein family6–8 (BOX 2). ISSs and ESSs are commonly bound by heterogeneous nuclear RNPs (hnRNPs; TABLE 1), which have one or more RNA-binding domains and protein–protein interaction domains9,10. ISEs are not as well characterized as the other three types of element, although recently several proteins, such as hnRNP F, hnRNP H, neuro-oncological ventral antigen 1 (NOVA1), NOVA2, FOX1 and FOX2 (also known as RBM9), have been shown to bind ISEs and to stimulate splicing11–14.
Box 2. SR proteins.
The SR (Ser–Arg) proteins are a family of nuclear factors that have many important roles in the splicing of mRNA precursors in metazoan organisms, functioning in both constitutive and alternative RNA splicing7. They are involved in many steps of splicing regulation, by binding exonic splicing enhancers (ESEs) through their RNA recognition motifs (RRMs) and mediating protein–protein41,42, and perhaps protein–RNA159, interactions through their RS (Arg–Ser repeat-containing) domains. All canonical SR proteins have common characteristics (see the table). They have a similar structure, with one or two ribonucleoprotein particle (RNP)-type RNA-binding domains at their amino termini and a variable-length domain enriched in Arg–Ser dipeptides at their carboxyl termini (the RS domain). RS domains are extensively phosphorylated and they function in splicing, usually as activators. Most SR proteins function as pivotal regulators in multiple aspects of mRNA metabolism, such as mRNA nuclear export160, nonsense-mediated mRNA decay161 and translation162. Numerous additional RS domain-containing proteins have been identified; proteins known to be involved in alternative splicing are listed in the table.
| Name* | Domains | Binding sequence | Target genes |
|---|---|---|---|
| Canonical SR proteins | |||
| SRp20 (SFRS3) | RRM and RS | GCUCCUCUUC | SRP20, CALCA and INSR |
| SC35 (SFRS2) | RRM and RS | UGCUGUU | ACHE and GRIA1–GRIA4 |
| ASF/SF2 (SFRS1) | RRM, RRMH and RS | RGAAGAAC | HIPK3, CAMK2D, HIV RNAs and GRIA1–GRIA4 |
| SRp40 (SFRS5) | RRM, RRMH and RS | AGGAGAAGGGA | HIPK3, PRKCB and FN1 |
| SRp55 (SFRS6) | RRM, RRMH and RS | GGCAGCACCUG | TNNT2 and CD44 |
| SRp75 (SFRS4) | RRM, RRMH and RS | GAAGGA | FN1, E1A and CD45 |
| 9G8 (SFRS7) | RRM, zinc finger and RS | (GAC)n | TAU, GNRH and SFRS7 |
| SRp30c (SFRS9) | RRM, RRMH and RS | CUGGAUU | BCL2L1, TAU and HNRNPA1 |
| SRp38 (FUSIP1) | RRM and RS | AAAGACAAA | GRIA2 and TRD |
| Other SR proteins | |||
| SRp54 | RRM and RS | ND | TAU |
| SRp46 (SFRS2B) | RRM and RS | ND | NA |
| RNPS1 | RRM and Ser-rich | ND | TRA2B |
| SRrp35 | RRM and RS | ND | NA |
| SRrp86 (SRrp508 and SFRS12) | RRM and RS | ND | NA |
| TRA2α | RRM and two Arg-rich | GAAARGARR | dsx |
| TRA2β | RRM and two RS | (GAA)n | SMN1, CD44 and TAU |
| RBM5 | RRM and RS | ND | CD95 |
| CAPER (RBM39) | RRM and RS | ND | VEGF |
Alternative names are provided in brackets. ACHE, acetylcholine; BCL2L1, BCL-2-like 1; CAMK2D, calcium/calmodulin-dependent protein kinase II-δ; CALCA, calcitonin-related polypeptide-α; CAPER, coactivator of activating protein 1 and oestrogen receptors; FN1, fibronectin 1; FUSIP, FUS-interacting serine-arginine-rich protein 1; GNRH, gonadotropin-releasing hormone; GRIA, glutamate receptor, ionotropic, AMPA; HIPK3, homeodomain-interacting protein kinase 3; HNRNPA1, human nuclear RNP A1; INSR, insulin receptor; PRKCB; protein kinase Cβ; RBM, RNA-binding protein; RNPS1, RNA-binding protein with Ser-rich domain 1; RRMH, RRM homology; NA, not applicable; ND, not determined; SFRS, splicing factor, Arg- and Ser-rich; SMN1, survival of motor neuron 1; TNTT2, troponin T type 2; TRA2, transformer 2; TRD, tradin; VEGF, vascular endothelial growth factor.
Table 1.
Ribonucleoproteins that are involved in pre-mRNA splicing
| Name | Other names | Domains* | Binding sequences | Target genes |
|---|---|---|---|---|
| hnRNP A1 | NA | RRM, RGG and G | UAGGGA/U | SMN2 and RAS |
| hnRNP A2 | NA | RRM, RGG and G | (UUAGGG)n | HIV tat and IKBKAP |
| hnRNP B1 | ||||
| hnRNP C1 | AUF1 | RRM | U rich | APP |
| hnRNP C2 | ||||
| hnRNP F | NA | RRM, RGG and GY | GGGA and G rich | PLP, SRC and BCL2L2 |
| hnRNP G | NA | RRM and SRGY | AAGU | SMN2 and TMP1 |
| hnRNP H | DSEF1 | RRM, RGG, GYR and GY | GGGA and G rich | PLP, HIV tat and BCL2L1 |
| hnRNP H′ | ||||
| hnRNP I | PTB | RRM | UCUU and CUCUCU | PTB, nPTB, SRC, CD95, TNTT2, CALCA and GRIN3B |
| hnRNP L | NA | RRM | C and A rich | NOS and CD45 |
| hnRNP LL | SRRF | RRM | C and A rich | CD45 |
| hnRNP M | NA | RRM and GY | ND | FGFR2 |
| hnRNP Q | NA | RRM and RGG | CC(A/C) | SMN2 |
Domains are enriched in particular amino acids, as indicated by their names. APP, amyloid-β precursor protein; BCL2L1, BCL-2-like 2; CALCA, calcitonin-related polypeptide-α; GRIN3B, glutamate receptor, ionotropic, NMDA 3B; FGFR2, fibroblast growth factor receptor 2; hnRNP, heterogeneous nuclear ribonucleoprotein particle; hnRNP LL, hnRNP L-like; IKBKAP, inhibitor of κ-light polypeptide gene enhancer in B cells, kinase complex-associated protein; NA, not applicable; ND, not determined; NOS, nitric oxide synthase; SMN2, survival motor neuron protein 2; PLP, proteolipid protein; PTB, polypyrimidine-tract binding protein; RRM, RNA recognition motif; TNTT2, troponin T type 2; TPM1, α-tropomyosin.
Choices of alternative splicing have long been thought to be made at the stages of splice site recognition and early spliceosome assembly, and indeed this is frequently the case1. However, several recent studies have shown that the decision can be made at different stages of spliceosome assembly, and even during conformational changes between the two transesterification steps15–17. In addition, there has been accumulating evidence showing the coupling of RNA transcription to splicing regulation18–22.
Alternative splicing contributes to genomic diversity and tissue specificity23. Comprehending tissue-specific alternative splicing requires an understanding of the regulatory network of protein–protein, protein–RNA and RNA–RNA interactions that are involved in this process1,24. Tissue-specific alternative splicing is thought to be controlled by differentially expressed splicing regulators1,25 and/or ubiquitously expressed splicing factors of different concentrations and/or activity26,27. In addition, tremendous progress has been made using high-throughput methods, which has not only revealed many more new alternative splicing events28–30, but has also accelerated the process of understanding its regulation in different tissues by examining the expression levels of protein regulators and helping to define cis-regulatory elements31,32. In this Review, we discuss mechanisms of alternative splicing control from different perspectives. At which stage of spliceosome assembly is the decision of whether to include an alternative exon made? What protein factors are involved? How does RNA polymerase II (RNAP II) function in alternative splicing regulation? Are the kinetics of spliceosome assembly important for this process? Which mechanisms control tissue-specific alternative splicing? We address these questions and conclude by summarizing the diversity of the mechanisms of alternative splicing regulation and speculate on the future directions of alternative splicing research.
Splice site recognition and selection
Most human genes contain multiple exons, and the average length of exons (50–250 bp) is much shorter than that of intervening sequences (frequently thousands of bps). Early stages of spliceosome assembly occur around the exons owing to the large size of introns33. This type of exon-centred splice site recognition is referred to as ‘exon definition’ (REF. 34) (BOX 1). Exon definition must eventually be converted to intron definition, which occurs by cross-intron interactions between the U1 and U2 snRNPs35,36. The best-studied mechanisms of alternative splicing regulation involve controlling splice site recognition by facilitating or interfering with the binding of the U1 or U2 snRNP to the splice sites.
Facilitating splice site recognition
SR proteins have important roles in facilitating splice site recognition. For example, they recruit the U1 snRNP to the 5′ splice site and the U2AF complex and U2 snRNP to the 3′ splice site by binding to an ESE and directly interacting with protein targets37–40 (FIG. 1a). These interactions are mediated by their RS (Arg–Ser repeat-containing) domains41,42, which have to be properly phosphorylated and dephosphorylated1,43,44. SR proteins also cooperate with other positive regulatory factors to form larger splicing enhancing complexes by interacting with other RS domain-containing proteins, such as transformer 2 (TRA2) and the SR-related nuclear matrix proteins SRm160 (also known as SRRM1) and SRm300 (also known as SRRM2)45–47 (FIG. 1a). Binding and recruiting can also be achieved by intronic binding proteins: for example, T cell-restricted intracellular antigen 1 (TIA1) binds a U-rich sequence downstream of weak 5′ splice sites to recruit the U1 snRNP48,49; and Src-associated in mitosis 68 kDa protein (SAM68; also known as KHDRBS1) binds and recruits U2AF to the 3′splice site of exon V5 of the transmembrane glycoprotein CD44 pre-mRNA50.
Figure 1. Mechanisms of alternative splicing by splice site selection.

