

Abstract
The carbonyl unit of the title compound, C13H10Cl2N2O, lies on a twofold rotation axis. The ring is aligned at 51.6 (1)° with respect to the N—C(=O)—N fragment. The two –NH– fragments of one molecule form hydrogen bonds [2.845 (2) Å] to the C=O fragment of an adjacent molecule, giving rise to the formation of a linear hydrogen-bonded chain.
Related literature
For isostructural N,N′-bis(4-bromophenyl)urea, see: Lin et al. (2004 ▶). N,N′-Bis-(4-chlorophenyl)urea has been isolated as a co-crystal with a phthalazinium chloride; see: Wamhoff et al. (1994 ▶). For the self-condensation of 4-chlorophenyl isocyanate to yield the title symmetrical urea, see: Fu et al. (2007 ▶); Jimenez Blanco et al. (1999 ▶).
Experimental
Crystal data
C13H10Cl2N2O
M r = 281.13
Monoclinic,
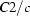
a = 27.093 (3) Å
b = 4.5768 (5) Å
c = 9.901 (1) Å
β = 96.389 (2)°
V = 1220.1 (2) Å3
Z = 4
Mo Kα radiation
μ = 0.52 mm−1
T = 100 (2) K
0.20 × 0.20 × 0.10 mm
Data collection
Bruker SMART APEX diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.862, T max = 0.950
3703 measured reflections
1386 independent reflections
1210 reflections with I > 2σ(I)
R int = 0.020
Refinement
R[F 2 > 2σ(F 2)] = 0.031
wR(F 2) = 0.096
S = 1.11
1386 reflections
87 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.31 e Å−3
Δρmin = −0.29 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: X-SEED (Barbour, 2001 ▶); software used to prepare material for publication: publCIF (Westrip, 2008 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808011069/tk2256sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808011069/tk2256Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O1i | 0.87 (1) | 2.05 (1) | 2.845 (2) | 152 (2) |
Symmetry code: (i) 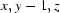 .
.
Acknowledgments
We thank the University of Malaya for funding this study (SF022155/2007 A) and also for the purchase of the diffractometer.
supplementary crystallographic information
Comment
The title compound, a symmetrical urea derivative, was the unexpected product from the reaction of 4-chlorophenyl isocyanate with p-tolylsulfonic acid in ethanol. The carbonyl unit of (Cl-4-C6H4)NH–C(=O)–NH(C6H4-4-Cl) lies on a twofold rotation axis, Fig. 1, that relates one aromatic ring to the other. The ring is aligned at 51.6 (1) ° with respect to the N–C(=O)–N fragment. The two –NH– fragments of one molecule forms hydrogen bonds to the C=O fragment of an adjacent molecule, giving rise to the formation of a linear hydrogen-bonded chain (Table 1). The compound has previously been synthesized from the self-condensation of 4-chlorophenyl isocyanate in acetone (Fu et al., 2007) and in water catalyzed by pyridine (Jimenez Blanco et al., 1999).
Experimental
4-Chlorophenyl isocyanate (1.0 g, 6.5 mmol) and p-toluenesulfonic acid (1.2 g, 6.5 mmol) were heated in ethanol (100 ml) for 1 h. The solution was filtered; evaporation of the solvent gave plates of the symmetrical urea.
Refinement
Carbon-bound H-atoms were placed in calculated positions (C—H 0.95 Å) and were included in the refinement in the riding model approximation, with U(H) set to 1.2Ueq(C).
The amino H-atom was located in a difference Fourier map, and was refined with a distance restraint of N–H 0.88±0.01 Å; its temperature factor was freely refined.
Figures
Fig. 1.

The molecular structure of (I) showing the atom-numbering scheme and 70% probability displacement ellipsoids. Hydrogen atoms are drawn as spheres of arbitrary radius. The unlablled atoms related by a 2-fold axis of symmetry.
