

Abstract
Summary
Bacterial binding to human platelets is an important step in the pathogenesis of infective endocarditis. Streptococcus gordonii can mediate its platelet attachment through a cell wall glycoprotein termed GspB (“gordonii surface protein B”). GspB export is mediated by a seven component accessory Sec system, containing two homologues of the general secretory pathway (SecA2 & SecY2) and five accessory Sec proteins (Asps 1 – 5). Here we show that the Asps are required for optimal export of GspB independent of the glycosylation process. Furthermore, yeast two-hybrid screening of the accessory Sec system revealed interactions occurring between Asp3 and the other components of the system. Asp3 was shown to bind SecA2, Asp1, Asp2 and itself. Mutagenesis of Asp3 identified N and C-terminal regions that are essential for GspB transport, and conserved residues within the C-terminal domain mediated Asp3 binding to other accessory Sec components. The loss of binding by Asp3 also resulted in an impaired ability of S. gordonii to secrete GspB. These studies indicate that Asp3 is a central element mediating multiple interactions among accessory Sec components that are essential for GspB transport to the cell surface.
Keywords: accessory Sec, secretion, glycoprotein, Streptococcus gordonii, Asp
Introduction
Protein transport in bacteria is an essential process for growth, replication and nutrient acquisition (Thanassi and Hultgren, 2000). Among pathogenic bacteria, transport also serves to express virulence determinants on the cell surface or in the extracellular milieu, thereby conferring a survival advantage. A common theme that has emerged among such organisms is the use of a specialized secretion system that is dedicated to the export of specific virulence factors or effector proteins. Typically, such export systems differ from the general secretory pathway (Sec) in their structural features, and contain components necessary for targeting, trafficking and membrane passage that specifically accommodate the unique properties of the exported substrate (Galan and Wolf-Watz, 2006; Lee and Schneewind, 2001; Tampakaki et al., 2004).
The Gram-positive bacterium Streptococcus gordonii, a predominant cause of infective endocarditis (IE), contains such a specialized export pathway (the accessory Sec system) that is dedicated exclusively to the transport of the serine-rich glycoprotein GspB (Bensing and Sullam, 2002). This adhesin, together with other GspB homologues in streptococci, has been identified as an important platelet-binding protein on the bacterial surface (Bensing and Sullam, 2002; Jakubovics et al., 2005; Plummer et al., 2005). The glycoprotein mediates bacterium platelet contact via a non-glycosylated region enriched in basic amino acids that binds to the platelet membrane receptor glycoprotein (GP) Ibα, specifically interacting with sialic acid (N-acetyl neuraminic acid) moieties (Bensing et al., 2004b; Takamatsu et al., 2005a). More recently, GspB has also been shown to bind to the salivary glycoproteins mucin (MG2) and agglutinin, suggesting this adhesin may exhibit high affinity for other sialic acid-containing proteins (Takamatsu et al., 2006). Consistent with its platelet-binding properties, a gspB deletion strain was shown to be markedly reduced in virulence, when tested in an animal model of endocarditis, confirming the important contribution of this adhesin to the pathogenesis of this disease (Xiong et al., 2008).
GspB is synthesized as a 286 kDa preprotein within the cell, but becomes heavily glycosylated prior to export (Bensing et al., 2004a). It is encoded within a locus that also encodes a seven component accessory Sec system (Takamatsu et al., 2004b). A unique feature of this alternative secretion system is the presence of SecY and SecA homologues (SecY2 and SecA2). Both SecY and SecA are conserved components of the general Sec pathway that are found in all bacteria. SecY is the central component of a membrane protein complex, termed the “translocon,” which forms a channel for protein passage across the cell membrane (Breyton et al., 2002; Clemons et al., 2004; Veenendaal et al., 2004). SecA is a motor protein that both binds the targeted preprotein and generates energy for the active transport process, thereby linking the export substrate with the SecY-based translocon (Driessen and Nouwen, 2007; Rusch and Kendall, 2007; Zimmer et al., 2008). The presence of such homologues within the accessory Sec system suggests that a similar Sec-like mechanism may be utilized for the transport of this glycoprotein. Indeed, recent studies have shown that SecA2 is an ATPase that shares certain structural features with SecA, further indicating that these two pathways may function alike (Bensing and Sullam, 2009).
Despite the similarity of the core translocating components, the remaining five proteins of the accessory Sec system (Accessory Sec protein [Asp] 1 – 5) exhibit no significant similarity to proteins of known function. Although expression of the Asps, together with SecY2 and SecA2, is essential for GspB export (Takamatsu et al., 2004b, Takamatsu et al., 2005b), it is unclear how these accessory Sec components facilitate export of the native glycoprotein. One possibility is that the Asps function similarly to other members of the canonical Sec system. Extensive studies of this pathway within Escherichia coli have identified numerous factors that interact with SecY or SecA to mediate secretion. The cytosolic effector protein SecB binds some preproteins (preventing premature folding), and facilitates the interaction of these substrates with SecA (Kim and Kendall, 2000; Zhou and Xu, 2005). Other proteins are either components of the translocon (e.g., SecE and SecG), or enhance transport via their association with the export channel (e.g., SecD, SecF, and YajC) (Baba et al., 1994) (Nouwen and Driessen, 2005). Thus, there exists in the canonical Sec System a requirement for ancillary proteins that enhance, stabilize, or tailor protein export by interacting with the core translocation machinery. It is conceivable that the Asps have analogous roles in the export of GspB.
Of note, studies in Streptococcus parasanguinis indicate that the homologues of Asp1 and Asp3 (Gap1 and Gap3, respectively) may be glycosyltransferases that modify Fap1 (a homologue of GspB) (Li et al., 2008; Peng et al., 2008). Although Gap1 and Gap3 have not been shown directly to have enzymatic activity, disruption of these proteins is associated with altered glycosylation of Fap1 (Li et al., 2008). Changes in GspB glycosylation have not been observed upon mutagenesis of Asp1 and Asp3. However, the above findings in S. parasanguinis raise the question of whether the Asps have a direct role in export. It is conceivable that the loss of GspB export seen with disruption of Asp1 and Asp3 is an indirect or secondary effect on transport stemming from the altered glycosylation of GspB. To address these issues, we directly examined the role of the Asps in mediating GspB transport. We show that the Asps are essential for the export of a non-glycosylated GspB variant. Furthermore, we have identified binding occurring among the Asps themselves, as well as with SecA2 (the likely translocase motor) and determined that these accessory Sec protein interactions are coordinated mainly through the Asp3 protein. We also provide evidence that these protein-protein interactions are needed for efficient GspB export. Thus, we propose that these accessory Sec components have a primary function in the transport of this glycoprotein.
Results
Accessory Sec proteins are required for the export of a non-glycosylated variant of GspB
As noted above, disruption of any asp abolishes the export of native GspB. In view of the finding that some Asp homologues of S. parasanguinis may be involved in glycosylation, it was unclear whether the requirement of the Asps for GspB export was secondary to any role they might have in glycosylation. To address this issue, we assessed the impact of disrupting each Asp on the transport of a GspB variant (GspB736flag) in S. gordonii strain M99. This truncated GspB has the same requirements for transport as the native protein, but it is freely secreted into the culture medium, making it a more convenient and accurate substrate for measuring export. Non-glycosylated GspB736flag was produced by expressing this protein in a GtfA- background (GtfA is required for the glycosylation of GspB (Takamatsu et al., 2004a)).
In control studies, glycosylated GspB736flag was efficiently exported in the parent strain PS919, with high levels of the protein being detected in the culture supernatant, and only trace amounts in protoplasts (Fig. 1A, lane 1). No export was seen in the GtfA+/SecA2- variant (Fig. 1A, lane 2), demonstrating that export of the glycosylated substrate is wholly dependent on the accessory Sec pathway. Similarly, export of glycosylated GspB736flag was abolished or markedly reduced by disruption of Asp1, 2, 3, 4, or Asp5, as has been reported previously for native GspB (Bensing et al., 2005). Of note, the apparent MW of GspB736flag within protoplasts (~150 kDa) was 10 kDa larger than exported GspB736flag, indicative of a lack of signal peptide cleavage. Furthermore, the size of this preprotein was comparable among the asp and secA2 mutants, indicating that no major change in glycosylation of the substrate had occurred.
Figure 1. Export of glycosylated and non-glycosylated variants of GspB736flag by accessory Sec deletion strains.
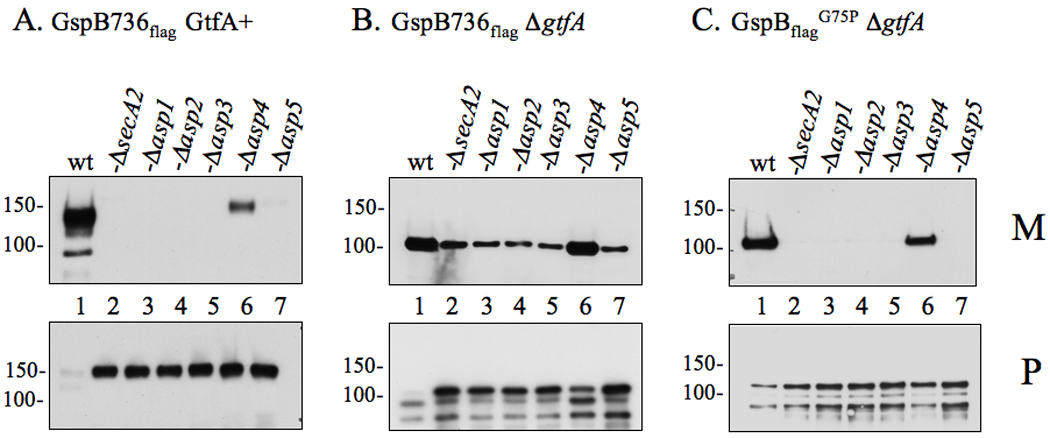
Proteins obtained from culture media (M) or lysed protoplasts (P) were separated by SDS-PAGE (3–8%) and probed with anti-FLAG antibody for GspB736flag detection. (A) Export of GspB736flag by the parent strain PS919 (Lane 1) or its accessory Sec deletion variants, PS920 (Lane 2), PS942 (Lane 3), PS944 (Lane 4), PS945 (Lane 5), PS925 (Lane 6), PS924 (Lane 7). (B) Export of nonglycosylated GspB736flag by PS1057 (Lane 1), or by accessory Sec/gtfA double deletion strains PS1063 (Lane 2), PS1060 (Lane 3), PS1061 (Lane 4), PS1062 (Lane 5), PS1059 (Lane 6), PS1058 (Lane 7). (C) Export of nonglycosylated GspB736flagG75P by PS1765 (Lane 1), or by accessory Sec/gtfA double deletion strains PS1208 (Lane 2), PS1768 (Lane 3), PS1769 (Lane 4), PS1770 (Lane 5), PS1771 (Lane 6), PS1772 (Lane 7). Glycosylated GspB is visible as a protein of approximately 140 or 150 kDa in the media or protoplasts respectively, while the non-glycosylated form is detected as a protein of approximately 100 or 110 kDa in size in the media or protoplasts respectively. Breakdown products of both forms of GspB are visible as smaller proteins accumulating within the protoplasts.