Schematic depicting mechanisms of splicing activation. a | SR (Ser–Arg) proteins bind to exonic splicing enhancers (ESEs) to stimulate the binding of U2AF to the upstream 3′ splice site (ss) or the binding of the U1 small nuclear ribonucleoprotein (snRNP) to the downstream 5′ ss. SR proteins function with other splicing co-activators, such as transformer 2 (TRA2) and the SR-related nuclear matrix proteins SRm160–SRm300. T cell-restricted intracellular antigen 1 (TIA1) binds to U-rich sequences (intronic splicing enhancers (ISEs)) immediately downstream of 5′ splice sites to facilitate U1 binding. CELF (CUGBP- and ETR3-like factor) proteins, such as ETR3, bind to similar sequences as polypyrimidine-tract binding protein (PTB), thereby activating splicing by competing with PTB. b | Fox1 and Fox2 inhibit the inclusion of CALCA (calcitonin-related polypeptide-α) exon 4 by blocking the binding of splicing factor 1 (SF1) to the branch point (top panel) and of TRA2 and SRp55 to ESEs (bottom panel), thereby inhibiting spliceosome assembly at two stages, the E′ and E complexes. The arrow indicates that SRp55 and TRA2 promote binding of the U2AF complex. c | Single-nucleotide differences in SMN2 (survival of motor neuron 2) compared with SMN1 create binding sites for heterogeneous nuclear ribonucleoprotein particle A1 (hnRNP A1) (or hnRNP A2) in exon 7 and in the downstream intron in SMN2 pre-mRNA. hnRNP A1 (or hnRNP A2) may then inhibit the formation or stabilization of the U2 snRNP complex, either directly or indirectly by blocking the activity of the downstream TRA2-dependent ESE. Note that it has also been suggested that the base change in exon 7 destroys an ASF/SF2-dependent ESE163,164 (but see also REF. 69).
The SR protein SRp38 (also known as TASR51, NSSR52, FUSIP1 and SRrp40 (REF. 53)) has been characterized as a general splicing repressor that is activated by dephos-phorylation54,55 (see below). However, recent studies indicate that it also functions as a sequence-dependent splicing activator when phosphorylated39. It was found that in vitro SRp38 activates the formation and splicing of the A complex by facilitating the recruitment of the U1 and U2 snRNPs to the pre-mRNA and stabilizing the 5′ splice site and branch site recognition (FIG. 2a). Notably, SRp38, unlike other SR proteins, cannot complement cytoplasmic S100 extracts (which contain all factors required for splicing except SR proteins) to activate splicing56. Although an A-like complex is formed in the presence of S100 and SRp38, it is stalled or inactive and requires a specific (and currently unknown) coactivator to proceed39 (FIG. 2a). In vivo, SRp38 was shown to favour inclusion of the Flip exon of the GRIA2 (glutamate receptor, ionotropic, AMPA 2; also known as GLURB) pre-mRNA52, whereas the mutually exclusive Flop exon is included when SRp38 is absent39. Interestingly, both exons contain SRp38-binding sites, and it was proposed that the intracellular concentrations of SRp38 as well as differential binding to the exons (that is, stronger binding to the Flip exon) influence the decision to include either the Flip or Flop exon39.
Figure 2. Phosphorylation switches the general splicing repressor SRp38 into a sequence-specific activator.

a | Phosphorylated SRp38 (Ser–Arg protein 38) activates splicing by recruiting the U1 and U2 small nuclear ribonucleoprotein particles (snRNPs) to splice sites (ss). SRp38 binds SRp38-dependent exonic splicing enhancers (ESEs) in target transcripts and facilitates the association of U1 and U2 snRNPs with the pre-mRNA to stabilize 5′ ss and branch site recognition by interacting with U1 and U2 snRNPs, respectively. However, the spliceosomal A complex formed is stalled in S100 extract, in which an SRp38-specific cofactor from NF40-60 is absent, which is required to proceed through the splicing pathway. b | SRp38 enhances the inclusion of the Flip exon of GRIA2 (glutamate receptor, ionotropic, AMPA 2) pre-mRNA relative to the mutually exclusive Flop exon. Both exons contain SRp38-binding sites (indicated by black bars under exon 14 (Flop) and exon 15 (Flip)), but the site in Flip is stronger (indicated by the thicker bar), and Flip inclusion is therefore favoured in the presence of SRp38. c | Protein phosphatase 1 (PP1) dephosphorylates SRp38 on heat shock. Under normal conditions, phosphorylated SRp38 is associated with 14-3-3 proteins, which help to protect SRp38 from dephosphorylation, and PP1 activity is inhibited by PP1-associated proteins, including nuclear inhibitor of PP1 (NIPP1). During heat shock, PP1 dissociates from NIPP1 and directly binds to and dephosphorylates SRp38, which has dissociated from 14-3-3 proteins. Part a of the figure modified, with permission, from Nature Struct. Mol. Biol. REF.39 © (2008) Macmillan Publishers Ltd. All rights reserved. Part c of the figure modified, with permission, from REF.151 © (2007) Elsevier.
Inhibiting splice site recognition
Inhibition of splice site recognition can be achieved in many ways. First, when splicing silencers are located close to splice sites or to splicing enhancers, inhibition can occur by sterically blocking the access of snRNPs or of positive regulatory factors. For example, polypyrimidine-tract binding protein (PTB; also known as PTB1 and hnRNP I), binds the polypyrimidine tract and blocks the binding of U2AF to regulated exons57–59. In addition, hnRNP A1 binds ISSs that are located upstream of exon 3 in HIV Tat pre-mRNA and prevents binding of the U2 snRNP60. Finally, tissue-specific splicing factors FOX1 and FOX2 inhibit the formation of the E′ complex by binding to an intronic sequence to prevent SF1 from binding to the branch site of CALCA (calcitonin-related polypeptide-α) pre-mRNA61 (FIG. 1b).
Splicing inhibitors also sterically block the binding of activators to enhancers. Hu/ELAV family proteins inhibit U1 snRNP binding by competing with the binding of TIA1 to an AU-rich sequence downstream of the 5′ splice site of exon 23a of neurofibromatosis type 1 pre-mRNA62. FOX1 and FOX2 also inhibit E complex formation by binding to an exonic sequence in the CALCA pre-mRNA close to the ESE that TRA2 and SRp55 bind to, preventing the recruitment of U2AF by the activators61 (FIG. 1b). Finally, hnRNP A1 binds an ESS upstream of the TRA2-dependent ESE in exon 7 in the SMN2 (survival of motor neuron 2) pre-mRNA, possibly inhibiting the formation or stabilization of the U2 snRNP complex63,64 (FIG. 1c).
Some silencers can be over 100–200 bp away from enhancers, and a simple ‘bind and block’ model thus cannot explain their inhibitory effect. One explanation for the activity of such splicing inhibitors is that they function by masking splice site recognition through multimerization along the RNA58. Another model proposes that the alternative exon might be ‘looped out’ in a process involving protein–protein interactions between RNA-binding proteins bound at sites spanning the alternative exon58,65–67, and that this loop formation may sterically interfere with further spliceosome assembly, even though splice site recognition may not be inhibited67. For example, hnRNP A1 binds to elements upstream and downstream of exon 7B in its own pre-mRNA to promote skipping of exon 7B68. hnRNP A1 has also been shown to bind to exonic and intronic silencers in SMN2 exon 7 and intron 7, and it is proposed that an interaction between hnRNP A1 molecules is required to fully suppress the inclusion of SMN2 exon 7 (REFS 64,69,70). In addition, PTB has been shown to bind sites flanking the SRC N1 exon, and mutating one of the PTB sites affects the binding of PTB to the other site58,71.
Combinatorial effects of activators and inhibitors
Splicing of individual pre-mRNAs is frequently controlled by combinatorial or competitive effects of both activators and inhibitors. The final decision of whether an alternative exon is included is determined by the concentration or activity of each type of regulator, often by SR proteins and hnRNPs72–74. For example, the SR protein 9G8 (also known as SFRS7) and hnRNP F and hnRNP H regulate the splicing of α-tropomyosin exon 2 by competing for binding to the same element75; hnRNP A1 and the SR proteins ASF/SF2 (also known as SFRS1) and SC35 (also known as SFRS2) have antagonistic functions in splicing of β-tropomyosin exon 6B76; and CELF (CUGBP- and ETR3-like factor)-family proteins ETR3 (also known as CELF2) and CUGBP1 activate the splicing of exon 5 of TNTT2 (troponin T type 2; also known as cTNT) by displacing PTB77. A recent study showed that at least some Drosophila melanogaster SR proteins and hnRNPs do not have as many common targets as had been thought78. Using small interfering RNAs (siRNAs) to deplete individual splicing factors followed by splicing-sensitive microarray analysis, the authors compared genes regulated in opposite directions by hnRNPs and two SR proteins (dASF and BC52). Surprisingly, less than 5% of the genes overlap. However, a systematic analysis of more SR and hnRNPs will be necessary to decide whether antagonizing effects between these proteins is a main mode of alternative splicing regulation.
Position-dependent splicing regulation
The nature of the activity of cis-acting elements and their cognate binding proteins in some cases depends on their position relative to regulated exons. Several proteins, such as NOVA1, NOVA2, FOX1, FOX2, hnRNP L, hnRNP L-like, hnRNP F and hnRNP H, have been shown to act as either repressors or activators depending on the location of their binding site11–14,79–83. For example, NOVA1 binds to an ISE in GABRG2 (GABA A receptor, γ2) pre-mRNA and promotes inclusion of exon 9 (REF. 84), but it binds to the ESS in the alternative exon 4 of its own pre-mRNA and prevents exon 4 from being included81. Similarly to NOVA1, hnRNP L can activate or repress upstream alternative exons, and this probably depends on the location of its binding site relative to the regulated 5′ splice site12. hnRNP H promotes the formation of ATP-dependent spliceosomal complexes when it binds to G-rich sequences (G runs) downstream of the 5′ splice site85, but it inhibits splicing when the G-rich sequences are located in exons86. By using information from mRNA transcripts that are know to be targeted by NOVA1 and NOVA2 and by searching for YCAY clusters (which are NOVA1 and NOVA2 binding sequences) around regulated exons, one group11 drew an ‘mRNA map’ that includes the location of NOVA1 and NOVA2 binding sites and the consequence of each binding event. This map provides insight into the mechanisms underlying the effects of NOVA1 and NOVA2 on splicing. For example, binding of NOVA1 and NOVA2 to an ESS inhibits the formation of the pre-spliceosomal E complex by altering its composition before the binding of hnRNPs and inhibits the binding of U1 snRNP11. By contrast, NOVA1 and NOVA2 binding to an ISE downstream of the alternative exon promotes the formation of spliceosomal complexes A, B and C11. A new technique that combines CLIP and high-throughput sequencing, known as HITS-CLIP80 (CLIP-seq)13,87,88, not only verified the reproducibility of this mRNA map, but also provided genome-wide information on the target genes of NOVA1, NOVA2 and FOX2 as well as possibly mechanisms by which these proteins are regulated80.