Crystal data
| C13H10Cl2N2O | F000 = 576 |
| Mr = 281.13 | Dx = 1.530 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 1510 reflections |
| a = 27.093 (3) Å | θ = 3.0–28.2º |
| b = 4.5768 (5) Å | µ = 0.52 mm−1 |
| c = 9.901 (1) Å | T = 100 (2) K |
| β = 96.389 (2)º | Block, colorless |
| V = 1220.1 (2) Å3 | 0.20 × 0.20 × 0.10 mm |
| Z = 4 |
Data collection
| Bruker SMART APEXII diffractometer | 1386 independent reflections |
| Radiation source: fine-focus sealed tube | 1210 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.020 |
| T = 100(2) K | θmax = 27.5º |
| ω scans | θmin = 1.5º |
| Absorption correction: multi-scan(SADABS; Sheldrick, 1996) | h = −34→27 |
| Tmin = 0.862, Tmax = 0.950 | k = −5→5 |
| 3703 measured reflections | l = −10→12 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.031 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.096 | w = 1/[σ2(Fo2) + (0.0445P)2 + 1.607P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.11 | (Δ/σ)max < 0.001 |
| 1386 reflections | Δρmax = 0.31 e Å−3 |
| 87 parameters | Δρmin = −0.29 e Å−3 |
| 1 restraint | Extinction correction: none |
| Primary atom site location: structure-invariant direct methods |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.293344 (15) | 0.99597 (10) | 0.33207 (4) | 0.02417 (17) | |
| O1 | 0.5000 | 0.9101 (4) | 0.7500 | 0.0163 (4) | |
| N1 | 0.46380 (5) | 0.4789 (3) | 0.67795 (15) | 0.0149 (3) | |
| H1 | 0.4640 (8) | 0.292 (2) | 0.691 (2) | 0.024 (5)* | |
| C1 | 0.5000 | 0.6399 (5) | 0.7500 | 0.0130 (4) | |
| C2 | 0.42311 (6) | 0.6073 (3) | 0.59591 (15) | 0.0131 (3) | |
| C3 | 0.43093 (6) | 0.8150 (4) | 0.49760 (16) | 0.0152 (3) | |
| H3 | 0.4638 | 0.8730 | 0.4854 | 0.018* | |
| C4 | 0.39102 (6) | 0.9373 (4) | 0.41754 (17) | 0.0175 (4) | |
| H4 | 0.3963 | 1.0823 | 0.3520 | 0.021* | |
| C5 | 0.34334 (6) | 0.8455 (4) | 0.43438 (16) | 0.0162 (3) | |
| C6 | 0.33491 (6) | 0.6357 (4) | 0.52957 (17) | 0.0183 (4) | |
| H6 | 0.3021 | 0.5729 | 0.5391 | 0.022* | |
| C7 | 0.37498 (6) | 0.5180 (4) | 0.61096 (17) | 0.0176 (4) | |
| H7 | 0.3695 | 0.3754 | 0.6774 | 0.021* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0159 (2) | 0.0299 (3) | 0.0249 (3) | 0.00200 (17) | −0.00584 (17) | 0.00510 (17) |
| O1 | 0.0173 (8) | 0.0094 (8) | 0.0207 (8) | 0.000 | −0.0037 (6) | 0.000 |
| N1 | 0.0146 (7) | 0.0082 (6) | 0.0208 (7) | −0.0002 (5) | −0.0028 (6) | 0.0007 (5) |
| C1 | 0.0128 (10) | 0.0123 (11) | 0.0141 (10) | 0.000 | 0.0025 (8) | 0.000 |
| C2 | 0.0141 (7) | 0.0104 (7) | 0.0142 (7) | 0.0001 (6) | −0.0006 (6) | −0.0027 (6) |
| C3 | 0.0124 (7) | 0.0162 (8) | 0.0167 (8) | −0.0028 (6) | 0.0004 (6) | −0.0013 (6) |