We then examined the impact of mutating the Asp proteins on the export of non-glycosylated GspB736flag (apparent MW ~100 kDa). Transport of this substrate was also highly reduced in the secA2, asp1, 2, 3, or asp5 deletion strains (Fig 1B, Lane 2 – 5, & 7). Although relatively minor amounts of GspB export were detected from these strains, we have previously demonstrated that this residual export of non-glycosylated GspB736flag occurs via the canonical Sec pathway, albeit inefficiently (Bensing et al., 2007; Bensing and Sullam, 2009). Further evidence of impaired export could be seen in the protoplasts from these double deletion mutants, which had an accumulation of non-glycosylated preGspB736flag (Fig 1B, Fig 2–Fig 5 & Fig 7), while little to no protein was observed in protoplasts of the gtfA single gene knockout mutant (Fig 1B, Lane 1). Export of GspB736flag was also reduced in the asp4 deletion strain (Fig 1B, Lane 6) though not to the extent observed with the other asp deletion strains.
Figure 2. Protein-protein binding among accessory Sec components as identified by yeast two-hybrid analysis.

(A) Diploid yeast cells containing the indicated plasmid pairs (bait or prey) were grown on selective media containing galactose and X-gal. Cells containing a positive bait and prey interaction were visualized as blue growing colonies. (B) Quantitative β-galactosidase assays of Asp3 binding pairs.
Figure 5. Effects of Asp3 domain disruption upon the export of GspB736flag.
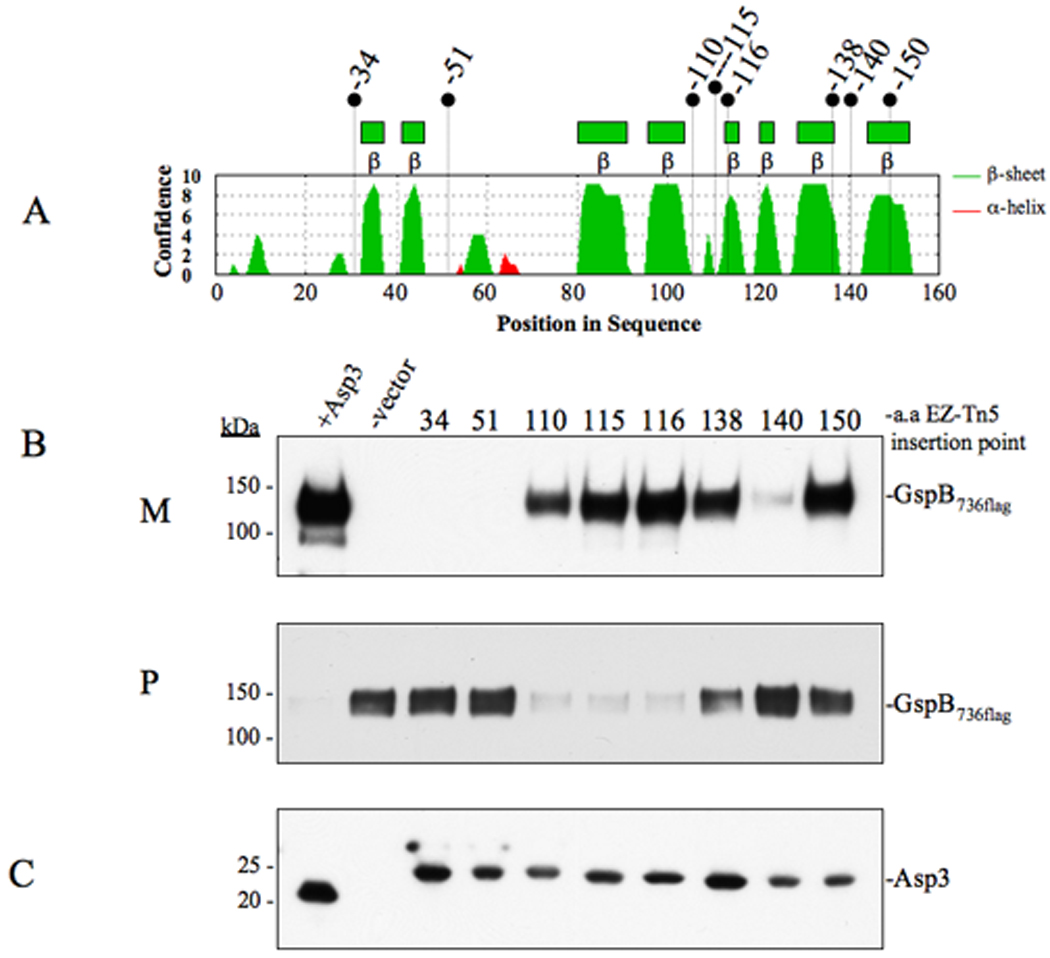
(A) Secondary structure analysis of Asp3. Locations of EZ-Tn5 insertions are shown above. (B) Western blot analysis of GspB736flag export from PS1244 derivative strains carrying in-frame insertions within Asp3. Culture media (M) and protoplasts (P) were collected from exponentially growing strains and prepared as described in the methods and materials. Proteins were separated by SDS-PAGE (3–8%) and following Western blot transfer, GspB736flag was probed with anti-flag antibody. (C) Western blot detection of Asp3 and the in-frame insertion mutants. Proteins from the protoplast fraction were separated by SDS-PAGE (4–12%) and probed with anti-Asp3 antibody.
Figure 7. Disruption of protein binding between Asp3 mutants and accessory Sec components.
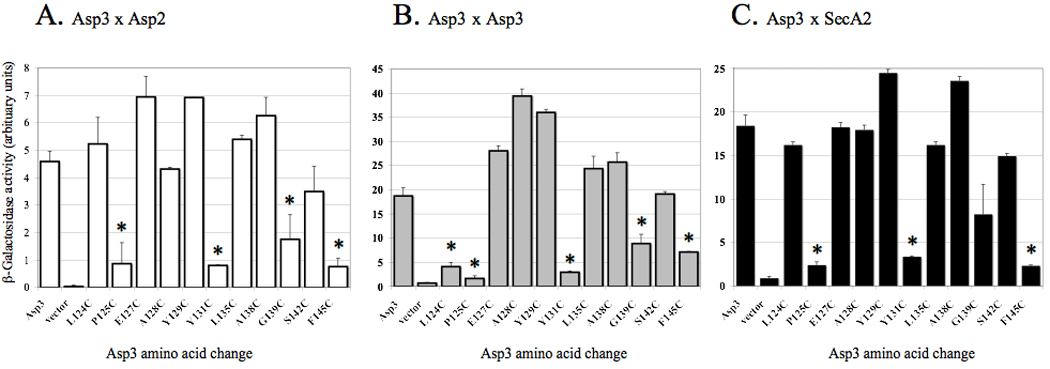
Binding of Asp3 cysteine replacement mutants with (A) Asp2, (B) Asp3 and (C) SecA2 was assessed by yeast two-hybrid assays. Diploid yeast cells containing the indicated Asp3 mutant and interacting partner were grown on selective media containing galactose and assayed for β-galactosidase activity using ONPG as the substrate. The values shown are the geometric means ± SD of triplicate samples. * = P<0.01, for mutant versus WT.
To more directly test the role of the Asps in export of non-glycosylated GspB, we took advantage of a previously characterized GspB736flag variant that contains a Gly75-Pro75 [GspB736flag G75P] substitution within the hydrophobic core of the signal peptide, resulting in exclusive routing to the accessory Sec pathway (Bensing et al., 2007). Export of this variant was absent in the asp1, 2, 3 and asp5 deletion mutants, with GspB accumulation clearly visible within the protoplasts of these strains. (Fig 1C, Lane 2 – 5, & 7). Again, a partial reduction of GspB736flag G75P export was seen in the asp4 deletion strain (Fig 1C, Lane 6). Collectively these findings demonstrate that all of the accessory Sec proteins are directly involved in the export GspB independent of the glycosylation process.
Asps interact with components of the accessory Sec system
The finding that Asps 1–5 are all required for optimal GspB export suggested that these components may physically interact with each other, or with other members of the accessory Sec system, to mediate export. To examine this possibility, we used a directed yeast two-hybrid (YTH) screening assay to identify physical interactions between these proteins, and with other members of the export pathway. All accessory Sec components were cloned as N-terminal fusions to either the E. coli B42D transcriptional activator domain within the prey plasmid pjsc401, or to the lexA DNA-binding domain within the bait plasmid pEG202. Saccharomyces cereversiae diploid strains containing both a bait and prey plasmid were assayed for β-galactosidase activity, a phenotype indicative of bait and prey binding. When examined by this method, no direct binding was identified between preGspB and any component of the accessory Sec system. However, interactions were identified among the accessory Sec components (Fig 2A). Interestingly, Asp3 was shown to bind Asp1, Asp2, SecA2, as well as with itself. We also detected Asp4-Asp4 binding by this assay. The reciprocal prey-bait (B42D–LexA) interactions were observed for Asp2-Asp3, but not for Asp1-Asp3 or SecA2-Asp3 protein pairs. Poor expression of Asp1 as a B42D fusion was noted, which in part could explain the lack of reciprocal binding (data not shown). Asp3-based interactions were further confirmed by quantitative β-galactosidase assays (Fig 2B).
Asp3 mediates multiple protein-protein interactions within the accessory Sec system
The findings from our YTH screen suggested that Asp3 was a central component for binding between the accessory Sec proteins. To confirm the observed Asp3-based interactions, we co-expressed His6Asp3 with either flagAsp1, flagAsp2, SecA2 or GST in E. coli, and assessed whether each protein would co-purify with His6Asp3 on a Ni2+ affinity column. As expected, GST failed to co-purify when co-expressed with His6Asp3, with all of the former protein being exclusively detected in the flow-through fraction (Fig. 3A). Moreover, none of the Flag-tagged Asps or SecA2 bound to Ni2+ resin alone (data not shown). However, all three accessory Sec components co-purified with His6Asp3 following elution from the column (Fig. 3A). As an additional method to confirm Asp3 binding to accessory Sec components, lysates of whole cells expressing Flag-tagged Asps, MalE.SecA2 and MalE were probed by far Western blotting with His6Asp3. This protein was found to bind to bands corresponding to flagAsp2 & flagAsp3, with faint band seen corresponding to MalE.SecA2 (Fig 3B). However, His6Asp3 failed to bind to flagAsp1 suggesting that the requirements of this interaction was not maintained in this assay. In aggregate, these data indicate that the observed binding of Asp3 with Asp1, Asp2, SecA2 and itself is not due to nonspecific associations, but rather, represent specific Asp3-mediated interactions.