Why does the position of splicing regulatory elements determine the action of cognate splicing factors? It is possible that enhancers are positioned so that when the splicing factors bind to them, the splice sites of the alternative exon are better presented to the splicing machinery by changing the local mRNA structure. By contrast, silencing elements function in the opposite manner, by competing with components of the splicing machinery or by changing the structure of the mRNA to impede splice site recognition.
Roles for RNA in alternative splicing regulation
The selection of the splice site can also be influenced by secondary structures in the pre-mRNA. Perhaps the most striking example of this is the complex alternative splicing that is observed in D. melanogaster Dscam pre-mRNA. The exon 6 cluster of Dscam consists of 48 mutually exclusive exons. Pairing between a conserved sequence located downstream of constitutive exon 5 (the docking site) and another conserved sequence, a variant of which is located upstream of each exon 6 variant (the selector sequence), allows the inclusion of only one exon 6 variant89; the other variants are excluded by the binding of hrp36, a D. melanogaster hnRNP A homologue, to the selector sequence90.
Secondary structures can affect alternative splicing by masking splice sites91 or by binding sites for splicing factors92,93. For example, a stem and loop secondary structure was shown to sequester alternative exon 6B of the chicken β-tropomyosin pre-mRNA, leading to its exclusion94. The IDX exon of RAS can form a secondary structure with an ISS (RASISS1), preventing the binding of hnRNP H to RASISS1. Unwinding this secondary structure by the RNA helicase P68 exposes the binding site, partially explaining the exclusion of IDX93. Riboswitches, which control gene expression in prokaryotes95, also have the potential to modulate alternative splicing. Splicing of the NMT1 (N-tetradecanoyltransferase 1) pre-mRNA in Neurosporacrassa was found to respond to the coenzyme thiamine pyrophosphate through a riboswitch-like structure96. It remains to be seen, however, whether this intriguing mechanism operates in higher eukaryotes. In mammals, small nucleolar RNAs (snoRNAs) have also been implicated in alternative splicing regulation97. For example, the snoRNA HBII 52 regulates the alternative splicing of HTR2C pre-mRNA by binding to a silencing element in exon Vb to promote its inclusion98.
Regulation by U1 and U2 snRNP pairing
After the 5′ and 3′ splice sites are recognized and exons are defined, exon definition must be converted to intron definition, which involves cross-intron interaction between U1 and U2 snRNPs, to form a functional spliceosome. Precisely when this occurs and when the commitment to splice site pairing happens have been intensively investigated over the past few years. Several studies have shown that the commitment to splicing of at least some alternative exons occurs during splice site pairing in the A complex35,36. For example, ATP hydrolysis is required for splice site pairing, which locks splice sites into a splicing pattern after U2 snRNP binding to the branch site36. Additional studies, discussed below, also provided evidence that binding of U1 and U2 snRNPs to splice sites to define an exon does not necessarily commit the exon to splicing15,16,99.
Regulation by protein factors
A new splicing inhibition mechanism was demonstrated recently by a study showing that binding of hnRNP L to an ESS can inhibit the pairing of U1 and U2 snRNPs15. An ATP-dependent spliceosome-like complex, known as A-like exon-definition complex (AEC), was found to form across alternative exon 4 of the CD45 pre-mRNA, even when its inclusion was inhibited. The AEC contains U1 and U2 snRNPs and displays the same gel mobility as the A complex, but progression into the B complex is inhibited. They proposed a model in which an A-like complex forms across exons when hnRNP L is not present, after which the U4/U6–U5 tri-snRNP complex is recruited to the intron-defined A complex to form the B complex. However, when hnRNP L is present, an hnRNP L-containing AEC prevents the U1 or U2 snRNPs bound to the splice sites of exon 4 from cross-intron pairing with the adjacent U2 or U1, resulting in exon 4 skipping. There are two possible ways in which the binding of hnRNP L might interfere with snRNP pairing. One is that binding of hnRNP L physically shields the interaction between the snRNPs. Another possibility is that hnRNP L induces a change in the conformation of the pre-mRNA that prevents cross-intron pairing of the snRNP-bound alternative exon. It is intriguing that the AEC, at least superficially, resembles the stalled A complex formed by SRp38 in S100 extract (see above). Although the importance of this is unknown, the following example suggests that such complexes may be more widespread than realized.
PTB is another inhibitory splicing factor that has been shown to function, in some cases, by blocking the transition from exon definition to intron definition16,65 (FIG. 3a). One group16 studied the mechanism of PTB inhibition by comparing the active and inactive spliceosomal complexes from neuronal WERI-1 cell nuclear extracts, in which the SRC N1 exon is included, and from HeLa cell nuclear extracts, in which the SRC N1 exon is excluded. PTB is highly expressed by HeLa cells, whereas a less repressive brain paralogue, nPTB (also known as PTB2 and brPTB), is expressed by WERI-1 cells100–103 (see below). Similarly to the example provided by hnRNP L, ATP-dependent exon definition complexes form in nuclear extracts from both cell lines. The protein compositions of the exon definition E and A complexes (EDE and EDA, respectively) that formed on constitutive exon 4 on nuclear extracts from both cell lines were similar, and PTB was found only in complexes formed in HeLa nuclear extracts. By contrast, the EDE and EDA complexes that formed on the substrate containing both exon N1 and exon 4 in WERI-1 nuclear extracts and in HeLa nuclear extracts differed in their properties and protein compositions. The WERI-1 EDE can move on to functional A, B and C complexes following ATP addition, whereas the EDE formed in HeLa nuclear extracts can only localize to a ‘dead end’ A complex. Several proteins only exist in the functional A complex formed in WERI-1 nuclear extracts, such as the PRP19 complex104,105 and SRm160–SRm300 complex46,106, which are might be important for 3′ and 5′ splice site bridging and exon N1 inclusion, and are excluded from the spliceosome by PTB. This study provides a new mechanism for how different protein compositions in different tissues can help to determine the alternative splicing pattern through a silencing factor and its interactions with other splicing factors to prevent intron definition. This study also reveals for the first time that the protein composition of different exon and intron definition complexes can vary, and thus begins to decipher a new mechanism for alternative splicing regulation and tissue specificity.
Figure 3. Mechanisms of alternative splicing regulation at the transition from exon definition to intron definition.

a | Polypyrimidine-tract binding protein (PTB) inhibits the inclusion of SRC exon N1 by inhibiting the interactions of the U1 and U2 small nuclear ribonucleoprotein particles (snRNPs) and intron definition. In both WERI-1 and HeLa cells, the N1 exon is defined by the binding of U1 snRNP to the 5′ splice site (ss) and of U2 snRNP to the branch point. In WERI-1 cells, in the absence of PTB, U1 and U2 snRNPs bound to the N1 exon interact with the U2 and U1 snRNP on adjacent constitutive exons, respectively, thereby allowing efficient spliceosome assembly on introns flanking exon N1. In HeLa cells, PTB binds to sequences flanking exon N1 and prevents the cross-intron interactions that occur in WERI-cells, thereby excluding exon N1. b | RBM5 (RNA-binding protein 5) regulates alternative splicing of CD95 by inhibiting the inclusion of exon 6. It does so not at the stage of U1 and U2 snRNP binding, but instead by promoting tri-snRNP assembly on the intron-defined spliceosomal complex between exon 5 and exon 7, while blocking tri-snRNP recruitment to complexes that would result in inclusion of exon 6. Double-headed arrows indicate intron and exon definition.
RBM5 (RNA-binding protein 5; also known as LUCA15 and H37) is a putative tumour suppressor protein107,108 that promotes the exclusion of exon 6 of CD95 (also known as FAS) pre-mRNA109. RBM5 was found to interact with sequences in exon 6 but to not affect the association of U1 and U2 snRNPs to the adjacent splice sites. Instead, RBM5 inhibited the incorporation of the U4/U6–U5 tri-snRNP complex on the introns flanking exon 6, thereby blocking the maturation of pre-spliceosomes. RBM5 also promoted the pairing of U1 and U2 at the distal splice sites, contributing further to the exclusion of exon 6 (REF. 109) (FIG. 3b).
In addition to alternative splicing regulation by the inhibition of intron definition, it is also possible that alternative splicing is stimulated by the activation of intron definition. In vitro experiments with substrates containing expanded introns have shown that the presence of binding sites for hnRNPs near intron boundaries can facilitate splicing82. This presumably involves cross-intron interactions between hnRNPs that help to bring together the ends of the introns, indicating a possible positive regulatory role for hnRNPs in splicing.
Cis-acting elements that affect U1 and U2 snRNP pairing by modulating 5′ splice site competition have recently been identified. In one study an in vitro screen was carried out for splicing silencers (ESSs and ISSs) that alter the selection of the 5′ splice site by choosing a distal, weak 5′ splice site over a proximal, strong 5′ splice site99. The isolated silencers did not affect whether U1 snRNP bound to the 5′ splice site, but instead somehow altered the conformation of the proximal U1 snRNP–5′ splice site complex so that it lost its advantage to compete with the distal U1 snRNP–5′ splice site complex for the pairing with the U2 snRNP–3′ splice site complex. This study suggests a new mechanism by which silencers can subtly affect splice site selection that does not involve sequestering splice sites, but instead entails changing the conformation of the snRNP–pre-mRNA complex. Moreover, the silencers do not affect the rate-limiting step of splicing but instead affect how well U1 and U2 snRNPs pair, which in turn can influence splice site choice.
