

Abstract
The title compound, C10H5NO5, features an intramolecular O—H⋯O hydrogen bond, forming a six-membered ring with an S(6) ring motif. The nitro group makes a dihedral angle of 71.66 (5)° with the plane of the benzene ring to which it is bound. The two rings are almost coplanar, with a dihedral angle of 4.44 (5)°. Short intermolecular distances between the centroids of the six-membered rings [3.7188 (6)–3.8299 (6) Å] indicate the existence of π–π interactions. The interesting features of the crystal structure are the short intermolecular O⋯O and O⋯N interactions. The crystal packing is stabilized by intramolecular O—H⋯O and intermolecular C—H⋯O (×3) hydrogen bonds, and C—H⋯π interactions.
Related literature
For related literature on hydrogen-bond motifs, see Bernstein et al. (1995 ▶). For values of bond lengths, see Allen et al. (1987 ▶). For related literature, see, for example: Guingant & Barreto (1987 ▶); Larsen et al. (1996 ▶); Krohn et al. (2004 ▶); Krohn et al. (2004 ▶); Cui et al. (2006 ▶); Anuradha et al. (2006 ▶).
Experimental
Crystal data
C10H5NO5
M r = 219.15
Monoclinic,
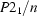
a = 8.6809 (2) Å
b = 8.4250 (2) Å
c = 12.1845 (3) Å
β = 93.946 (1)°
V = 889.02 (4) Å3
Z = 4
Mo Kα radiation
μ = 0.14 mm−1
T = 100.0 (1) K
0.35 × 0.14 × 0.13 mm
Data collection
Bruker SMART APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2005 ▶) T min = 0.900, T max = 0.982
22792 measured reflections
3028 independent reflections
2493 reflections with I > 2σ(I)
R int = 0.041
Refinement
R[F 2 > 2σ(F 2)] = 0.041
wR(F 2) = 0.119
S = 1.11
3028 reflections
165 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.50 e Å−3
Δρmin = −0.23 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: APEX2; data reduction: SAINT (Bruker, 2005 ▶); program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2003 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808021594/at2591sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808021594/at2591Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected interatomic and centroid–centroid distances (Å).
Cg1 and Cg2 are the centroids of the C1–C5/C10 and C5–C10 rings, respectively.
| Cg1⋯Cg2i | 3.7188 (6) |
| Cg1⋯Cg2i | 3.8299 (6) |
| O2⋯O5i | 2.9940 (11) |
| O5⋯O5ii | 3.0367 (11) |
| O5⋯N1ii | 3.0608 (11) |
Symmetry codes: (i) 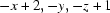 ; (ii)
; (ii) 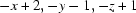 .
.
Table 2. Hydrogen-bond geometry (Å, °).
Cg1 is the centroid of the C1–C5/C10 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1O1⋯O2 | 0.889 (18) | 1.769 (19) | 2.5695 (10) | 148.5 (16) |
| C2—H2⋯O3iii | 0.969 (15) | 2.547 (16) | 3.1853 (12) | 123.4 (12) |
| C3—H3⋯O5ii | 0.970 (15) | 2.577 (15) | 3.3827 (13) | 140.6 (11) |
| C7—H7⋯O1iv | 0.982 (16) | 2.561 (16) | 3.1851 (13) | 121.4 (12) |
| C8—H8⋯Cg1v | 0.950 (15) | 2.976 (14) | 3.6548 (11) | 129.5 (11) |
Symmetry codes: (ii) ; (iii) 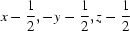 ; (iv)
; (iv) 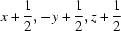 ; (v)
; (v) 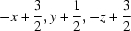 .
.