Figure 3. Asp3 forms complexes with Asp1, Asp2, Asp3 and SecA2.
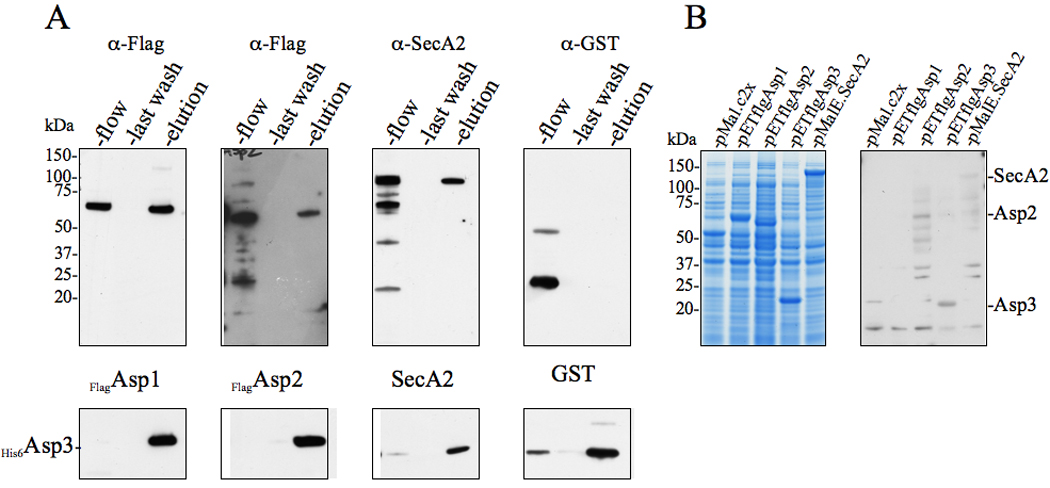
(A) Lysates of E. coli BL21 (DE3) co-expressing His6Asp3 and Flag-tagged Asp1, Asp2, or SecA2 were applied to Ni2+ agarose, followed by washing of the columns and elution of the retained material. Lysates of cells co-expressing His6Asp3 and GST served as negative controls for nonspecific binding. The various fractions (flow, last wash, elution) were probed by Western blotting for proteins co-purifying with His6Asp3. Eluted His6Asp3 was detected using anti-His6 antibody (lower panel) and co-purifying proteins were detected with anti-Flag, anti-SecA2 or anti-GST antibodies (upper panel).
(B) E. coli BL21 (DE3) lysates expressing MalE, flagAsp1, flagAsp2 flagAsp3 or MalE.SecA2 were electrophoresed in SDS-PAGE (4–12%) and either stained with Coomassie blue (left panel), or transferred to membranes and probed with His6Asp3 (right panel). Binding of Asp3 to the immobilized proteins was detected with anti-His6 antibody.
Asp3 co-purifies with other accessory Sec members in vivo
To assess whether the above interactions of Asp3 could be detected in vivo, we looked for accessory Sec members that co-purified with Asp3 when it was isolated from S. gordonii. Initial immunoblot analysis of lysed M99 whole cell extracts indicated that Asp3 was produced in vivo at very low levels. In order to more clearly detect Asp3 within the cell, His6Asp3 was used to complement PS1244 (an asp3 deletion strain). Lysed cells were passed through a column previously cross-linked with anti-Asp3 antibodies. As expected, His6Asp3 was recovered in the eluted fraction (Fig 4A), as identified by immunoblot analysis. Consistent with our previous co-expression and co-purification in E. coli, Asp1 was also detected within the eluted fraction, demonstrating that this accessory Sec member binds with Asp3 in vivo (Fig. 4A). We did not detect Asp2 and SecA2 within the eluted fraction. However, this may reflect the very low levels of these proteins within M99, since we have been unable to detect by immunoblotting the presence of SecA2 within whole cell extracts of this organism (data not shown). In addition, PSORTb analysis identified the sub cellular location of Asp3 and Asp1 to be within the cytoplasm, while Asp2 is predicted to be membrane localized. Collectively, these findings may explain why only the Asp3-Asp1 interaction was identified in vivo.
Figure 4. Effects of Asp3-Asp1 interaction upon the export of GspB736flag .
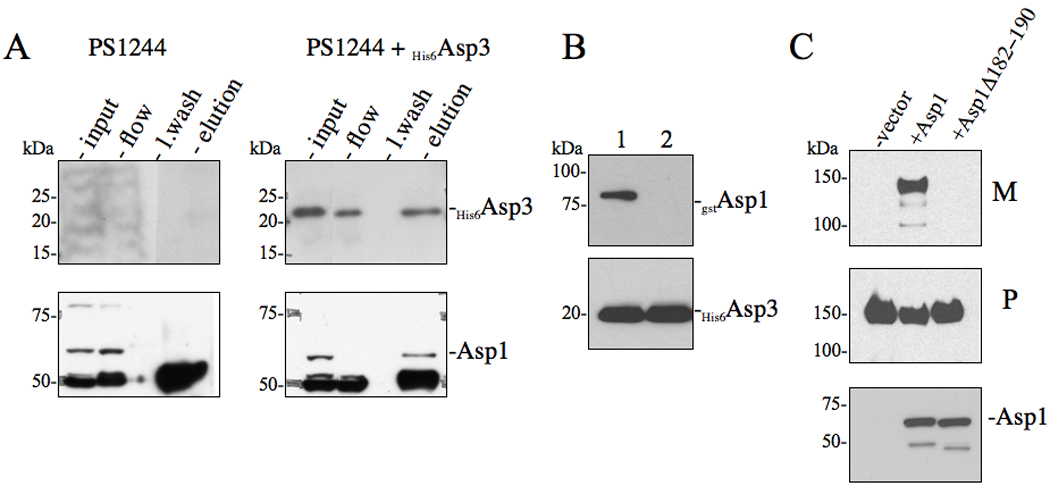
(A) in vivo co-immunoprecipitation of Asp3 with accessory Sec components. S. gordonii PS1244 was complemented in trans with His6Asp3 or vector alone. Clarified lysates were applied to anti-Asp3 resin, followed by washing of the column and elution of the retained material. The various fractions (flow, last wash, elution) were probed by Western blotting for proteins co-purifying with His6Asp3. Eluted His6Asp3 was detected using anti-His6 antibody (upper panel) and the co-purifying protein was detected with anti-Asp1 antisera. (B) Co-expression co-purification of Asp3-Asp1 complexes. Lysates of E. coli BL21 (DE3) co-expressing His6Asp3 with either (1) gstAsp1 or (2) gstAsp1 (Δ182−190) were applied to Ni2+ agarose, followed by washing of the columns and elution of the retained material. Eluted material was probed by Western blotting for proteins co-purifying with His6Asp3. Eluted His6Asp3 was detected using anti-His6 antibody (lower panel) and co-purifying Asp1 was detected with anti-GST antisera (upper panel). (C) Western blot analysis of GspB736flag export from S. gordonii PS1242 derivative strains carrying internal deletions within Asp1. Culture media (M) and protoplasts (P) were collected from exponentially growing strains and prepared as described in the methods and materials. Proteins were separated by SDS-PAGE (3–8%) and following Western blot transfer, GspB736flag was probed with anti-flag antibodies.
Asp1-Asp3 interaction is also necessary for GspB secretion
Previously, homologues of Asp1 and Asp3 (Gap1 and Gap3) within S. parasanguinis were shown to also interact, and this binding was essential for the correct glycosylation of the srr glycoprotein Fap1 (Li et al., 2008). Our findings that the Asps are essential for GspB export prompted us to test whether the binding of Asp1 with Asp3 was functionally relevant for the transport process. A conserved nine amino acid residue stretch within Gap1 (182–190 AA) was shown to mediate its interaction with Gap3 (Li et al., 2008). To determine whether the corresponding region of Asp1 mediates binding to Asp3, a gstAsp1 (Δ182–190 AA) mutant was constructed and co-expressed with His6Asp3 in E. coli. As previously seen with Gap1 (Δ182–190 AA), this mutant failed to co-purify with Asp3 (Fig 4B). To assess the significance of this interaction on export, the Asp1 (Δ182–190 AA) protein was used to complement an M99 variant (PS1242) containing a deletion of asp1. Analysis of the culture supernatant from this strain revealed that complementation with the 182–190 AA mutant protein failed to restore GspB736flag export (Fig 4C), indicating the Asp1-Asp3 interaction is needed for the successful transport of GspB by S. gordonii.
The C and N-terminal regions of Asp3 are essential for GspB secretion
Standard bioinformatic searches have not identified any domains of Asp3 that are similar to functionally characterized regions of other proteins. However, preliminary secondary structure analysis identified an extensive beta-sheet region within the Asp3 C-terminal region (Fig 5A), with no significant secondary structural features apparent at its N-terminus. To examine the role of the domains of this protein in export, we assessed the effects of random mutagenesis of asp3 on this process. A total of 8 mutations were generated, each containing an in-frame insertion of 19 codons within the coding sequence of asp3 (Fig 5A). These mutated forms of asp3 were used to complement an M99 variant (PS1244) containing a deletion of asp3. The resulting transformants were then tested for their ability to export GspB736flag. Western blotting of the complemented strains revealed that all eight variants of Asp3 were stably expressed at similar levels (Fig 5C). Analysis of the culture supernatants from each strain identified three insertions within Asp3 (at amino acids 34, 51 and 140) that abolished GspB736flag export (Fig 5B). Accumulation of GspB736flag was also clearly visible within the protoplasts of these insertion mutants, indicating a bona fide transport defect due to asp3 mutagenesis (Fig 5B). Insertion at amino acids 138 and 150 appeared to have a minor effect upon export, with a partial export defect manifesting in an accumulation of GspB736flag within the protoplast. In contrast, insertions within the central region of Asp3 (at amino acids 110, 115, 116) had little to no effect upon secretion. These findings suggest that specific domains in the N and C-terminus of Asp3 are essential for the secretion process.