Transcription-coupled alternative splicing
Two models have been proposed to explain the role of RNAP II in the regulation of alternative splicing110. The first model is known as the recruitment model, in which RNAP II and transcription factors interact, directly or indirectly, with splicing factors22,111,112, thereby increasing or decreasing the efficiency of splicing. One study has shown that the structure of the promoter can affect the splicing pattern of the FN1 (fibronectin 1) pre-mRNA by facilitating the differential recruitment of ASF/SF2 (REF. 113). It is conceivable that this reflects the ability of different transcription factors to influence the recruitment of distinct splicing factors to the nascent pre-mRNA, resulting in the inclusion or exclusion of the alternative exon. For example, PGC1 (peroxisome proliferator-activated receptor-γ coactivator 1), a transcription co-activator that is recruited to target genes by specific transcription factors, can modulate the alternative splicing of nascent RNA transcripts by interacting with other splicing factors through its RS domain114.
Another study showed that differential recruitment of transcription co-activators to progesterone- and oestrogen-responsive elements upstream of reporter genes, such as CD44, alters the alternative splicing of the resultant mRNA transcripts112. Specifically, recruitment of activating signal cointegrator 1(ASC1) and ASC2 and their associated proteins to these elements was found to both activate the transcription of the reporter genes and affect splicing, but in opposite ways. The ASC1-associated protein CAPER (coactivator of activating protein 1 and oestrogen receptors; also known as RBM39) contains an RS domain and two RRM domains, similarly to SR proteins, whereas the ASC2-associated protein COAA (also known as RBM14) is structurally related to hnRNP A1. It was suggested that the antagonistic effects of the ASC1 and ASC2 complexes are mediated by these factors.
A second, kinetic model proposes that the rate of transcription elongation influences the inclusion of alternative exons by affecting whether the splicing machinery is recruited sufficiently quickly for spliceosome assembly and splicing to occur. In support of this model, an RNAP II with a reduced elongation rate caused by a point mutation was found to greatly stimulate the inclusion of an alternative exon that has weak splice sites115. Specifically, it was found that slow transcription favours the inclusion of FN1 EDI exon, which was excluded when transcription was more rapid. Consistent with this, a recent study showed that changes in the RNAP II elongation rate following ultraviolet irradiation could lead to changes in alternative splicing that occur in response to DNA damage116.
One way of changing the transcription rate is through changing the phosphorylation status of RNAP II. The carboxy-terminal domain of the largest subunit of RNAP II consists of up to 52 tandem repeats of the heptapeptide consensus sequence YSPTSPS117. Excess phosphorylation on Ser5 of the C-terminal domain is associated with RNAP II stalling downstream of the promoter region, whereas phosphorylation on Ser2 is associated with elongation through the gene117,118. One group18 showed that a subunit of the human SWI–SNF (switching-defective–sucrose non-fermenting) complex, BRM, regulates changes in the alternative splicing of CD44 pre-mRNA that are stimulated following T cell activation. On T cell stimulation, RNAP II phosphorylated on Ser5 pauses at the variant exon region of CD44 by a mechanism requiring its association with BRM. Interestingly, this also results in increased association of BRM with components of the splicing machinery and splicing factor SAM68, leading to inclusion of the V5 exon. This study not only provides direct support of the kinetic model, but also shows that the mechanism for alternative splicing regulation by transcription can result from a combination of transcription elongation-related effects and differential recruitment of splicing factors.
Alternative splicing and tissue specificity
Alternative splicing regulation by tissue-specific splicing factors
Alternative splicing has an important role in defining tissue specificity. Recent high-throughput studies have shown that, of the human tissues examined, 50% or more of alternative splicing isoforms are differently expressed among tissues29, indicating that most alternative splicing is subject to tissue-specific regulation.
Tissue-specific alternative splicing events can be explained in part by tissue-specific expression of splicing factors, and the corresponding regulation of their target mRNA transcripts31,119,120. In keeping with this, numerous tissue-specific alternative splicing regulators have now been identified (TABLE 2). Among all human tissues, brain is the most functionally diverse tissue, as it has the highest occurrence of tissue-specific alternative splicing isoforms. Accordingly, several brain-specific factors have been identified, such as nPTB101,121, NOVA1, NOVA2 (REFS 81,84,122) and Hu/Elav proteins123–125. In addition, region- and cell type-specific expression of most of the >300 RNA-binding proteins examined was observed in proliferating and post-mitotic mouse brain cells126. PTB is expressed in neural progenitor cells, but its expression levels are greatly downregulated in differentiated neurons, where nPTB is upregulated100,101. Recent experiments provided evidence that the PTB-to-nPTB switch provides a post-transcriptional mechanism that is important for programming neuronal differentiation101. Using microarray analysis, it was shown that siRNA-mediated PTB depletion in N2A neuroblastoma cells caused the upregulation of nPTB and an altered alternative splicing pattern. Most of the observed changes were also detected when P19 cells (derived from an embryonal carcinoma) were differentiated into neuronal cells. In neuronal cells, expression of nPTB and downregulation of PTB explains ~25% of nervous system-specific alternative splicing101. However, the molecular mechanism that allows nervous system-specific alternative exons to be included in neuronal cells even when bound by nPTB is still unclear. It may reflect differences in the ability of nPTB and PTB to interact with other splicing factors and/or in the presence or absence of other splicing factors (for example, in the presence of Nova proteins) in neuronal and non-neuronal cells.
Table 2.
Tissue-specific alternative splicing factors
| Name | Other names | Binding domain | Binding motif | Tissue expression | Target genes |
|---|---|---|---|---|---|
| nPTB | brPTB and PTBP2 | RRM | CUCUCU | Neurons, myoblasts and testes | BIN1, GLYRA2, ATP2B1, MEF2, NASP, SPAG9 and SRC |
| NOVA1 | NA | KH | YCAY | Neurons of the hindbrain and spinal cord | GABRG2, GLYRA2 and NOVA1 |
| NOVA2 | NA | KH | YCAY | Neurons of the cortex, hippocampus and dorsal spinal cord | KCNJ, APLP2, GPHN, JNK2, NEO, GRIN1 and PLCB4 |
| FOX1 | A2BP1 | RRM | (U)GCAUG | Muscle, heart and neurons | ACTN, EWSR1, FGFR2, FN1 and SRC |
| FOX2 | RBM9 | RRM | (U)GCAUG | Muscle, heart and neurons | EWS, FGFR2, FN1 and SRC |
| RBM35a | ESRP1 | RRM | GU rich | Epithelial cells | FGFR2, CD44, CTNND1 and ENAH |
| RBM35b | ESRP2 | RRM | GU rich | Epithelial cells | FGFR2, CD44, CTNND1 and ENAH |
| TIA1 | mTIA1 | RRM | U rich | Brain, spleen and testes | MYPT1, CD95, CALCA, FGFR2, TIAR, IL8, VEGF, NF1 and COL2A1 |
| TIAR | TIAL1 and mTIAR | RRM | U rich | Brain, spleen, lung, liver and testes | TIA1, CALCA, TIAR, NF1 and CD95 |
| SLM2 | KHDRBS3 and TSTAR | KH | UAAA | Brain, tests and heart | CD44 and VEGFA |
| Quaking | QK and QKL | KH | ACUAAY[…]UAAY | Brain | MAG and PLP |
| HUB | HUC, HUD and ELAV2 | RRM | AU rich | Neurons | CALCA, CD95 and NF1 |
| MBNL | NA | CCCH zinc finger domain | YGCU(U/G)Y | Muscles, uterus and ovaries | TNTT2, INSR, CLCN1 and TNNT3 |
| CELF1 | BRUNOL2 | RRM | U and G rich | Brain | TNTT2 and INSR |
| ETR3 | CELF2 and BRUNOL3 | RRM | U and G rich | Heart, skeletal muscle and brain | TNTT2, TAU and COX2 |
| CELF4 | BRUNOL4 | RRM | U and G rich | Muscle | MTMR1 and TNTT2 |
| CELF5 | BRUNOL5 and NAPOR | RRM | U and G rich | Heart, skeletal muscle and brain | ACTN, TNTT2 and GRIN1 |
| CELF6 | BRUNOL6 | RRM | U and G rich | Kidney, brain and testes | TNTT2 |
A2BP1, ataxin 2-binding protein 1; ACTN, α-actinin; APLP2, amyloid-β precursor-like protein 2; ATP2B1, ATPase, Ca2+ transporting, plasma membrane 1; BIN1, bridging integrator 1; CALCA, calcitonin-related polypeptide-α; CELF, CUGBP- and ETR3-like factor; CLCN1, chloride channel 1; COL2A1, collagen, type II, α1; COX2, cytochrome c oxidase II; CTNND1, catenin δ1, EWSR1, Ewing sarcoma breakpoint region 1; FGFR2, fibroblast growth factor receptor 2; FN1, fibronectin 1; GABRG2, GABA A receptor, γ2; GLYRA2, glycine receptor, α2 subunit; GPHN, gephyrin; GRIN1, glutamate receptor, ionotropic, NMDA 3B; IL8, interleukin-8; INSR, insulin receptor; JNK2, Jun N-terminal kinase 2; KCNJ, potassium inwardly-rectifying channel, subfamily; KHDRBS3, KH domain-containing, RNA-binding, signal transduction-associated protein 3; MAG, myelin associated glycoprotein; MBNL, muscleblind; MEF2, myocyte enhancing factor 2; MTMR1, myotubularin-related protein 1; NASP, nuclear autoantigenic sperm protein; NEO, neogenin; NF1, neurofibromin 1; NOVA, neuro-oncological ventral antigen; PLCB4, phospholipase C β4; PLP, proteolipid protein; PTB, polypyrimidine-tract binding protein; RBM, RNA-binding protein; RRM, RNA recognition motif; SLM2, SAM68-like mammalian protein 2; SPAG9, sperm associated antigen 9; TIA1, T cell-restricted intracellular antigen 1; TIAR, TIA1-related protein; TNTT2, troponin T type 2; VEGF, vascular endothelial growth factor.