Acknowledgments
HO thanks the Malaysian government for the FRGS fund (grant No. 203/PKIMIA/671026). DTCT thanks Universiti Sains Malaysia for financial support. HKF and RK thank the Malaysian government and Universiti Sains Malaysia for the Science Fund grant No. 305/PFIZIK/613312. RK thanks Universiti Sains Malaysia for a post-doctoral research fellowship.
supplementary crystallographic information
Comment
5-Hydroxy-1,4-naphthoquinone (juglone) and its 5-acetoxy-2-bromo analogue is the essential dienophile in the highly convergent and regiospecific Diels-Alder synthesis of ochromycinone (Guingant & Barreto, 1987; Larsen et al., 1996; Krohn et al., 2004) a type of natural anthraquinone which exhibits remarkable antibiotic and antitumour activities (Krohn et al., 2004; Cui et al., 2006). Our aim is to prepare aromatic ring substituted juglone analogues for the purpose of synthesizing new ochromycinone analogues. The title compound was prepared by the direct nitration of juglone with nickel(II) nitrate. The method outlined previously (Anuradha et al., 2006) predicted a ortho-nitro product. However the product that we obtained is a para-nitro product.
Compound (I), ( Fig. 1), features an intramolecular O—H···O hydrogen bond to form a six-membered ring, producing a S(6) ring motif (Bernstein et al., 1995). The bond lenghts and angles are within the normal ranges (Allen et al., 1987). The two phenyl rings are almost coplanar with the dihedral angle of 4.44 (5)°. The nitro group is not coplanar with the benzene ring and its orientation is indicated by the dihedral angle of 71.66 (5)° with the plane of the benzene ring to which it is bound. The short intermolecular distances between the centroids of six-membered rings [3.7188 (6) - 3.8299 (6) Å] prove existence of π–π interactions (Table 1). The interesting feature of the crystal structure is the short intermolecular O···O and O···N interactions (Table 1). The crystal packing,(Fig. 2), of the compound shows one-dimensional infinite chains along the b axis.The crystal packing is stabilized by the intramolecular O—H···O, intermolecular C—H···O hydrogen bonds, π–π, and C—H···π interactions.
Experimental
8-Nitro-5-hydroxy-1,4-naphthoquinone was prepared from 5-hydroxy-1,4-naphthoquinone by the protocol outlined by (Anuradha et al., 2006). Single crystals of 8-nitro-5-hydroxy-1,4-naphthoquinone was obtained by slow evaporation of a chloroform solution at 286 K° C.
Refinement
All of the H-atoms were located from the difference Fourier map and refined freely.
Figures
Fig. 1.

The molecular structure of the title compound, showing 50% probability displacement ellipsoids and the atomic numbering. Intramolecular hydrogen bond is drawn as a dashed line.
Fig. 2.

The crystal packing of (I) shows a one-dimensional infinite chain along the [010] direction when viewed down the a-axis. Intramolecular and intermolecular interactions are drawn as dashed lines.