Conserved residues in the C-terminus of Asp3 are required for interactions with accessory Sec components
The identification of domains within Asp3 essential for GspB export led us to hypothesize that these regions may play a part in mediating the binding of this protein with other accessory Sec components. To characterize the domains of Asp3 responsible for binding, each of the mutated asp3 coding regions was re-cloned into pjsc401 for testing within our YTH screen. However little or no expression of these Asp3 variants was seen in S. cerevisiae, preventing a direct correlative analysis of GspB export and Asp3 binding. When expressed in E. coli, these Asp3 variants were found to be highly degraded, which also precluded their use.
As an alternative approach, we used site directed mutagenesis in combination with our YTH screen to identify critical residues mediating Asp3 binding. We focused on the C-terminus because, unlike the N-terminus, it is an entirely uncharacterized region of the protein, not associated with glycosylation or other processes. In addition, our transposon studies indicated that the binding region of this C-terminal domain is located within a relatively short segment, since insertions at AA 138 or 150 had only a minor effect on export, while an insertion at AA 140 markedly reduced substrate transport. This finding indicated more precise mapping of the region would be amenable to analysis by single amino acid substitution. Sequence alignment of the C-terminal region of Asp3 and its homologs identified a stretch of conserved residues that flanked amino acid 140 (Fig 6A). Within this region, 11 conserved amino acids underwent cysteine residue replacement. As a negative control, we also replaced conserved residues at neighboring insertions sites that were predicted to have no effect upon export.
Figure 6. C-terminus alignment of Asp3 and its homologues.
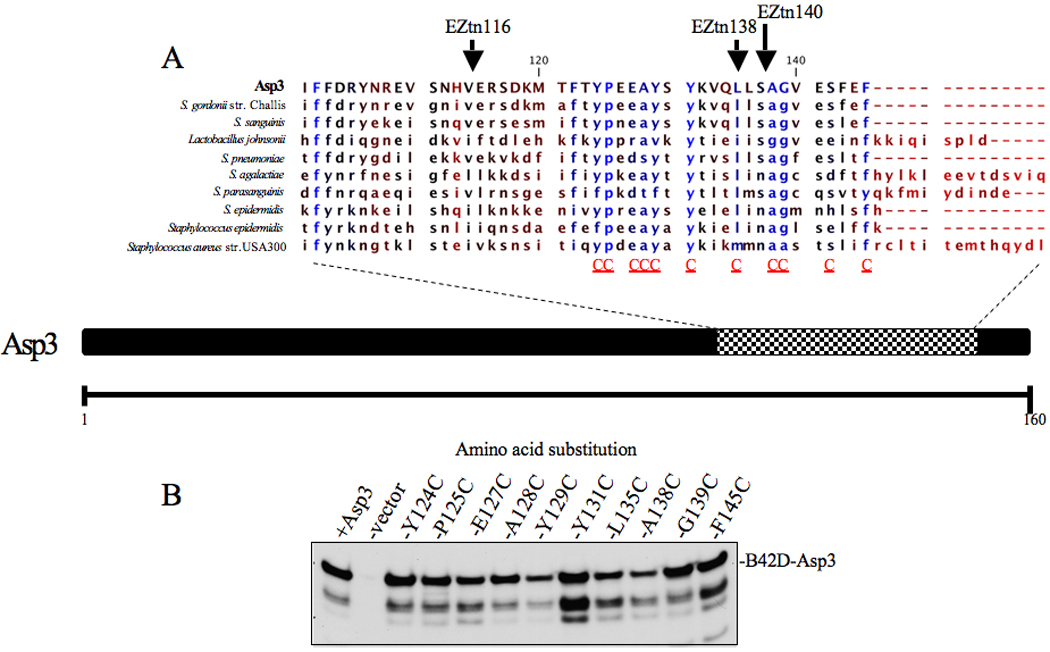
(A) Conserved amino acid residues present among Asp3 homologues are shown in blue. Residues targeted for cysteine replacement are indicated by a red C below the sequence alignment of the C-terminus. Previously generated Asp3 EZ-Tn5 insertions are shown above the amino acid insertion point. (B) Lysates of S. cerversiae cells over-expressing Asp3 cysteine replacement mutants were separated by SDS-PAGE (4–12%) transferred to nitrocellulose and probed with Asp3 (bottom panel).
The asp3 variants were cloned into pjsc401 and their gene products compared for their binding properties. When assessed by Western blotting all of the Asp3 variants were expressed, although Y131C did show greater fragmentation (Fig 6B). The effect of each mutation upon Asp3 binding was then tested, using our yeast two-hybrid screen. Of the Asp3-interacting partners identified previously, the greatest levels of binding were observed between the Asp3-Asp2, Asp3-Asp3 and Asp3-SecA2 interacting pairs (Fig 2B). We therefore focused upon these interactions to characterize the key residues of Asp3 mediating binding. Of the 11 site-directed mutants tested, E127, A128, Y129, L135, A138 and S142 failed to show any significant change in binding to their accessory Sec partners. However, cysteine replacement of Y124 significantly reduced the binding among Asp3-Asp3 interaction (Fig 7B). In addition, cysteine replacement of residues P125, Y131, G139 and F145 resulted in a significant reduction in Asp3 binding to all tested partners (Asp2, Asp3 and SecA2) (Fig 7A – C), indicating that these residues within the C-terminus are required for efficient Asp3 binding.
Conserved residues required for Asp3 binding are also needed for GspB export
We next sought to examine whether the ability of Asp3 to bind to one or more accessory Sec components (as detected by our YTH method) was important for GspB transport. Wild-type asp3 and the cysteine substitution variants were used to complement an M99 mutant (PS1244) in which asp3 had been deleted. Media supernatants and protoplasts from the resulting transformants were examined for GspB736flag export or intracellular accumulation. Western blotting of the complemented strains revealed that all forms of Asp3 were stably expressed (Figure 8C). The majority of the cysteine replacements (P125, E127, A128, Y129, Y131, S142 and including Y124 and L135) had no significant impact upon GspB736flag export, with the protein being transported normally into the media. However, cysteine replacement at Y124, G139 and F145 resulted in a pronounced reduction in GspB736flag export, with a marked reduction of the protein in the media supernatants, and a clear accumulation of the substrate within protoplasts (Fig 8A, P, Lane 3, 11 & 13). Although cysteine replacement at Y131 produced no discernable difference in the amount of GspB736flag exported to the media as compared with wild-type, it was associated with a retention of GspB within the protoplast fraction, suggestive of an impaired transport process (Fig 8A, P, Lane 4). Analysis of the these Western blots by densitometry confirmed that cysteine substitutions at Y124, G139 and F145 of Asp3 resulted in a clear reduction in GspB736flag export to the culture media, with a corresponding accumulation of the preprotein within the protoplasts (8B). Substitution at Y131 was associated with a slight reduction in GspB736flag within the medium, and a moderate accumulation of substrate for Y131 was seen, suggestive of an export defect.
Figure 8. Effect of Asp3 cysteine replacement mutagenesis on export of GspB736flag .
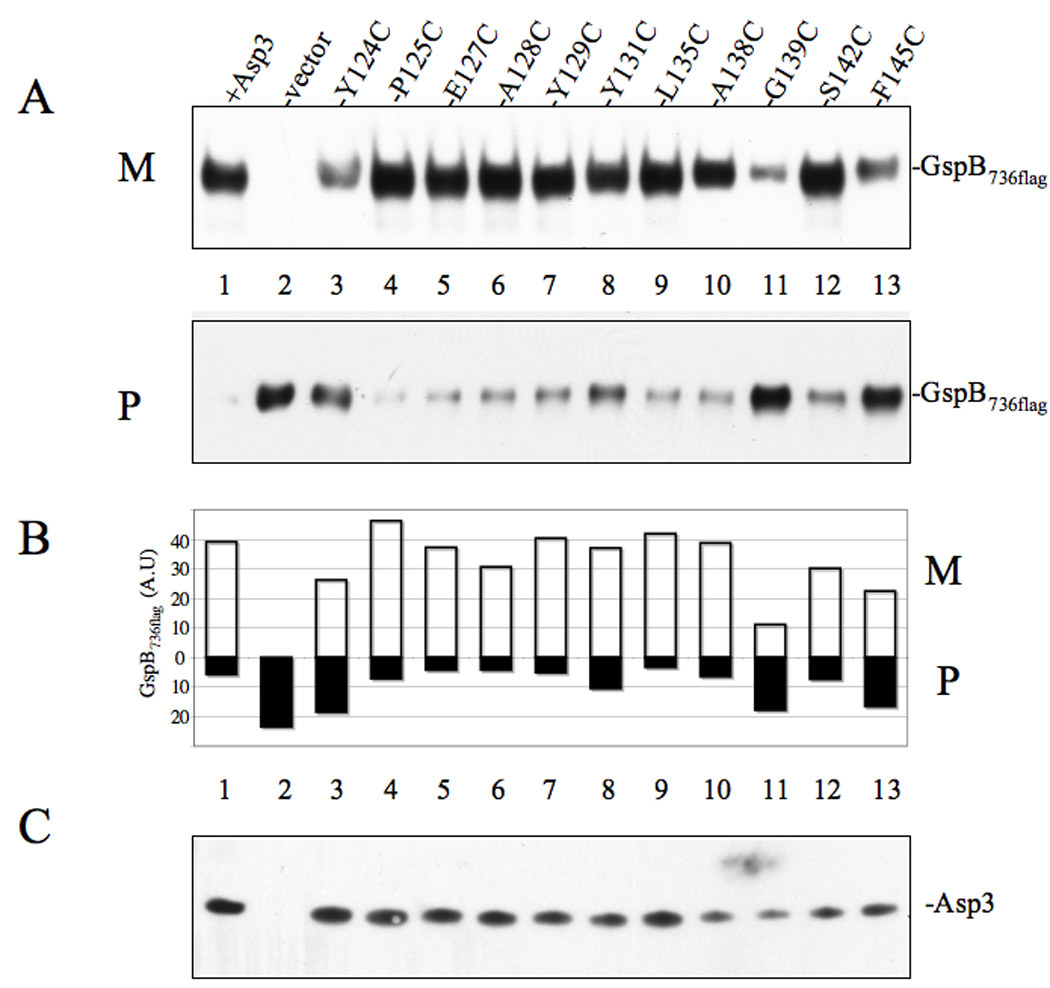
(A) Western blot analysis of GspB736flag export from PS1244 strains expressing either Asp3 or the indicated Asp3 cysteine replacement mutant. Culture media (M) and protoplasts (P) were collected from exponentially growing strains and prepared as described in the methods and materials. Proteins were separated by SDS-PAGE (3–8%) and following Western blot transfer, GspB736flag were probed with anti-flag antibodies. (B) Densitometry analysis of GspB736flag levels within media (M) or protoplasts (P). The X-axis represents GspB736flag levels based on band intensity analysis via Li-Cor imaging. (C) Asp3 levels within protoplasts, as measured by Western blotting with anti-Asp3 polyclonal antiserum.