Other brain-specific factors, including Nova proteins, may be involved in fine tuning the programming of different types of neuronal cell. NOVA1 and NOVA2 are differentially expressed in post-natal mouse brain, with NOVA2 being highly expressed in the neocortex and hippocampus, and NOVA1 being expressed primarily in the hindbrain and spinal cord127. NOVA1-null mice die shortly after birth from a motor defect that is associated with apoptotic cell death of spinal and brainstem neurons. Furthermore, studies in mice in which NOVA2 has been conditionally knocked out showed that NOVA2 regulates ~7% of brain-specific splicing in the neocortex and that NOVA2-dependent alternative splicing regulates the expression of mRNA transcripts that encode synaptic functions122. These findings indicate that Nova proteins regulate alternative splicing events that result in transcripts with specific functions in the brain. Similarly to the roles of PTB and nPTB in helping to define non-neuronal and neuronal tissues, the expression of NOVA1 and NOVA2 may contribute to different functions of different brain regions. However, owing to the limited number of exons analysed and the overlap between PTB or nPTB and Nova targets, more detailed studies are needed to obtain a complete understanding of tissue-specific alternative splicing regulation by these factors.
Tissue-specific alternative splicing factors have recently been shown to be important in controlling the expression of epithelial cell-specific exons. One study128 identified two paralogues, RBM35a (also known as ESRP1) and RBM35b (also known as ESRP2), that are important for the inclusion of epithelial cell-specific exons in several mRNA transcripts128. In addition, downregulation of RBM35a was found to coincide with the loss of epithelial splicing during the epithelial-to-mesenchymal cell transition, and ectopic expression of RBM35a in mesenchymal cells restored epithelial splicing. These data show that RBM35a and RBM35b contribute to defining the distinguishing characteristics of epithelial cells.
Alternative splicing regulation by constitutive splicing factors
SR proteins were originally discovered by biochemical methods as general, or non-sequence-specific, splicing activators7. However, more recent findings indicate that individual SR proteins can act as specific alternative splicing regulators in different cell types and tissues. Disruption of the genes that encode ASF/SF2 and SC35 specifically in the heart have shown that they have important but distinct roles in tissue development129,130. In addition, a recent study showed that mice with complete ablation of SRp38 survived through early embryogenesis and, strikingly, displayed only cardiac defects; these mice showed differences in alternative splicing131.
Core spliceosomal proteins (CSPs) are also involved in alternative splicing regulation. Analysis of microarray-based expression profiles from mouse, chimpanzee and human tissues revealed that snRNPs are differentially expressed in particular tissues132. This is consistent with results from an RNA interference (RNAi) screen in D. melanogaster, which showed that changing levels of CSPs leads to changes in alternative splicing26. These CSPs include components of U1, U2 and U4/U6 snRNPs, as well as the U2AF heterodimer. Further evidence was provided by RNAi knockdown of the isoforms of U2AF35, U2AF35a and/or U2AF35b, and a subunit of splicing factor SF3B, SAP155 (also known as SF3B1), in human cells44,133,134. Knockdown of these CSPs was found to affect only alternative splicing of a subset of transcripts: for example, mRNAs encoding cell cycle phosphatases in the case of U2AF35 and 5′ splice site selection of BCL2L1 (BCL-2-like 1; also known as BCLX) pre-mRNA in response to ceramide in the case of SAP155. In addition, evidence from budding yeast that showed differences in splicing patterns in response to different kinds of stress also suggests that CSPs are involved in alternative splicing regulation135.
SMN, which is part of the SMN complex, has recently been shown to regulate alternative splicing in many mouse tissues27. The SMN complex is important for efficient assembly of snRNPs, and depletion of SMN in HeLa cells leads to a decrease in snRNP levels. SMN-deficient mice showed tissue-specific alterations in snRNAs, and different snRNPs were affected in different tissues, leading to an altered stoichiometry of snRNPs27. In addition, microarray analysis of total RNA samples from different tissues of SMN-deficient mice revealed changes in several alternative splicing events in various tissues27. The mechanism of alternative splicing alteration by SMN deficiency is unknown, but it is probable that the resulting changes in snRNP levels directly affected alternative splicing of specific pre-mRNAs. It will be important to understand how, and whether, these changes in alternative splicing contribute to spinal muscular atrophy, which is caused by SMN deficiency136,137.
Alternative splicing regulation by post-translational modifications of splicing factors
Differences in protein expression levels of either tissue-specific splicing regulators or CSPs may not fully explain how cells can change alternative splicing patterns rapidly, for example in response to cellular stress. Mounting evidence has shown that post-translational modification of splicing factors can affect alternative splicing. The best studied modification is phosphorylation, and consistent with this, several well-studied cell signalling pathways have been shown to be involved in alternative splicing regulation (reviewed in REFS 138,139). Phosphorylation has been shown to affect the local concentration of splicing factors that are adjacent to pre-mRNA substrates, by altering their intracellular localization140–144, protein–protein43 and protein–RNA interactions50,145 and even intrinsic splicing activity39,54.
Phosphorylation can change the ability of splicing factors to interact with other proteins or mRNA substrates, leading to changes in splice site selection. Phosphorylation of RS domains in SR proteins affects their interaction with CSPs43 and is necessary for sequence-specific mRNA binding in vitro146. Phosphorylation of TIA1 and TIA1-related protein (TIAR) by Fas-activated serine/threonine kinase (FASTK) enhances TIA1 and TIAR-mediated recruitment of U1 snRNP to a suboptimal 5′ splice site, leading to the inclusion of CD95 exon 6 (REF. 147). Tyrosine phosphorylation of SAM68 by Fyn protein kinase favours the formation of the anti-apoptotic BCLXL (B cell lymphoma XL) mRNA by interfering with the interaction between SAM68 and hnRNP A1, and with the interaction of both proteins with the pre-mRNA148. Phosphorylation of RS motifs of PTB-associated splicing factor PSF (also known as SPFQ) by SR kinases inhibits the binding of PSF to mRNA145.
Phosphorylation can also change the intracellular localization of splicing factors. Osmotic shock stresses cells and activates the signalling pathway involving MEK3 (also known as MAPKK3), MEK6 (also known as MAPKK6) and p38, which leads to the relocalization of hnRNP A1 to the cytoplasm as a result of hyperphosphorylation. This change in hnRNP A1 localization can alter the alternative splicing pattern of an adenovirus E1A reporter transcript141,144. Furthermore, ischaemia triggers changes in Ca2+ concentration, leading to hyperphosphorylation of a TRA2 isoform, TRA2β1, and to its localization to the cytoplasm143. In addition, protein kinase A phosphorylates PTB on Ser16, which leads to its translocation to the cytoplasm149,150.
Phosphorylation status can also, in one case, determine whether a splicing factor functions as a splicing repressor or activator. SRp38 acts as a global splicing repressor when dephosphorylated in the M phase of the cell cycle and following heat shock54,55. However, it becomes a sequence-specific activator when phosphorylated39. Shi and Manley151 elucidated the detailed regulatory mechanism of SRp38 phosphorylation in response to heat shock. Specifically, at normal temperatures two mechanisms ensure that SRp38 remains phosphorylated (FIG. 2c): first, SRp38 is bound and protected by 14-3-3 proteins, and second, protein phosphatase 1 (PP1), which has been shown to target SRp38, is masked by associated proteins, including nuclear inhibitor of PP1 (NIPP1), which had previously been implicated in splicing control152,153. At increased temperatures 14-3-3 proteins dissociate from SRp38, and PP1 is released from NIPP1, thereby freeing PP1 and allowing it to dephosphorylate SRp38. Unlike other SR proteins, SRp38 is a poor substrate for the SR protein kinases CLK1 (CDC-like kinase; also known as STY) and SRPK1. Therefore, after dephosphorylation SRp38 remains dephosphorylated and can thus repress splicing events, unlike other SR proteins, which are rapidly rephosphorylated.
Conclusions and perspectives
The studies described here reveal the complexity of alternative splicing regulation. Alternative splicing can be regulated at different steps of spliceosome assembly by different splicing factors, both general and specific, and by many mechanisms that rely on cis-acting elements. Although alternative exons are shorter than constitutive exons and are flanked by longer introns, alternative exons are more conserved than constitutive exons, especially the exon–intron junctions, and these conserved regions often extend into flanking introns for 80–100 nucleotides154, where cis-regulatory elements are embedded. Correct alternative splicing also depends on the stoichiometry and interactions of positive and negative regulatory proteins, including CSPs. Each cell type has a unique repertoire of SR proteins and hnRNPs, and moderate changes in their relative stoichiometry can have great effects on the pattern of alternative splicing1,63. It is possible that changes in the stoichiometry of snRNPs perturb the complex network of splicing factors and the interactions between splicing factors and CSPs. Therefore, alternative splicing regulatory networks have such an exquisite architecture that perturbation of any single step can lead to alternative splicing misregulation.
Diverse mechanisms are used to ensure tissue and cell type-specific splicing regulation. Accumulating evidence has shown that alternative splicing plays an important part in defining tissue specificity. The action of sequence- specific transcription factors has been thought to be the most robust way of defining tissue specificity155. Importantly, >2,500 transcription factors have been identified in humans156, whereas the reported number of sequence-specific alternative splicing factors is <50. Given that it now seems that alternative splicing is as prevalent and perhaps as important a mechanism as transcriptional control, what might be the explanation for this? One possibility is that many more splicing regulators remain to be discovered. However, the total number of putative RBPs in mice has been estimated to be less than 400 (REF. 126), and some fraction of these proteins will not be involved in splicing. Another possibility is that there are fundamental differences in how splicing and transcription are regulated. For example, individual splicing regulators control much larger groups of genes than specific transcription factors. This is consistent with the large numbers of neuronal transcripts that are thought to be controlled by PTB and Nova proteins80,101,103,122. It is also probable that considerable regulation is achieved by combinations of abundant regulators with limited sequence-specificity (that is, the SR proteins and hnRNPs), which act in concert to regulate different mRNA transcripts in different tissues depending on their relative concentrations.