| C4 | 0.0184 (8) | 0.0178 (8) | 0.0159 (8) | −0.0013 (6) | −0.0001 (6) | 0.0016 (6) |
| C5 | 0.0138 (8) | 0.0189 (8) | 0.0152 (8) | 0.0018 (6) | −0.0023 (6) | −0.0017 (6) |
| C6 | 0.0118 (8) | 0.0233 (9) | 0.0199 (8) | −0.0016 (6) | 0.0024 (6) | −0.0009 (7) |
| C7 | 0.0176 (8) | 0.0174 (8) | 0.0178 (8) | −0.0019 (6) | 0.0026 (6) | 0.0026 (6) |
Geometric parameters (Å, °)
| Cl1—C5 | 1.741 (2) | C3—C4 | 1.386 (2) |
| O1—C1 | 1.237 (3) | C3—H3 | 0.9500 |
| N1—C1 | 1.363 (2) | C4—C5 | 1.386 (2) |
| N1—C2 | 1.422 (2) | C4—H4 | 0.9500 |
| N1—H1 | 0.87 (1) | C5—C6 | 1.382 (2) |
| C1—N1i | 1.363 (2) | C6—C7 | 1.387 (2) |
| C2—C7 | 1.390 (2) | C6—H6 | 0.9500 |
| C2—C3 | 1.393 (2) | C7—H7 | 0.9500 |
| C1—N1—C2 | 122.9 (1) | C5—C4—C3 | 119.2 (2) |
| C1—N1—H1 | 118 (1) | C5—C4—H4 | 120.4 |
| C2—N1—H1 | 119 (1) | C3—C4—H4 | 120.4 |
| O1—C1—N1 | 122.7 (1) | C4—C5—C6 | 121.3 (2) |
| O1—C1—N1i | 122.7 (1) | C4—C5—Cl1 | 119.0 (1) |
| N1—C1—N1i | 114.6 (2) | C6—C5—Cl1 | 119.65 (13) |
| C7—C2—C3 | 119.5 (2) | C7—C6—C5 | 119.21 (15) |
| C7—C2—N1 | 119.6 (1) | C7—C6—H6 | 120.4 |
| C3—C2—N1 | 120.8 (1) | C5—C6—H6 | 120.4 |
| C4—C3—C2 | 120.4 (2) | C6—C7—C2 | 120.42 (15) |
| C4—C3—H3 | 119.8 | C6—C7—H7 | 119.8 |
| C2—C3—H3 | 119.8 | C2—C7—H7 | 119.8 |
| C2—N1—C1—O1 | 0.4 (2) | C3—C4—C5—C6 | 0.3 (3) |
| C2—N1—C1—N1i | −179.6 (2) | C3—C4—C5—Cl1 | −179.1 (1) |
| C1—N1—C2—C7 | −129.4 (2) | C4—C5—C6—C7 | 0.9 (3) |
| C1—N1—C2—C3 | 52.6 (2) | Cl1—C5—C6—C7 | −179.8 (1) |
| C7—C2—C3—C4 | 1.6 (2) | C5—C6—C7—C2 | −0.8 (3) |
| N1—C2—C3—C4 | 179.6 (2) | C3—C2—C7—C6 | −0.5 (2) |
| C2—C3—C4—C5 | −1.5 (2) | N1—C2—C7—C6 | −178.5 (2) |
Symmetry codes: (i) −x+1, y, −z+3/2.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O1ii | 0.87 (1) | 2.05 (1) | 2.845 (2) | 152 (2) |
Symmetry codes: (ii) x, y−1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: TK2256).
References
- Barbour, L. J. (2001). J. Supramol. Chem.1, 189–191.
- Bruker (2007). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Fu, J.-L., Wang, Z. & Zhu, H. (2007). Huaxue Shiji, 29, 187–188.
- Jimenez Blanco, J. L., Saitz Barria, C., Benito, J. M., Ortiz Mellet, C., Fuentes, J., Santoyo-Gonzalez, F. & Garcia Fernandez, J. (1999). Synthesis, pp. 1907–1914.
- Lin, Q., Zhang, Y.-M., Wei, T.-B. & Wang, H. (2004). Acta Cryst. E60, o696–o698.
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wamhoff, H., Bamberg, C., Hermann, S. & Nieger, M. (1994). J. Org. Chem.59, 3985–3993.
- Westrip, S. P. (2008). publCIF In preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808011069/tk2256sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808011069/tk2256Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report