Discussion
GspB is encoded within a pathogenicity island that additionally contains four genes responsible for its post-translational glycosylation (Gly, Nss, GtfA and GtfB), together with seven genes encoding accessory Sec components required for its surface presentation (Asps1–5, SecY2 and SecA2) (Bensing and Sullam, 2002; Takamatsu et al., 2005b). This multi-gene locus is dedicated to the synthesis, glycosylation and transport of GspB (Bensing and Sullam, 2002). The specific roles of each accessory Sec component have yet to be defined, although the presence of a SecA and a SecY homologue suggests that a Sec-like mechanism of export for GspB may be utilized.
The accessory Sec proteins (Asps) share no homology to any protein of characterized function and remain the least understood aspect of GspB transport. Previous studies in S. parasanguinis had shown that disruption of the asp1 and asp3 homologues (gap1 and gap3) resulted in higher MW forms of Fap1 with altered glycosylation. This suggested that perhaps any changes in export associated with mutagenesis of these genes in S. gordonii might be secondary to aberrant glycosylation of the substrate. To clarify this issue, we examined the role of the Asp1–5 in a glycosylation – deficient background, such that any effects of glycosylation on export would be eliminated. We found that Asps1–3 and Asp5 were absolutely required for the export of GspB736flag. These finding were further substantiated through experiments using another GspB variant (GspB736flag G75P), in which a Gly75-Pro75 conversion within the signal peptide results in exclusive routing to the accessory Sec pathway (Bensing et al., 2007). Loss of Asp4 expression also produced a significant reduction in the transport of the glycosylated or non-glycosylated substrate, though this process was not abolished. Of note, deletion of asp4 within a glycosylation-negative background had a lesser effect upon the transport of GspB736flag. These findings suggest that Asp4 may not be essential for GspB transport, but its presence permits better accommodation of the glycosylated substrate by the accessory Sec system. Although we cannot rule out that some accessory Sec components may also have a role in the glycosylation of GspB, the observation that Asp1 – 5 are required for optimal export of a non-glycosylated GspB variant demonstrates that these proteins have a direct role in the export process.
The requirement of numerous accessory Sec components for GspB export prompted us to determine whether these proteins worked in concert. Examination of protein-protein interactions through YTH, far Western, co-expression and co-purification analyses showed that Asp3 could form complexes with Asp1, Asp2 and SecA2. The binding of Asp1 and Asp3 is consistent with the recently described interaction identified between Gap1 and Gap3 within S. parasanguinis (Li et al., 2008). These findings indicate that Asp3 is a central component in mediating protein-protein binding among other members of the accessory Sec system.
To test the role of Asp3 binding upon GspB transport in vivo, we introduced mutant forms of Asp3 into a Δasp3 S. gordonii strain. Domain disruption at either the N or C-terminus of Asp3 resulted in a significant reduction of GspB transport, suggesting both regions contribute towards the export process. Site directed mutations within the C-terminus identified conserved residues (Y124, P125, Y131, G139 & F145) whose replacement with cysteine resulted in a reduction of Asp3 binding ability, as measured by the YTH assay. Of these mutations, Y124, G139 & F145 resulted in a clear loss of GspB transport to the extracellular milieu, with a concordant accumulation of the substrate within the protoplast fraction, thus establishing a link between Asp3 binding and successful GspB export. Although each individual cysteine replacement mutant did not produce the total loss in GspB transport seen with transposon insertion at AA 140, it is more than likely that these amino acids have a cumulative role in mediating export. For some mutations, the changes in export were fairly subtle. For example, the Y131 mutation produced a partial export defect that was manifested primarily as a moderate accumulation of GspB736flag within the protoplasts. The P125 mutation was the only mutation to show a decrease in binding but produce no clear alteration in export, but this could be due to technical limitations of our YTH assay. Nonetheless, the finding that the majority of C-terminus mutants showed a significant loss in binding that correlated with an export defect confirms that GspB transport is affected when Asp3 binding to one or more accessory Sec components is disrupted.
Of the numerous Asp3-based interactions identified, C-terminal mutations at P125, Y131, G139 and F145 resulted in a decrease in binding of Asp2, Asp3 and SecA2. This collective reduction in binding could be attributed to the ability of Asp3 to self-associate and that Asp3 self-association is prerequisite for all other interactions to occur. Alternatively, the Asp3 binding partners may share a common binding site. Of note, however, the Y124 mutant resulted in a loss of Asp3 binding only with itself, indicating that not all residues required for export contribute towards a common binding site for all accessory Sec components. It is also possible that these amino acids are essential for maintaining the native conformation of the protein, such that substitutions at these residues produced changes in secondary structure that affected binding at other sites. However, these single amino acid changes generated in Asp3 led to only a partial reduction in GspB transport, arguing against a grossly altered protein conformation. Furthermore, a series of neighboring substitutions had no effect upon Asp3-based interactions, suggesting that the identified key residues within this region are specific for Asp3 binding.
Our finding that all of the Asps are required for optimal GspB export suggests that these components are directly linked to the export process of this glycoprotein, and that Asp3 binding to other accessory Sec components is needed for optimal secretion. However, the precise roles the Asps have in the transport process remain to be defined. Studies of the canonical Sec system suggest three possible stages of export that may be subject to Asp involvement. First, one or more of the Asps may interact directly with the preprotein, functioning as molecular chaperones to prevent premature aggregation and to maintain the substrate in an export competent state (Lecker et al., 1989). We did not detect any interaction between GspB and members of the accessory Sec system, suggesting that these components do not bind GspB directly. However, a number of recognized technical limitations may have hindered our ability to detect GspB binding, including low levels of protein synthesis, abnormal protein folding, or the lack of protein targeting to the yeast nucleus. Moreover, if the interaction of GspB with the Asps is a multifactorial process, where more than one Asp binds to GspB to initiate export, such binding would not have been detected in our YTH screen, since this method only detects binary interactions.
A second possibility is that the Asps could modulate SecA2 function. Precedent for this can be found in the canonical Sec system, where binding of SecB to SecA enhances the interaction of the latter with its preprotein substrate, thereby facilitating export (Fekkes et al., 1997). Recently, our group has shown that SecA2 is an ATPase that shares several structural features with SecA, suggesting it may have a similar role in GspB transport (Bensing and Sullam, 2009). In view of our finding that Asp3 binds SecA2, it is possible that this interaction may also facilitate preprotein binding to SecA2.
A third possibility is that the Asps may interact with SecY2 to form a larger, membrane-associated translocation complex, similar to the translocon of the canonical Sec system. SecY readily interacts with two additional transmembrane proteins, SecE and SecG, to form the channel for substrate export. Like SecE and SecG, Asp4 and Asp5 are predicted to have at least one transmembrane domain and exhibit a similar topology (Takamatsu et al., 2005b). Moreover, the finding that Asp4 is partially dispensable, while Asp5 is required for export of GspB, resembles the requirements for SecG and SecE, respectively, in canonical export (Veenendaal et al., 2004). Thus, it is possible that these or other Asps constitute a structural part of the protein channel. A further possibility is that the Asps bind peripherally to the protein channel to influence translocation as has been described for FtsY, or the SecDF(YajC) protein complex. FtsY behaves as a docking intermediate between SecY and the nascent pre-protein in complex with the signal recognition particle, while association of the SecDF(YajC) protein complex with the protein channel enhances the translocation rate (Driessen and Nouwen, 2007). Although no interactions were seen between SecY2 and the Asps within our YTH screening, this may reflect the propensity of this assay to detect interactions among soluble, cytosolic proteins, as compared to membrane-associated proteins.
The gene organization within the GspB pathogenicity island is highly conserved among numerous streptococci and staphylococci expressing GspB homologues on their cell surface (Rose et al., 2008; Seifert et al., 2006; Siboo et al., 2008; Takamatsu et al., 2004b). Interestingly, the greatest variation seen within this pathogenicity island lies in the number of glycosyltranserase genes and additional sugar modification genes present. Indeed, Streptococcus pneumoniae contains up to eight glycosytransferase genes within this operon, with only two such genes seen in Staphylococcus aureus, which presumably is reflected in differential sugar modification of the respective serine-rich surface protein (Takamatsu et al., 2004b). However, Asp1–3 appear to be the most conserved elements, forming an integral core along with SecA2 and SecY2. Furthermore, the requirement of these proteins in other bacteria for export of their respective GspB homologue (Siboo et al., 2008) suggests that these accessory Sec proteins are universally required members of this transport system. Thus, the findings presented here are likely to have broad functional relevance for the transport of the entire family of serine-rich glycoproteins.
Methods and Materials
Strains and growth conditions
All strains and plasmids used in this study are listed in Table 1. S. gordonii strains were grown in Todd Hewitt broth (Difco Laboratories). Escherichia coli DH5α served as a host for cloning purposes. This strain was grown at 37°C under aeration in Luria broth (LB). When appropriate, antibiotics were added to the media at the following concentrations, unless stated otherwise: ampicillin; 50 µg ml−1 for E. coli; chloramphenicol, 34 µg ml−1 for E. coli and 5 µg ml−1 for S. gordonii; kanamycin, 30 µg ml−1 for E. coli; erythromycin 500 µg ml−1 for E. coli and 60 µg ml−1 for S. gordonii.
Table 1.
Strains and Plasmids used in this study.