Important goals of future studies of alternative splicing regulation include understanding how regulators switch key splicing events during development and in response to environmental stimuli, and how misregulation of alternative splicing leads to disease. Complete characterization of tissue-specific patterns of expression is of great importance to defining mechanisms of alternative splicing regulation in different cell types. De novo identification of regulatory motifs31 and HITS-CLIP80 are two complementary approaches to achieve this goal. However, until now only a limited number of alternative splicing regulators and tissues have been studied by these methods. In addition to Nova proteins and FOX2, CLIP-seq also provided a landscape of potential ASF/SF2 target mRNA transcripts and characterized a purine-rich consensus motif87,88 that is nearly identical to a consensus sequence obtained previously by an in vitro SELEX (systematic evolution of ligands by exponential enrichment) approach157. A database that includes more comprehensive information on the expression patterns of proteins that regulate alternative splicing, definition of potential target mRNA transcripts and positions of binding motifs on these transcripts will facilitate searches for regulatory proteins that control specific splicing events, such as of genes that are involved in disease, and possibly provide insights into underlying mechanisms.
Owing to the dynamic nature of spliceosome assembly and the potential for regulation at multiple points, detailed proteomic analysis will be important in completely elucidating the molecular mechanisms of alternative splicing regulation. For example, how do RNA-binding proteins interact with other factors and core splicing factors? When and where do splicing factors function to regulate the spliceosome? How do post-translational modifications influence these events? Obtaining a full understanding of the mechanisms underlying alternative splicing and its role in defining tissue specificity will require multiple approaches and methods.
Acknowledgments
Work from the authors’ laboratory was supported in part by grants from the National Institutes of Health. We thank C. David for comments on the manuscript.
- Small nuclear ribonucleoprotein particle
(snRNP). A protein, including U1, U2, U4, U5 and U6, which contains U-rich small nuclear RNAs (snRNAs) and both small nuclear ribonucleoprotein (snRNP)-specific and common proteins, and is a core component of the spliceosome
- Branch point
A nucleotide, usually an adenosine, within a variably conserved branch point sequence upstream of the 3′ splice site, the 2′ hydroxyl group of which attacks the 5′ splice site in the first step of splicing
- SR (Ser–Arg) protein family
A family of nuclear factors that have many important roles in splicing mRNA precursors in metazoan organisms, functioning in both constitutive and alternative splicing
- Heterologous nuclear RNP
(hnRNP). A pre-mRNA- or mRNA-binding protein that associates with transcripts during or after transcription and influences their function and fate. Some hnRNPs shuttle in and out of nuclei, whereas others are constitutively nuclear
- Alternative exon
An exon that is included in mature mRNA in certain cellular contexts but excluded in others
- RS (Arg–Ser repeat-containing) domain
A protein domain that is variable in length and enriched in Arg–Ser dipeptides and seems to be involved in protein–protein and protein–RNA interactions
- Hu/ELAV family protein
A protein belonging to a family of nervous system-specific RNA-binding proteins that specifically bind to AU-rich sequences
- CLIP
A method that combines cross-linking and immunoprecipitation to identify in vivo targets of RNA-binding proteins
- RRM domain
(RNA recognition motif domain). A protein domain that is frequently involved in sequence-specific single-stranded RNA binding. Also known as an RNP-type RNA-binding domain
- 14-3-3 protein
A protein belonging to a family of conserved proteins that bind to phosphorylated serine and threonine residues and that are encoded by seven genes in most mammals. They bind diverse regulatory proteins, including kinases, phosphatases and transmembrane receptors
- SELEX
A technique to determine the DNA or RNA sequence that is specifically recognized by a protein. The method involves multiple rounds of binding to an initially random sequence until a high-affinity consensus sequence emerges
Footnotes
DATABASES
UniProtKB: http://www.uniprot.org
Fox1|Fox2|hnRNP A1|hnRNP F|hnRNP H|hnRNP L|NOVA1|NOVA2|nPTB|PTB|RBMS|SAM68|SRp38
FURTHER INFORMATION
James L. Manley’s homepage: http://www.columbia.edu/cu/biology/faculty/manley
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–787. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- 4.Zamore PD, Green MR. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci USA. 1989;86:9243–9247. doi: 10.1073/pnas.86.23.9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson KK, Green MR. Mammalian U2 snRNP has a sequence-specific RNA-binding activity. Genes Dev. 1989;3:1562–1571. doi: 10.1101/gad.3.10.1562. [DOI] [PubMed] [Google Scholar]
- 6.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tacke R, Manley JL. Determinants of SR protein specificity. Curr Opin Cell Biol. 1999;11:358–362. doi: 10.1016/S0955-0674(99)80050-7. [DOI] [PubMed] [Google Scholar]
- 8.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 9.Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 10.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nature Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 11.Ule J, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 12.Hui J, et al. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J. 2005;24:1988–1998. doi: 10.1038/sj.emboj.7600677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeo GW, et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nature Struct Mol Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mauger DM, Lin C, Garcia-Blanco MA. hnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol Cell Biol. 2008;28:5403–5419. doi: 10.1128/MCB.00739-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.House AE, Lynch KW. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nature Struct Mol Biol. 2006;13:937–944. doi: 10.1038/nsmb1149. [DOI] [PubMed] [Google Scholar]
- 16.Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nature Struct Mol Biol. 2008;15:183–191. doi: 10.1038/nsmb.1375. This study shows that PTB inhibits SRC exon N1 inclusion by preventing the transition from an exon-definition to an intron-definition complex and analyses the protein composition of different complexes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lallena MJ, Chalmers KJ, Llamazares S, Lamond AI, Valcarcel J. Splicing regulation at the second catalytic step by Sex-lethal involves 3′ splice site recognition by SPF45. Cell. 2002;109:285–296. doi: 10.1016/s0092-8674(02)00730-4. [DOI] [PubMed] [Google Scholar]
- 18.Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nature Struct Mol Biol. 2006;13:22–29. doi: 10.1038/nsmb1030. This study shows that BRM promotes the inclusion of variable exons of CD44 pre-mRNA by stalling RNAP II at the variable exon-containing region of the CD44 gene. It also shows that BRM interacts with splicing factor SAM68. [DOI] [PubMed] [Google Scholar]
- 19.de la Mata M, Kornblihtt AR. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nature Struct Mol Biol. 2006;13:973–980. doi: 10.1038/nsmb1155. [DOI] [PubMed] [Google Scholar]
- 20.Sims RJ, 3rd, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin S, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD. The splicing factor SC35 has an active role in transcriptional elongation. Nature Struct Mol Biol. 2008;15:819–826. doi: 10.1038/nsmb.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moldon A, et al. Promoter-driven splicing regulation in fission yeast. Nature. 2008;455:997–1000. doi: 10.1038/nature07325. [DOI] [PubMed] [Google Scholar]
- 23.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 24.Blencowe BJ, Graveley BR, editors. Alternative Splicing in the Postgenomic Era. Springer; the Netherlands: 2007. [Google Scholar]
- 25.Grabowski PJ, Black DL. Alternative RNA splicing in the nervous system. Prog Neurobiol. 2001;65:289–308. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- 26.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. This paper shows that SMN deficiency regulates snRNP levels in a tissue-specific manner, and this was reflected in altered alternative splicing patterns in different mouse tissues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 29.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 31.Castle JC, et al. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nature Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. A transcriptome study that analyses alternative splicing events from 48 tissues and identifies tissue-specific regulatory motifs and cognate binding proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sultan M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 33.Sterner DA, Carlo T, Berget SM. Architectural limits on split genes. Proc Natl Acad Sci USA. 1996;93:15081–15085. doi: 10.1073/pnas.93.26.15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berget SM. Exon recognition in vertebrate splicing. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 35.Lim SR, Hertel KJ. Commitment to splice site pairing coincides with A complex formation. MolCell. 2004;15:477–483. doi: 10.1016/j.molcel.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 36.Kotlajich MV, Crabb TL, Hertel KJ. Spliceosome assembly pathways for different types of alternative splicing converge during commitment to splice site pairing in the A complex. Mol Cell Biol. 2009;29:1072–1082. doi: 10.1128/MCB.01071-08. This study shows that the commitment to splicing of some alternative exons occurs during splice site pairing in the A complex and that ATP hydrolysis is required for splice site paring, thereby locking splice sites into a splicing pattern after U2 snRNP binding to the branch site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bourgeois CF, Popielarz M, Hildwein G, Stevenin J. Identification of a bidirectional splicing enhancer: differential involvement of SR proteins in 5′ or 3′ splice site activation. Mol Cell Biol. 1999;19:7347–7356. doi: 10.1128/mcb.19.11.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuo P, Maniatis T. The splicing factor U2AF35 mediates critical protein–protein interactions in constitutive and enhancer-dependent splicing. Genes Dev. 1996;10:1356–1368. doi: 10.1101/gad.10.11.1356. [DOI] [PubMed] [Google Scholar]
- 39.Feng Y, Chen M, Manley JL. Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nature Struct Mol Biol. 2008;15:1040–1048. doi: 10.1038/nsmb.1485. This paper shows that phosphorylation switches SRp38 from a general repressor to a sequence-specific activator that functions by recruiting and stabilizing U1 and U2 snRNP at splice sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graveley BR, Hertel KJ, Maniatis T. The role of U2AF35 and U2AF65 in enhancer-dependent splicing. RNA. 2001;7:806–818. doi: 10.1017/s1355838201010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohtz JD, et al. Protein–protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 42.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 43.Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997;11:334–344. doi: 10.1101/gad.11.3.334. [DOI] [PubMed] [Google Scholar]