Crystal data
| C10H5NO5 | F000 = 448 |
| Mr = 219.15 | Dx = 1.637 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 5257 reflections |
| a = 8.6809 (2) Å | θ = 2.8–31.8º |
| b = 8.4250 (2) Å | µ = 0.14 mm−1 |
| c = 12.1845 (3) Å | T = 100.0 (1) K |
| β = 93.9460 (10)º | Block, brown |
| V = 889.02 (4) Å3 | 0.35 × 0.14 × 0.13 mm |
| Z = 4 |
Data collection
| Bruker SMART APEXII CCD area-detector diffractometer | 3028 independent reflections |
| Radiation source: fine-focus sealed tube | 2493 reflections with I > 2σ(I) |
| Monochromator: graphite | Rint = 0.041 |
| T = 100.0(1) K | θmax = 31.8º |
| φ and ω scans | θmin = 2.8º |
| Absorption correction: multi-scan(SADABS; Bruker, 2005) | h = −12→12 |
| Tmin = 0.901, Tmax = 0.982 | k = −12→12 |
| 22792 measured reflections | l = −18→17 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.041 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.119 | w = 1/[σ2(Fo2) + (0.0671P)2 + 0.1177P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.11 | (Δ/σ)max = 0.001 |
| 3028 reflections | Δρmax = 0.50 e Å−3 |
| 165 parameters | Δρmin = −0.22 e Å−3 |
| Primary atom site location: structure-invariant direct methods | Extinction correction: none |
Special details
| Experimental. The low-temperature data was collected with the Oxford Cyrosystem Cobra low-temperature attachment. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.60929 (9) | 0.14448 (9) | 0.47684 (6) | 0.01684 (17) | |
| O2 | 0.70729 (9) | 0.29533 (9) | 0.65117 (6) | 0.01718 (17) | |
| O3 | 1.13635 (9) | −0.14728 (9) | 0.74975 (6) | 0.01826 (17) | |
| O4 | 0.97394 (10) | −0.42986 (9) | 0.67891 (6) | 0.02146 (19) | |
| O5 | 1.11804 (9) | −0.37894 (9) | 0.54429 (6) | 0.01912 (18) | |
| N1 | 1.01058 (10) | −0.34927 (10) | 0.60140 (7) | 0.01427 (18) | |
| C1 | 0.71200 (11) | 0.03181 (12) | 0.50785 (8) | 0.01280 (18) | |
| C2 | 0.71889 (11) | −0.10405 (12) | 0.44177 (8) | 0.01457 (19) | |
| C3 | 0.81830 (11) | −0.22605 (12) | 0.47321 (8) | 0.01431 (19) | |
| C4 | 0.91370 (11) | −0.21081 (11) | 0.56968 (8) | 0.01219 (18) | |
| C5 | 0.91680 (11) | −0.07504 (11) | 0.63367 (7) | 0.01156 (18) | |
| C6 | 1.03288 (11) | −0.05166 (12) | 0.72840 (8) | 0.01326 (19) | |
| C7 | 1.02414 (12) | 0.09636 (13) | 0.79222 (8) | 0.0172 (2) | |
| C8 | 0.91880 (12) | 0.20847 (12) | 0.76649 (8) | 0.0168 (2) | |
| C9 | 0.80517 (11) | 0.19086 (12) | 0.67159 (8) | 0.01387 (19) | |
| C10 | 0.81225 (11) | 0.04769 (11) | 0.60319 (8) | 0.01189 (18) | |
| H8 | 0.9139 (16) | 0.3051 (18) | 0.8063 (12) | 0.022 (4)* | |
| H2 | 0.6513 (17) | −0.1106 (19) | 0.3752 (12) | 0.025 (4)* | |
| H3 | 0.8222 (16) | −0.3231 (18) | 0.4307 (12) | 0.020 (3)* | |