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| Strain | ||
| S. gordonii | ||
| M99 | Parent strain | (Sullam et al., 1987) |
| PS919 | M99 gspB∷pB736flag | (Bensing et al., 2005) |
| PS920 | M99 gspB∷pB736flag, ΔsecA2∷spec | (Bensing et al., 2005) |
| PS942 | M99 gspB∷pB736flag, Δasp1∷spec | (Bensing et al., 2005) |
| PS944 | M99 gspB∷pB736flag, Δasp2∷spec | (Bensing et al., 2005) |
| PS945 | M99 gspB∷pB736flag, Δasp3∷spec | (Bensing et al., 2005) |
| PS925 | M99 gspB∷pB736flag, Δasp4∷spec | (Bensing et al., 2005) |
| PS924 | M99 gspB∷pB736flag, Δasp5∷spec | (Bensing et al., 2005) |
| PS1057 | PS919 ΔgtfA∷cat | This study |
| PS1063 | PS920 ΔgtfA∷cat | This study |
| PS1060 | PS942 ΔgtfA∷cat | This study |
| PS1061 | PS944 ΔgtfA∷cat | This study |
| PS1062 | PS945 ΔgtfA∷cat | This study |
| PS1059 | PS925 ΔgtfA∷cat | This study |
| PS1058 | PS924 ΔgtfA∷cat | This study |
| PS1244 | M99 gspB∷pB736flagCflag, Δasp3∷spec | This study |
| PS1208 | M99 gspB∷pB736flagC/G75P ΔgtfA∷cat ΔsecA2 | (Bensing et al., 2007) |
| PS1740 | M99 ΔgspB | This study |
| PS1764 | PS1740 gspB∷pB736flagC/G75P | This study |
| PS1765 | PS1764 gspB∷pB736flagC/G75P ΔgtfA∷cat | This study |
| PS1768 | PS1765 gspB∷pB736flagC/G75P ΔgtfA∷cat Δasp1∷spec | This study |
| PS1769 | PS1765 gspB∷pB736flagC/G75P ΔgtfA∷cat Δasp2∷spec | This study |
| PS1770 | PS1765 gspB∷pB736flagC/G75P ΔgtfA∷cat Δasp3∷spec | This study |
| PS1771 | PS1765 gspB∷pB736flagC/G75P ΔgtfA∷cat Δasp4∷spec | This study |
| PS1772 | PS1765 gspB∷pB736flagC/G75P ΔgtfA∷cat Δasp5∷spec | This study |
| E. coli | ||
| DH5-α | General cloning strain | Invitrogen |
| BL21λDE3 | F-ompT hsdSB(rB-mB)gal dcm (DE3) | Novagen |
| S. cerevisiae | ||
| W303a | MATa; ura3-1; leu2–3, | (MacGurn et al., 2005) |
| EGY48 | MATα; trp1; ura3; his3; LEU2∷pLexAop1-LEU2 | (MacGurn et al., 2005) |
| Plasmids | ||
| pET28b | E. coli T7 expression vector, Kanr | Novagen |
| pET His6asp3 | Vector over-expressing His6Asp3 protein, Kanr | This study |
| pACYCDuet | E. coli T7 expression vector, Cmr | Novagen |
| pACYCFlgAsp1 | Vector over-expressing flagAsp1 protein, Cmr | This study |
| pACYCFlgAsp2 | Vector over-expressing flagAsp2 protein, Cmr | This study |
| pACYCFlgAsp3 | Vector over-expressing flagAsp3 protein, Cmr | This study |
| pMal.c2X | Maltose-binding fusion expression plasmid | New England Biolabs |
| pMal.SecA2 | MalE-SecA2 expression plasmid | (Bensing and Sullam, 2009) |
| pEG202 | LexA bait plasmid, HIS3, Ampr | (MacGurn et al., 2005) |
| pEG202gspB | pEG202 + gspB (1–736 codons) | This study |
| pEG202asp1 | pEG202 + asp1 | This study |
| pEG202asp2 | pEG202 + asp2 | This study |
| pEG202asp3 | pEG202 + asp3 | This study |
| pEG202secA2 | pEG202 + secA2 | This study |
| pEG202asp4 | pEG202 + asp4 | This study |
| pEG202asp5 | pEG202 + asp5 | This study |
| pJSC401 | B42D prey plasmid, TRP1, Ampr | (MacGurn et al., 2005) |
| pJSC401gspB | pJSC401 + gspB (1–736 codons) | This study |
| pJSC401secY2 | pJSC401 + secY2 | This study |
| pJSC401asp1 | pJSC401 + asp1 | This study |
| pJSC401asp2 | pJSC401 + asp2 | This study |
| pJSC401asp3 | pJSC401 + asp3 | This study |
| pJSC401secA2 | pJSC401 + secA2 | This study |
| pJSC401asp4 | pJSC401 + asp4 | This study |
| pJSC401asp5 | pJSC401 + asp5 | This study |
| pC326 | E. coli cloning vector, ColE1, Cmr | (Mitchell et al., 2007) |
| pMSP3545 | Nisin expression vector | (Bryan et al., 2000) |
| pMSP3545asp3 | pMSP3545 + asp3 | This study |
| pMSP3545asp3∷tn104 | pMSP3545 + asp3∷tn104 | This study |
| pMSP3545asp3∷tn152 | pMSP3545 + asp3∷tn152 | This study |
| pMSP3545asp3∷tn332 | pMSP3545 + asp3∷tn332 | This study |
| pMSP3545asp3∷tn347 | pMSP3545 + asp3∷tn347 | This study |
| pMSP3545asp3∷tn349 | pMSP3545 + asp3∷tn349 | This study |
| pMSP3545asp3∷tn414 | pMSP3545 + asp3∷tn414 | This study |
| pMSP3545asp3∷tn422 | pMSP3545 + asp3∷tn422 | This study |
| pMSP3545asp3∷tn452 | pMSP3545 + asp3∷tn3452 | This study |
Ermr, erthromycin resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance; Cmr, chloramphenicol resistance; Specr, spectinomycin resistance.
DNA manipulations
Routine molecular biology techniques for cloning, sequencing and PCR amplification were performed by methods described elsewhere (Sambrook and Russel, 2001). Chromosomal DNA was isolated from S. gordonii according to Madoff et al., 1996. Plasmid DNA was isolated from E. coli using plasmid miniprep columns (Qiagen). DNA restriction and modification enzymes were used according to manufacturer’s recommendations (NEB). E. coli cells were routinely transformed following CaCl2 treatment (Sambrook and Russel, 2001), while S. gordonii was transformed as described previously (Bensing and Sullam, 2002).
Construction of S. gordonii gtfA/asp double deletion mutants
Accessory Sec genes were deleted from S. gordonii strains as described previously (Bensing et al., 2005). For previously created asp-deletion strains, a non-polar gtfA mutation was created by non-polar allelic exchange. For the analysis of Asp-dependent export of non-glycoslyated GspB736flag G75P, a chloramphenicol-sensitive derivative of M99 that expresses GspB736flag G75P (strain PS1764) was first generated as follows. Plasmid pSKΔB, which carries a 880 bp HindIII fragment from the region just upstream of gspB adjacent to a 900 bp NsiI-BglII fragment from the 3’end of gspB, was used to transform PS846 (Bensing et al., 2005). This resulted in a markerless deletion of gspB. Transformants were screened for the loss of chloramphenicol resistance (indicating replacement of the chromosomally-integrated pEVP3 by double cross-over between the flanking upstream and downstream segments). Replacement of pEVP3 by the upstream and downstream fragments in one such transformant (PS1740) was confirmed by PCR analysis of chromosomal DNA. Plasmid pB736flagR/G75P (Bensing et al., 2007) was then used to transform PS1740. A non-polar mutation of gtfA in the resulting strain (PS1764) was generated as described (Takamatsu et al., 2004b), except using a chloramphenicol resistance cassette from pC326 (Mitchell et al., 2007). Non-polar mutations in asp1 through asp5 were then generated by transformation of PS1765 with the corresponding knock-out vectors as described (Takamatsu et al., 2004b).
Bioinformatic searches
Predictions of protein secondary structure were made using Quick 2D (Max-Planck Institute for Development Biology: http://toolkit.tuebingen.mpg.de/quick2_d), a prediction server that utilizes PSIPRED, JNET, Prof(Rost), Prof(Ouali), Coils, MEMSAT2, HMMTOP, DISOPRED2 and VSL2 analysis to compile secondary structural features of a protein. Predictions of accessory Sec components cell localization was made using PSORTb v 3.0.2 (http://www.psort.org/psrotb).
Asp3 protein purification
Plasmid pET28b (Novagen) was used for the synthesis of N-terminally His-tagged Asp3, which was constructed as follows. A DNA fragment containing the entire coding sequence of asp3 was PCR amplified from S. gordonii strain M99 chromosomal DNA, using Vent High Fidelity Polymerase (NEB) and primers 5’-GCATATGAAGATTCAAAAACATAAGGAA and 5- CGTTCTCGAGTAACCATTTGACTCCTCTAAA (underlining indicates either an Nde I or Xho I restriction site). asp3 was amplified to incorporate 5’ Nde I and 3’Xho I restriction sites in order to facilitate an in-frame insertion of a His6-tag at the N-terminus. The resulting PCR product was digested with the appropriate restriction endonuclease pair and ligated to similarly digested pET28b and transformed directly into E. coli BL21(DE3)™ (Novagen). The identity of the cloned DNA fragment was verified through restriction digest analysis and DNA sequencing. For induction of His6Asp3 protein expression, the appropriate E. coli BL21(DE3)™ strain was grown in LB medium at 37°C supplemented with kanamycin. At an OD600 of 0.6, isopropyl-β-D-thiogalactopyranoside (IPTG) was added at a final concentration of 1 mM during 3 h at 25°C to induce expression. His6Asp3 tagged protein was purified under native conditions by affinity chromatography, using Ni-NTA agarose (Qiagen). Purified Asp3 protein was reconstituted in storage buffer (25 mM HEPES pH 7.4, 150 mM NaCl and 5 % glycerol) and stored in aliquots at −80°C until required for use.
Yeast two-hybrid analysis
For construction of LexA-bait and B42D–prey fusions, the complete amplicons of asp1, asp2, asp3, asp4, asp5, secA2 and secY2 were cloned into the EcoRI and XhoI sites into pEG202 and pjsc401. A truncated version of gspB encoding the first 736 amino acids of GspB was used in these studies, as its protein product does not aggregate when over-expressed in vivo, and, like native GspB, is dependent on the accessory Sec system for export (Bensing et al., 2005). B42D–prey constructs were transformed into S. cerevisiae strain w303-1a (ura3-1, leu2–3, his3, trp1, ade2-1) and selected on complete synthetic medium (CSM) lacking tryptophan (Trp), while bait constructs were transformed into S. cerevisiae strain EGY48 (trp1, his3, ura3, 1LexA-operator-LEU2) and selected on CSM lacking both uracil (Ura) and histidine (His). Expression of the hybrid fusions in yeast transformants was confirmed by immunoblot analysis using either anti-LexA, or anti-HA antibodies (a hemagglutinin-tag is present on the B42D protein). Bait and prey hybrid strains were mated, and diploid strains selected on CSM -Ura, -His and -Trp. The resulting diploid strains were re-plated onto CSM agar lacking Ura, His and Trp but containing galactose and 4 µg/ml X-gal (5-bromo-4-chloro-3-indoyl-β-galactosidase).
Quantitative β-galactosidase activity assays were performed as previously described by (Serebriiskii et al., 2005). In brief, exponentially growing diploid strains containing D-(+)-galactose in the appropriate media were added to an equal volume of 2x Z buffer containing 2 mg/ml of 2-nitrophenyl β- D-glucopyranoside (ONPG) and 50% Y-PER (Pierce) lysis reagent. Activity was calculated as 1000(A420)/(reaction time × culture volume × A600).