- 44.Pacheco TR, Coelho MB, Desterro JM, Mollet I, Carmo-Fonseca M. In vivo requirement of the small subunit of U2AF for recognition of a weak 3′ splice site. Mol Cell Biol. 2006;26:8183–90. doi: 10.1128/MCB.00350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longman D, et al. Multiple interactions between SRm160 and SR family proteins in enhancer-dependent splicing and development of C. elegans. Curr Biol. 2001;11:1923–1933. doi: 10.1016/s0960-9822(01)00589-9. [DOI] [PubMed] [Google Scholar]
- 46.Blencowe BJ, et al. The SRm160/300 splicing coactivator subunits. RNA. 2000;6:111–120. doi: 10.1017/s1355838200991982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tacke R, Manley JL. Functions of SR and Tra2 proteins in pre-mRNA splicing regulation. Proc Soc Exp Biol Med. 1999;220:59–63. doi: 10.1046/j.1525-1373.1999.d01-10.x. [DOI] [PubMed] [Google Scholar]
- 48.Izquierdo JM, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. MolCell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 49.Forch P, Puig O, Martinez C, Seraphin B, Valcarcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J. 2002;21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tisserant A, Konig H. Signal-regulated pre-mRNA occupancy by the general splicing factor U2AF. PLoS ONE. 2008;3:e1418. doi: 10.1371/journal.pone.0001418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Embree LJ, Tsai S, Hickstein DD. Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem. 1998;273:27761–27764. doi: 10.1074/jbc.273.43.27761. [DOI] [PubMed] [Google Scholar]
- 52.Komatsu M, Kominami E, Arahata K, Tsukahara T. Cloning and characterization of two neural-salient serine/arginine-rich (NSSR) proteins involved in the regulation of alternative splicing in neurones. Genes Cells. 1999;4:593–606. doi: 10.1046/j.1365-2443.1999.00286.x. [DOI] [PubMed] [Google Scholar]
- 53.Cowper AE, Caceres JF, Mayeda A, Screaton GR. Serine-arginine (SR) protein-like factors that antagonize authentic SR proteins and regulate alternative splicing. J Biol Chem. 2001;276:48908–48914. doi: 10.1074/jbc.M103967200. [DOI] [PubMed] [Google Scholar]
- 54.Shin C, Manley JL. The SR protein SRp38 represses splicing in M phase cells. Cell. 2002;111:407–417. doi: 10.1016/s0092-8674(02)01038-3. [DOI] [PubMed] [Google Scholar]
- 55.Shin C, Feng Y, Manley JL. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature. 2004;427:553–558. doi: 10.1038/nature02288. [DOI] [PubMed] [Google Scholar]
- 56.Krainer AR, Conway GC, Kozak D. Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 1990;4:1158–1171. doi: 10.1101/gad.4.7.1158. [DOI] [PubMed] [Google Scholar]
- 57.Singh R, Valcarcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 58.Spellman R, Smith CW. Novel modes of splicing repression by PTB. Trends Biochem Sci. 2006;31:73–76. doi: 10.1016/j.tibs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Sauliere J, Sureau A, Expert-Bezancon A, Marie J. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the β-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol Cell Biol. 2006;26:8755–8769. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tange TO, Damgaard CK, Guth S, Valcarcel J, Kjems J. The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001;20:5748–5758. doi: 10.1093/emboj/20.20.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou HL, Lou H. Repression of prespliceosome complex formation at two distinct steps by Fox-1/Fox-2 proteins. Mol Cell Biol. 2008;28:5507–5516. doi: 10.1128/MCB.00530-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu H, Hinman MN, Hasman RA, Mehta P, Lou H. Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mRNA. Mol Cell Biol. 2008;28:1240–1251. doi: 10.1128/MCB.01509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nature Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 64.Martins de Araujo M, Bonnal S, Hastings ML, Krainer AR, Valcarcel J. Differential 3′ splice site recognition of SMN1 and SMN2 transcripts by U2AF and U2 snRNP. RNA. 2009;15:515–523. doi: 10.1261/rna.1273209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma S, Falick AM, Black DL. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol Cell. 2005;19:485–496. doi: 10.1016/j.molcel.2005.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Damgaard CK, Tange TO, Kjems J. hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA. 2002;8:1401–1415. doi: 10.1017/s1355838202023075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nasim FU, Hutchison S, Cordeau M, Chabot B. High-affinity hnRNP A1 binding sites and duplex-forming inverted repeats have similar effects on 5′ splice site selection in support of a common looping out and repression mechanism. RNA. 2002;8:1078–1089. doi: 10.1017/s1355838202024056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hutchison S, LeBel C, Blanchette M, Chabot B. Distinct sets of adjacent heterogeneous nuclear ribonucleoprotein (hnRNP) A1/A2 binding sites control 5′ splice site selection in the hnRNP A1 mRNA precursor. J Biol Chem. 2002;277:29745–29752. doi: 10.1074/jbc.M203633200. [DOI] [PubMed] [Google Scholar]
- 69.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 70.Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104:3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chou MY, Underwood JG, Nikolic J, Luu MH, Black DL. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Mol Cell. 2000;5:949–957. doi: 10.1016/s1097-2765(00)80260-9. [DOI] [PubMed] [Google Scholar]
- 72.Mayeda A, Helfman DM, Krainer AR. Modulation of exon skipping and inclusion by heterogeneous nuclear ribonucleoprotein A1 and pre-mRNA splicing factor SF2/ASF. Mol Cell Biol. 1993;13:2993–3001. doi: 10.1128/mcb.13.5.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zahler AM, Damgaard CK, Kjems J, Caputi M. SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing. J Biol Chem. 2004;279:10077–10084. doi: 10.1074/jbc.M312743200. [DOI] [PubMed] [Google Scholar]
- 74.Zhu J, Krainer AR. Pre-mRNA splicing in the absence of an SR protein RS domain. Genes Dev. 2000;14:3166–3178. doi: 10.1101/gad.189500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Crawford JB, Patton JG. Activation of α-tropomyosin exon 2 is regulated by the SR protein 9G8 and heterogeneous nuclear ribonucleoproteins H and F. Mol Cell Biol. 2006;26:8791–802. doi: 10.1128/MCB.01677-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Expert-Bezancon A, et al. hnRNP A1 and the SR proteins ASF/SF2 and SC35 have antagonistic functions in splicing of β-tropomyosin exon 6B. J Biol Chem. 2004;279:38249–59. doi: 10.1074/jbc.M405377200. [DOI] [PubMed] [Google Scholar]
- 77.Charlet BN, Logan P, Singh G, Cooper TA. Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol Cell. 2002;9:649–658. doi: 10.1016/s1097-2765(02)00479-3. [DOI] [PubMed] [Google Scholar]
- 78.Blanchette M, et al. Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol Cell. 2009;33:438–449. doi: 10.1016/j.molcel.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hung LH, et al. Diverse roles of hnRNP L in mammalian mRNA processing: a combined microarray and RNAi analysis. RNA. 2008;14:284–296. doi: 10.1261/rna.725208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. This paper introduces a method that combines CLIP and high-throughput sequencing to identify targets of Nova proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dredge BK, Stefani G, Engelhard CC, Darnell RB. Nova autoregulation reveals dual functions in neuronal splicing. EMBO J. 2005;24:1608–1620. doi: 10.1038/sj.emboj.7600630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez-Contreras R, et al. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006;4:e21. doi: 10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dredge BK, Darnell RB. Nova regulates GABAA receptor γ2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Mol Cell Biol. 2003;23:4687–4700. doi: 10.1128/MCB.23.13.4687-4700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schaub MC, Lopez SR, Caputi M. Members of the heterogeneous nuclear ribonucleoprotein H family activate splicing of an HIV-1 splicing substrate by promoting formation of ATP-dependent spliceosomal complexes. J Biol Chem. 2007;282:13617–13626. doi: 10.1074/jbc.M700774200. [DOI] [PubMed] [Google Scholar]
- 86.Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J Biol Chem. 2001;276:43850–43859. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- 87.Sanford JR, et al. Identification of nuclear and cytoplasmic mRNA targets for the shuttling protein SF2/ASF. PLoS ONE. 2008;3:e3369. doi: 10.1371/journal.pone.0003369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanford JR, et al. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Olson S, et al. A regulator of Dscam mutually exclusive splicing fidelity. Nature Struct Mol Biol. 2007;14:1134–1140. doi: 10.1038/nsmb1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grover A, et al. 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 92.Hiller M, Zhang Z, Backofen R, Stamm S. Pre-mRNA secondary structures influence exon recognition. PLoS Genet. 2007;3:e204. doi: 10.1371/journal.pgen.0030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Camats M, Guil S, Kokolo M, Bach-Elias M. P68 RNA helicase (DDX5) alters activity of cis- and trans-acting factors of the alternative splicing of H-Ras. PLoS ONE. 2008;3:e2926. doi: 10.1371/journal.pone.0002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Libri D, Balvay L, Fiszman MY. In vivo splicing of the beta tropomyosin pre-mRNA: a role for branch point and donor site competition. Mol Cell Biol. 1992;12:3204–3215. doi: 10.1128/mcb.12.7.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henkin TM. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008;22:3383–3390. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 97.Kishore S, Stamm S. Regulation of alternative splicing by snoRNAs. Cold Spring Harb Symp Quant Biol. 2006;71:329–334. doi: 10.1101/sqb.2006.71.024. [DOI] [PubMed] [Google Scholar]
- 98.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 99.Yu Y, et al. Dynamic regulation of alternative splicing by silencers that modulate 5′ splice site competition. Cell. 2008;135:1224–1236. doi: 10.1016/j.cell.2008.10.046. The authors screen for splicing silencers that favour the inclusion of a distal 5′ splice site and provide evidence that the silencers work by changing the conformation of the pre-mRNA–U1 snRNP complex and promoting pairing of the distal U1 snRNP with U2 snRNP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Boutz PL, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. Using siRNA and microarray analysis, the authors show that the PTB-to-nPTB switch provides a post-transcriptional mechanism that is important for programming neuronal differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coutinho-Mansfield GC, Xue Y, Zhang Y, Fu XD. PTB/nPTB switch: a post-transcriptional mechanism for programming neuronal differentiation. Genes Dev. 2007;21:1573–1577. doi: 10.1101/gad.1575607. [DOI] [PubMed] [Google Scholar]