Co-expression and co-purification of proteins
DNA encoding asp1, asp2 or asp3 was cloned as an N-terminal flag-tag fusion, while secA2 was cloned untagged within the IPTG inducible plasmid pACYCDuet. E. coli BL21™ cells were simultaneously transformed with a mix of pETHis6asp3 and the pACYCDuet variants encoding the designated Flag-tagged accessory Sec component. These plasmids encode different antibiotic resistance markers and origins of replication, thus enabling dual plasmid maintenance within the cell. Recombinant colonies were selected on plates containing 15 µgml−1 kanamycin with 17 µg ml−1 chloramphenicol. Lysates of induced cells were probed with anti-Flag or anti-His antibodies to confirm the expression of each accessory Sec component. Expression and purification of Asp3-based complexes were carried out as described previously for His6Asp3. The recovered protein complexes were again probed with anti-Flag or anti-His antibodies to identify the presence or absence of each accessory Sec co-purifying partner. Clarified E. coli lysates expressing only Flag-tagged accessory Sec components were subject to Ni-NTA affinity chromatography, as a control for nonspecific binding to the resin.
For protein preparations, cells were collected by centrifugation 2 h post-IPTG induction. For His-tag protein purification, an identical strategy to that described for His6Asp protein purification was followed. To examine the absence or presence of co-purifying proteins, Ni-NTA agarose was allowed to settle in a Bio-Spin chromatography column (Bio-Rad), and an aliquot of the flow-through liquid (containing unbound material (flow) was saved for analysis. The resin was washed with 10 resin volumes of wash buffer (50 mM NaHPO4 pH 7.8, 300 mM NaCl, 25 mM imidazole, 0.5 % Triton X-100), and an aliquot from the last wash step was saved for immunoblot analysis (last wash). Bound proteins were eluted in the same volume of Elution buffer (50 mM NaHPO4 pH 8.0, 150 mM NaCl, 500 mM imidazole) (elution).
To analyze protein content from the above fractions, samples of each fraction were separated by SDS-PAGE, transferred to nitrocellulose membranes, and analyzed by Western blotting. Mouse anti-His antibody (GE Amersham) was used at a concentration of 1:3000, mouse anti-Flag antibody (Sigma) used at a concentration of 1:5000 and anti-SecA2 antibody was used at a concentration of 1:2000 (Bensing and Sullam, 2009). After incubation of blots with rabbit anti-mouse IgG-horseradish peroxidase (HRP) conjugate, immunoreactive bands were visualized using Supersignal substrate (Pierce).
Far Western blot analysis
E. coli BL21™ cells expressing flag-tagged Asps1–3 and MalE (Maltose binding protein) and MalE.SecA2 under IPTG induction were pelleted, normalized to an O.D600 of 0.1/10 µl, and lysed by boiling in 4x LDS sample buffer (Invitrogen). Proteins were separated by SDS-PAGE (4–12%) and transferred to nitrocellulose membranes. The membranes were blocked by incubating for 2 hr in PBS containing 5% w/v skimmed milk at room temperature, followed by a 2 h incubation in PBS/0.05% tween containing recombinant His6Asp protein (1 µM). Membranes were washed 3 times for 15 min in PBS/0.05% tween, and bound His6Asp3 was probed with mouse anti-His antibody (GE Amersham). Membranes were developed as previously described.
Densitometry analysis
To quantify differences in GspB transport, blots were incubated at room temperature (RT) with mouse anti-Flag antibody (Sigma) used at a concentration of 1:5000 for 2 hr, followed by another 90 min incubation with a 1:20,000 dilution of Alexa Fluor-680 anti-Mouse IgG (LI COR Biosciences). Immunoreactive bands were visualized using a LiCor infrared imager (LI COR Biosciences) at 700 nm. Band intensity was measured using Odyssey v3.0 software.
Preparation of antisera
Generation of anti-SecA2 antisera has been described previously (Bensing and Sullam, 2009). For anti-Asp1 and anti-Asp2 antisera production, rabbit polyclonal antiserum was raised against either purified soluble His6Asp1 or against polyacrylamide gel slices containing His6Asp2.
Co-immunoprecipitation of proteins from streptococci
Antisera against recombinant Asp3 was generated in female New Zealand White rabbits, and the IgG fraction was recovered by Protein A affinity chromatography. This purified IgG preparation was subsequently used to generate Asp3-specific affinity resin by covalent linkage to agarose using a Co-Immunoprecipitation kit (Pierce). Over night cultures of strain PS1244 and PS1244 complemented with His6Asp3 (expressed from pMSP3545) were diluted (1:5) in 50 ml THB and incubated in 5% CO2 at 37°C for 5 hours under nisin induction (250 ng ml−1). Cells were harvested by centrifugation, suspended in Co-IP lysis buffer (Pierce) and lysed through the use of a Zenith beadbeater using 2 × 45-second bursts at full speed. The cell lysates were centrifuged at 14,000 rpm for 10 min and the supernatants were transferred to a new tube.
Protein concentrations were determined using Protein Assay Kit (Pierce). Extracted proteins (400 µg) were mixed with 100 µl of anti-Asp3 resin slurry (containing approximately 100 µg of coupled Asp3-specific IgG) and incubated over night at 4°C under constant rotation. The anti-Asp3 resin was collected by centrifugation at 1,000 rpm for 2 min and washed three times with IP-lysis/wash buffer (Pierce). The proteins bound to the anti-Asp3 resin were eluted by boiling in SDS sample loading buffer and analyzed by SDS-PAGE and Western blotting with polyclonal antisera directed against Asp1, Asp2, Asp3 or SecA2 at a 1:2000 dilution.
Mutagenesis of asp3
An EZ-Tn5 <KAN-2> insertion kit (Epicenter Technologies) was used to create random, in-frame insertions of Tn5 within asp3. An Eco RI-Xho I restriction fragment of asp3 was cloned into pBluescriptΔNot I (a pBluescript KS- derivative with the Not I restriction site removed from the multiple cloning site). The resulting construct, pBluescriptKS-asp3, was subjected to EZ-Tn5 transposition, and the transposition mixture was used to transform E. coli. Colonies containing Tn5<KAN> insertions within asp3 were identified by PCR and the KAN cassette was removed through Not I restriction digest. Upon re-ligation, the plasmid contained asp3 with a random 19 codon in-frame insertion. The mutated Eco RI-Xho I asp3 gene fragment was amplified using primers ORF3C5 and ORF3C2 (Takamatsu et al., 2004b), digested with Nco I and Spe I and cloned into similarly digested pMSP3545. The resulting plasmids were sequenced to confirm the identity of the asp3 variant and transformed into S. gordonii strain PS1225 (Δasp3 deletion strain). From nisin-induced cells supernatants were collected from the media, and protoplasts were treated with mutanolysin and probed for secreted GspB736flag and Asp3 expression as described previously (Bensing et al., 2005).
Cysteine scanning mutagenesis
Cysteine replacement mutations within asp3 were made by a two-stage PCR procedure. For codon conversion to cysteine, overlapping primers (Table 2) were used with either primer asp3YTH-F 5’-CCGAATTCAAGATTCAAAAACATAAGGAAA or primer asp3YTH-R 5’-CGTTCTCGAGTTAACCATTTGACTCCTCTAAA (underlining indicates either a EcoRI or XhoI restriction site) to generate to overlapping DNA fragments spanning the entire asp3 open reading frame. The two DNA fragments were combined for the second stage PCR, and then amplified using primers asp3YTH-F and asp3YTH-R. Amplified products were digested with the appropriate restriction enzymes and ligated into similarly digested pjsc401. Cysteine-replacement mutations were verified by complete sequence analysis. The confirmed asp3 mutants were subsequently amplified and cloned within pMSP3545 using primers ORF3C5 and ORF3C2 as described above. Of note, sequencing of the Y124C mutant also identified a W11A replacement. However, individual analysis of a W11A mutant identified no changes upon GspB export or Asp3 binding (data not shown).
Table 2.
Oligonucleotides used for Cysteine scanning mutagenesis of asp3.
| Mutant | Mutagenic oligonucleotidea | Codon change |
|---|---|---|
| Y124C | Sense GATAAAATGACTTTTACTTGTCCCGAAGAAGCCTACAG | TAT⇥TGT |
| Antisense GTAGGCTTCTTCGGGACAAGTAAAAGTCATTTTATCCG | ||
| P125C | Sense AATGACTTTTACTTATTGCGAAGAAGCCTACAGCTATAAG | CCC⇥TGC |
| Antisense GCTGTAGGCTTCTTCGCAAAGTAAAAGTCATTTTATCCG | ||
| E127C | Sense TTTTACTTATCCCGAATGTGCCTACAGCTATAAGGTACAG | GAA⇥TGT |
| Antisense CTTATAGCTGTAGGCACATTCGGGATAAGTAAAAGTCA | ||
| A128C | Sense TACTTATCCCGAAGAATGCTACAGCTATAAGGTACAGCTTC | GCC⇥TGC |
| Antisense GTACCTTATAGCTGTAGCATTCTTCGGGATAAGTAAAAG | ||
| Y129C | Sense TATCCCGAAGAAGCCTGCAGCTATAAGGTACAGCTTCTTAG | TAC⇥TGC |
| Antisense GCTGTACCTTATAGCTGCAGGCTTCTTCGGGATAAG | ||
| Y131 | Sense CGAAGAAGCCTACAGCTGTAAGGTACAGCTTCTTAGTGC | TAT⇥TGT |
| Antisense AGAAGCTGTACCTTACAGCTGTAGGCTTCTTCGG | ||
| L135C | Sense CTATAAGGTACAGTGTAGTGCGGGTGTTGAGTC | CTT⇥TGT |
| Antisense CAACACCCGCACTACAAAGCTGTACCTTATAGCTGTAG | ||
| A138C | Sense GTACAGCTTCTTAGTTGCGGTGTTGAGTCTTTTGAGTT | GCG⇥TGC |
| Antisense CAAAAGACTCAACACCGCAACTAAGAAGCTGTACCTTATAG | ||
| G139C | Sense CAGCTTCTTAGTGCGTGTGTTGAGTCTTTTGAGTTTCAC | GGT⇥TGT |
| Antisense CTCAAAAGACTCAACACACGCACTAAGAAGCTGTACC | ||
| S142C | Sense GTGCGGGTGTTGAGTGTTTGAGTTTCACTGCTTAAGAAT | TCT⇥TGT |
| Antisense GCAGTGAAACTCAAAACACTCAACACCCGCACTAAG | ||
| F145C | Sense GAGTCTTTTGAGTGTCACTGCTTAAGAATTGAAG | TTT⇥TGT |
| Antisense CAATTCTTAAGCAGTGACACTCAAAAGACTCAACAC |
Sequence of mutagenic primers are shown in the 5’-3’ order, with altered codons in bold typeface.
Statistical analysis
All statistical analysis was carried out using Prism 4 statistical software package (GraphPad Software Inc.). For β-galactosidase activity, results were expressed as the geometric mean ± SD based on readings from triplicate samples, and compared for statistical significance by the unpaired t-test.
Acknowledgements
This study was supported by the Department of Veterans Affairs, the VA Merit Review program, and grants R01 AI41513 and R01 AI057433 from the National Institutes of Health. We thank Jason McGovern for his help with the yeast two-hybrid assay.
References
- Baba T, Taura T, Shimoike T, Akiyama Y, Yoshihisa T, Ito K. A cytoplasmic domain is important for the formation of a SecY-SecE translocator complex. Proc Natl Acad Sci U S A. 1994;91:4539–4543. doi: 10.1073/pnas.91.10.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensing BA, Sullam PM. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol. 2002;44:1081–1094. doi: 10.1046/j.1365-2958.2002.02949.x. [DOI] [PubMed] [Google Scholar]
- Bensing BA, Gibson BW, Sullam PM. The Streptococcus gordonii platelet binding protein GspB undergoes glycosylation independently of export. J Bacteriol. 2004a;186:638–645. doi: 10.1128/JB.186.3.638-645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensing BA, Lopez JA, Sullam PM. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun. 2004b;72:6528–6537. doi: 10.1128/IAI.72.11.6528-6537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensing BA, Takamatsu D, Sullam PM. Determinants of the streptococcal surface glycoprotein GspB that facilitate export by the accessory Sec system. Mol Microbiol. 2005;58:1468–1481. doi: 10.1111/j.1365-2958.2005.04919.x. [DOI] [PubMed] [Google Scholar]
- Bensing BA, Siboo IR, Sullam PM. Glycine residues in the hydrophobic core of the GspB signal sequence route export toward the accessory Sec pathway. J Bacteriol. 2007;189:3846–3854. doi: 10.1128/JB.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensing BA, Sullam PM. Characterization of Streptococcus gordonii SecA2 as a paralogue of SecA. J Bacteriol. 2009 doi: 10.1128/JB.00365-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyton C, Haase W, Rapoport TA, Kuhlbrandt W, Collinson I. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature. 2002;418:662–665. doi: 10.1038/nature00827. [DOI] [PubMed] [Google Scholar]
- Bryan EM, Bae T, Kleerebezem M, Dunny GM. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid. 2000;44:183–190. doi: 10.1006/plas.2000.1484. [DOI] [PubMed] [Google Scholar]
- Clemons WM, Jr, Menetret JF, Akey CW, Rapoport TA. Structural insight into the protein translocation channel. Curr Opin Struct Biol. 2004;14:390–396. doi: 10.1016/j.sbi.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Driessen AJ, Nouwen N. Protein Translocation Across the Bacterial Cytoplasmic Membrane. Annu Rev Biochem. 2007 doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- Fekkes P, van der Does C, Driessen AJ. The molecular chaperone SecB is released from the carboxy-terminus of SecA during initiation of precursor protein translocation. Embo J. 1997;16:6105–6113. doi: 10.1093/emboj/16.20.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- Jakubovics NS, Kerrigan SW, Nobbs AH, Stromberg N, van Dolleweerd CJ, Cox DM, Kelly CG, Jenkinson HF. Functions of cell surface-anchored antigen I/II family and Hsa polypeptides in interactions of Streptococcus gordonii with host receptors. Infect Immun. 2005;73:6629–6638. doi: 10.1128/IAI.73.10.6629-6638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kendall DA. Sec-dependent protein export and the involvement of the molecular chaperone SecB. Cell Stress Chaperones. 2000;5:267–275. doi: 10.1379/1466-1268(2000)005<0267:sdpeat>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker S, Lill R, Ziegelhoffer T, Georgopoulos C, Bassford PJ, Jr, Kumamoto CA, Wickner W. Three pure chaperone proteins of Escherichia coli--SecB, trigger factor and GroEL--form soluble complexes with precursor proteins in vitro. Embo J. 1989;8:2703–2709. doi: 10.1002/j.1460-2075.1989.tb08411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VT, Schneewind O. Protein secretion and the pathogenesisof bacterial infections. Genes Development. 2001;15:1725–1752. doi: 10.1101/gad.896801. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen Y, Huang X, Zhou M, Wu R, Dong S, Pritchard DG, Fives-Taylor P, Wu H. A conserved domain of previously unknown function in Gap1 mediates protein-protein interaction and is required for biogenesis of a serine-rich streptococcal adhesin. Mol Microbiol. 2008;70:1094–1104. doi: 10.1111/j.1365-2958.2008.06456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn JA, Raghavan S, Stanley SA, Cox JS. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol Microbiol. 2005;57:1653–1663. doi: 10.1111/j.1365-2958.2005.04800.x. [DOI] [PubMed] [Google Scholar]
- Madoff LC, Michel JL, Gong EW, Kling DE, Kasper DL. Group B streptococci escape host immunity by deletion of tandem repeat elements of the alpha C protein. Proc Natl Acad Sci U S A. 1996;93:4131–4136. doi: 10.1073/pnas.93.9.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J, Siboo IR, Takamatsu D, Chambers HF, Sullam PM. Mechanism of cell surface expression of the Streptococcus mitis platelet binding proteins PblA and PblB. Mol Microbiol. 2007;64:844–857. doi: 10.1111/j.1365-2958.2007.05703.x. [DOI] [PubMed] [Google Scholar]
- Nouwen N, Driessen AJ. Inactivation of protein translocation by cold-sensitive mutations in the yajC-secDF operon. J Bacteriol. 2005;187:6852–6855. doi: 10.1128/JB.187.19.6852-6855.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Wu H, Ruiz T, Chen Q, Zhou M, Sun B, Fives-Taylor P. Role of gap3 in Fap1 glycosylation, stability, in vitro adhesion, and fimbrial and biofilm formation of Streptococcus parasanguinis. Oral Microbiol Immunol. 2008;23:70–78. doi: 10.1111/j.1399-302X.2007.00401.x. [DOI] [PubMed] [Google Scholar]
- Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol. 2005;129:101–109. doi: 10.1111/j.1365-2141.2005.05421.x. [DOI] [PubMed] [Google Scholar]
- Rose L, Shivshankar P, Hinojosa E, Rodriguez A, Sanchez CJ, Orihuela CJ. Antibodies against PsrP, a novel Streptococcus pneumoniae adhesin, block adhesion and protect mice against pneumococcal challenge. J Infect Dis. 2008;198:375–383. doi: 10.1086/589775. [DOI] [PubMed] [Google Scholar]
- Rusch SL, Kendall DA. Oligomeric states of the SecA and SecYEG core components of the bacterial Sec translocon. Biochim Biophys Acta. 2007;1768:5–12. doi: 10.1016/j.bbamem.2006.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russel DW. Molecular cloning: a laboratory manual. 3rd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Seifert KN, Adderson EE, Whiting AA, Bohnsack JF, Crowley PJ, Brady LJ. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology. 2006;152:1029–1040. doi: 10.1099/mic.0.28516-0. [DOI] [PubMed] [Google Scholar]
- Serebriiskii IG, Fang R, Latypova E, Hopkins R, Vinson C, Joung JK, Golemis EA. A combined yeast/bacteria two-hybrid system: development and evaluation. Mol Cell Proteomics. 2005;4:819–826. doi: 10.1074/mcp.T500005-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siboo IR, Chaffin DO, Rubens CE, Sullam PM. Characterization of the accessory Sec system of Staphylococcus aureus. J Bacteriol. 2008;190:6188–6196. doi: 10.1128/JB.00300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullam PM, Valone FH, Mills J. Mechanisms of platelet aggregation by viridans group streptococci. Infect Immun. 1987;55:1743–1750. doi: 10.1128/iai.55.8.1743-1750.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu D, Bensing BA, Sullam PM. Four proteins encoded in the gspB-secY2A2 operon of Streptococcus gordonii mediate the intracellular glycosylation of the platelet-binding protein GspB. J Bacteriol. 2004a;186:7100–7111. doi: 10.1128/JB.186.21.7100-7111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu D, Bensing BA, Sullam PM. Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol Microbiol. 2004b;52:189–203. doi: 10.1111/j.1365-2958.2004.03978.x. [DOI] [PubMed] [Google Scholar]
- Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, Lopez JA, Griffiss JM, Sullam PM. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol. 2005a;58:380–392. doi: 10.1111/j.1365-2958.2005.04830.x. [DOI] [PubMed] [Google Scholar]
- Takamatsu D, Bensing BA, Sullam PM. Two additional components of the accessory sec system mediating export of the Streptococcus gordonii platelet-binding protein GspB. J Bacteriol. 2005b;187:3878–3883. doi: 10.1128/JB.187.11.3878-3883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu D, Bensing BA, Prakobphol A, Fisher SJ, Sullam PM. Binding of the streptococcal surface glycoproteins GspB and Hsa to human salivary proteins. Infect Immun. 2006;74:1933–1940. doi: 10.1128/IAI.74.3.1933-1940.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampakaki AP, Fadouloglou VE, Gazi AD, Panopoulos NJ, Kokkinidis M. Conserved features of type III secretion. Cell Microbiol. 2004;6:805–816. doi: 10.1111/j.1462-5822.2004.00432.x. [DOI] [PubMed] [Google Scholar]
- Thanassi DG, Hultgren SJ. Multiple pathways allow protein secretion across the bacterial outer membrane. Curr Opin Cell Biol. 2000;12:420–430. doi: 10.1016/s0955-0674(00)00111-3. [DOI] [PubMed] [Google Scholar]
- Veenendaal AK, van der Does C, Driessen AJ. The protein-conducting channel SecYEG. Biochim Biophys Acta. 2004;1694:81–95. doi: 10.1016/j.bbamcr.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Xiong YQ, Bensing BA, Bayer AS, Chambers HF, Sullam PM. Role of the serine-rich surface glycoprotein GspB of Streptococcus gordonii in the pathogenesis of infective endocarditis. Microb Pathog. 2008 doi: 10.1016/j.micpath.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Xu Z. The structural view of bacterial translocation-specific chaperone SecB: implications for function. Mol Microbiol. 2005;58:349–357. doi: 10.1111/j.1365-2958.2005.04842.x. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]