- 103.Spellman R, Llorian M, Smith CW. Cross regulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell. 2007;27:420–434. doi: 10.1016/j.molcel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohi MD, et al. Structural and functional analysis of essential pre-mRNA splicing factor Prp19p. Mol Cell Biol. 2005;25:451–460. doi: 10.1128/MCB.25.1.451-460.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R. Isolation of an active step I spliceosome and composition of its RNP core. Nature. 2008;452:846–850. doi: 10.1038/nature06842. [DOI] [PubMed] [Google Scholar]
- 106.Eldridge AG, Li Y, Sharp PA, Blencowe BJ. The SRm 160/300 splicing coactivator is required for exon-enhancer function. Proc Natl Acad Sci USA. 1999;96:6125–6130. doi: 10.1073/pnas.96.11.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Edamatsu H, Kaziro Y, Itoh H. LUCA15, a putative tumour suppressor gene encoding an RNA-binding nuclear protein, is down-regulated in ras-transformed Rat-1 cells. Genes Cells. 2000;5:849–858. doi: 10.1046/j.1365-2443.2000.00370.x. [DOI] [PubMed] [Google Scholar]
- 108.Mourtada-Maarabouni M, Sutherland LC, Williams GT. Candidate tumour suppressor LUCA-15 can regulate multiple apoptotic pathways. Apoptosis. 2002;7:421–432. doi: 10.1023/a:1020083008017. [DOI] [PubMed] [Google Scholar]
- 109.Bonnal S, et al. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell. 2008;32:81–95. doi: 10.1016/j.molcel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 110.Kornblihtt AR. Chromatin, transcript elongation and alternative splicing. Nature Struct Mol Biol. 2006;13:5–7. doi: 10.1038/nsmb0106-5. [DOI] [PubMed] [Google Scholar]
- 111.Das R, et al. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26:867–881. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 112.Auboeuf D, et al. Differential recruitment of nuclear receptor coactivators may determine alternative RNA splice site choice in target genes. Proc Natl Acad Sci USA. 2004;101:2270–2274. doi: 10.1073/pnas.0308133100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cramer P, et al. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell. 1999;4:251–258. doi: 10.1016/s1097-2765(00)80372-x. [DOI] [PubMed] [Google Scholar]
- 114.Monsalve M, et al. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell. 2000;6:307–316. doi: 10.1016/s1097-2765(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 115.de la Mata M, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 116.Munoz MJ, et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 2009;137:708–720. doi: 10.1016/j.cell.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 117.Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 118.Lin PS, Marshall NF, Dahmus ME. CTD phosphatase: role in RNA polymerase II cycling and the regulation of transcript elongation. Prog Nucleic Acid Res Mol Biol. 2002;72:333–365. doi: 10.1016/s0079-6603(02)72074-6. [DOI] [PubMed] [Google Scholar]
- 119.David CJ, Manley JL. The search for alternative splicing regulators: new approaches offer a path to a splicing code. Genes Dev. 2008;22:279–285. doi: 10.1101/gad.1643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ohno G, Hagiwara M, Kuroyanagi H. STAR family RNA-binding protein ASD-2 regulates developmental switching of mutually exclusive alternative splicing in vivo. Genes Dev. 2008;22:360–374. doi: 10.1101/gad.1620608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nature Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 122.Ule J, et al. Nova regulates brain-specific splicing to shape the synapse. Nature Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 123.Perrone-Bizzozero N, Bolognani F. Role of HuD and other RNA-binding proteins in neural development and plasticity. J Neurosci Res. 2002;68:121–126. doi: 10.1002/jnr.10175. [DOI] [PubMed] [Google Scholar]
- 124.Zhu H, Hasman RA, Barron VA, Luo G, Lou H. A nuclear function of Hu proteins as neuron-specific alternative RNA processing regulators. Mol Biol Cell. 2006;17:5105–5114. doi: 10.1091/mbc.E06-02-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Soller M, Li M, Haussmann IU. Regulation of the ELAV target ewg: insights from an evolutionary perspective. Biochem Soc Trans. 2008;36:502–504. doi: 10.1042/BST0360502. [DOI] [PubMed] [Google Scholar]
- 126.McKee AE, et al. A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Dev Biol. 2005;5:14. doi: 10.1186/1471-213X-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang YY, Yin GL, Darnell RB. The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc Natl Acad Sci USA. 1998;95:13254–13259. doi: 10.1073/pnas.95.22.13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. The authors identify ESRP1 and ESRP2 as epithelial cell-specific alternative splicing factors and find that they regulate several epithelial cell-specific exons. They also show that ESRP1 and ESRP2 expression levels correlate with the splicing pattern change that is observed during the epithelial-to-mesenchymal cell transition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ding JH, et al. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J. 2004;23:885–896. doi: 10.1038/sj.emboj.7600054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu X, et al. ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 131.Feng Y, et al. SRp38 regulates alternative splicing and is required for Ca2+ handling in the embryonic heart. Dev Cell. 2009;16:528–538. doi: 10.1016/j.devcel.2009.02.009. This study shows that mice lacking SRp38 die perinatally and have multiple cardiac defects, and that the mRNA encoding cardiac triadin is a direct target of SRp38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grosso AR, et al. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008;36:4823–4832. doi: 10.1093/nar/gkn463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Massiello A, Roesser JR, Chalfant CE. SAP155 binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5′ splice site selection of Bcl-x pre-mRNA. FASEB J. 2006;20:1680–1682. doi: 10.1096/fj.05-5021fje. [DOI] [PubMed] [Google Scholar]
- 134.Pacheco TR, Moita LF, Gomes AQ, Hacohen N, Carmo-Fonseca M. RNA interference knockdown of hU2AF35 impairs cell cycle progression and modulates alternative splicing of Cdc25 transcripts. Mol Biol Cell. 2006;17:4187–4199. doi: 10.1091/mbc.E06-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol Cell. 2007;27:928–937. doi: 10.1016/j.molcel.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 137.Gabanella F, et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shin C, Manley JL. Cell signalling and the control of pre-mRNA splicing. Nature Rev Mol Cell Biol. 2004;5:727–738. doi: 10.1038/nrm1467. [DOI] [PubMed] [Google Scholar]
- 139.Tarn WY. Cellular signals modulate alternative splicing. J Biomed Sci. 2007;14:517–522. doi: 10.1007/s11373-007-9161-7. [DOI] [PubMed] [Google Scholar]
- 140.Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci USA. 2004;101:9666–9670. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van der Houven van Oordt W, et al. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol. 2000;149:307–316. doi: 10.1083/jcb.149.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Habelhah H, et al. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nature Cell Biol. 2001;3:325–330. doi: 10.1038/35060131. [DOI] [PubMed] [Google Scholar]
- 143.Daoud R, et al. Ischemia induces a translocation of the splicing factor tra2-beta 1 and changes alternative splicing patterns in the brain. J Neurosci. 2002;22:5889–5899. doi: 10.1523/JNEUROSCI.22-14-05889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guil S, Long JC, Caceres JF. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol Cell Biol. 2006;26:5744–5758. doi: 10.1128/MCB.00224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang CJ, Tang Z, Lin RJ, Tucker PW. Phosphorylation by SR kinases regulates the binding of PTB-associated splicing factor (PSF) to the pre-mRNA polypyrimidine tract. FEBS Lett. 2007;581:223–232. doi: 10.1016/j.febslet.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tacke R, Chen Y, Manley JL. Sequence-specific RNA binding by an SR protein requires RS domain phosphorylation: creation of an SRp40-specific splicing enhancer. Proc Natl Acad Sci USA. 1997;94:1148–1153. doi: 10.1073/pnas.94.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Izquierdo JM, Valcarcel J. Fas-activated serine/threonine kinase (FAST K) synergizes with TIA-1/TIAR proteins to regulate Fas alternative splicing. J Biol Chem. 2007;282:1539–1543. doi: 10.1074/jbc.C600198200. [DOI] [PubMed] [Google Scholar]
- 148.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007;176:929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ma S, Liu G, Sun Y, Xie J. Relocalization of the polypyrimidine tract-binding protein during PKA-induced neurite growth. Biochim Biophys Acta. 2007;1773:912–923. doi: 10.1016/j.bbamcr.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 150.Xie J, Lee JA, Kress TL, Mowry KL, Black DL. Protein kinase A phosphorylation modulates transport of the polypyrimidine tract-binding protein. Proc Natl Acad Sci USA. 2003;100:8776–8781. doi: 10.1073/pnas.1432696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shi Y, Manley JL. A complex signaling pathway regulates SRp38 phosphorylation and pre-mRNA splicing in response to heat shock. Mol Cell. 2007;28:79–90. doi: 10.1016/j.molcel.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 152.Van Eynde A, et al. Molecular cloning of NIPP-1, a nuclear inhibitor of protein phosphatase-1, reveals homology with polypeptides involved in RNA processing. J Biol Chem. 1995;270:28068–28074. doi: 10.1074/jbc.270.47.28068. [DOI] [PubMed] [Google Scholar]
- 153.Cohen PT. Protein phosphatase 1 — targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- 154.Kim E, Goren A, Ast G. Alternative splicing: current perspectives. Bioessays. 2008;30:38–47. doi: 10.1002/bies.20692. [DOI] [PubMed] [Google Scholar]
- 155.Hallikas O, et al. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 156.Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA. Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol. 2004;14:283–291. doi: 10.1016/j.sbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 157.Tacke R, Manley JL. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995;14:3540–3551. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kent OA, Ritchie DB, Macmillan AM. Characterization of a U2AF-independent commitment complex (E′) in the mammalian spliceosome assembly pathway. Mol Cell Biol. 2005;25:233–40. doi: 10.1128/MCB.25.1.233-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shen H, Green MR. RS domains contact splicing signals and promote splicing by a common mechanism in yeast through humans. Genes Dev. 2006;20:1755–1765. doi: 10.1101/gad.1422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang Y, Gattoni R, Stevenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003;11:837–843. doi: 10.1016/s1097-2765(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 161.Zhang Z, Krainer AR. Involvement of SR proteins in mRNA surveillance. Mol Cell. 2004;16:597–607. doi: 10.1016/j.molcel.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 162.Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18:755–768. doi: 10.1101/gad.286404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nature Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 164.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]