| H7 | 1.1034 (18) | 0.1097 (19) | 0.8530 (13) | 0.029 (4)* | |
| H1O1 | 0.620 (2) | 0.223 (2) | 0.5255 (16) | 0.049 (5)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0161 (3) | 0.0151 (4) | 0.0187 (4) | 0.0031 (3) | −0.0031 (3) | 0.0017 (3) |
| O2 | 0.0197 (4) | 0.0144 (4) | 0.0176 (3) | 0.0040 (3) | 0.0029 (3) | 0.0005 (3) |
| O3 | 0.0190 (4) | 0.0170 (4) | 0.0179 (3) | 0.0025 (3) | −0.0051 (3) | 0.0005 (3) |
| O4 | 0.0285 (4) | 0.0150 (4) | 0.0210 (4) | 0.0010 (3) | 0.0023 (3) | 0.0054 (3) |
| O5 | 0.0161 (3) | 0.0181 (4) | 0.0234 (4) | 0.0026 (3) | 0.0032 (3) | −0.0025 (3) |
| N1 | 0.0159 (4) | 0.0110 (4) | 0.0155 (4) | −0.0001 (3) | −0.0015 (3) | −0.0010 (3) |
| C1 | 0.0114 (4) | 0.0132 (4) | 0.0136 (4) | 0.0000 (3) | −0.0003 (3) | 0.0021 (3) |
| C2 | 0.0142 (4) | 0.0156 (5) | 0.0135 (4) | −0.0011 (3) | −0.0018 (3) | −0.0004 (3) |
| C3 | 0.0152 (4) | 0.0138 (4) | 0.0138 (4) | −0.0011 (3) | 0.0000 (3) | −0.0021 (3) |
| C4 | 0.0122 (4) | 0.0110 (4) | 0.0133 (4) | 0.0007 (3) | 0.0005 (3) | 0.0008 (3) |
| C5 | 0.0120 (4) | 0.0112 (4) | 0.0114 (4) | −0.0010 (3) | 0.0002 (3) | −0.0001 (3) |
| C6 | 0.0143 (4) | 0.0130 (4) | 0.0122 (4) | −0.0011 (3) | −0.0012 (3) | 0.0005 (3) |
| C7 | 0.0203 (5) | 0.0157 (5) | 0.0150 (4) | −0.0008 (4) | −0.0030 (3) | −0.0026 (4) |
| C8 | 0.0204 (5) | 0.0144 (5) | 0.0153 (4) | −0.0006 (4) | −0.0005 (3) | −0.0035 (3) |
| C9 | 0.0155 (4) | 0.0123 (4) | 0.0141 (4) | −0.0002 (3) | 0.0031 (3) | 0.0002 (3) |
| C10 | 0.0123 (4) | 0.0110 (4) | 0.0124 (4) | 0.0001 (3) | 0.0011 (3) | 0.0004 (3) |
Geometric parameters (Å, °)
| O1—C1 | 1.3388 (11) | C3—C4 | 1.3964 (13) |
| O1—H1O1 | 0.89 (2) | C3—H3 | 0.970 (15) |
| O2—C9 | 1.2367 (12) | C4—C5 | 1.3836 (13) |
| O3—C6 | 1.2211 (12) | C5—C10 | 1.4087 (13) |
| O4—N1 | 1.2229 (11) | C5—C6 | 1.4921 (13) |
| O5—N1 | 1.2274 (11) | C6—C7 | 1.4744 (14) |
| N1—C4 | 1.4741 (12) | C7—C8 | 1.3369 (15) |
| C1—C2 | 1.4031 (14) | C7—H7 | 0.982 (16) |
| C1—C10 | 1.4091 (13) | C8—C9 | 1.4748 (14) |
| C2—C3 | 1.3793 (14) | C8—H8 | 0.950 (15) |
| C2—H2 | 0.969 (15) | C9—C10 | 1.4699 (13) |
| Cg1···Cg2i | 3.7188 (6) | O5···O5ii | 3.0367 (11) |
| Cg1···Cg2i | 3.8299 (6) | O5···N1ii | 3.0608 (11) |
| O2···O5i | 2.9940 (11) | ||
| C1—O1—H1O1 | 107.7 (12) | C4—C5—C6 | 122.07 (8) |
| O4—N1—O5 | 124.96 (9) | C10—C5—C6 | 119.72 (8) |
| O4—N1—C4 | 117.90 (8) | O3—C6—C7 | 120.63 (9) |
| O5—N1—C4 | 117.04 (8) | O3—C6—C5 | 121.69 (9) |
| O1—C1—C2 | 118.06 (8) | C7—C6—C5 | 117.58 (8) |
| O1—C1—C10 | 121.79 (9) | C8—C7—C6 | 122.22 (9) |
| C2—C1—C10 | 120.15 (9) | C8—C7—H7 | 121.9 (10) |
| C3—C2—C1 | 119.94 (9) | C6—C7—H7 | 115.8 (9) |
| C3—C2—H2 | 121.4 (9) | C7—C8—C9 | 121.51 (9) |
| C1—C2—H2 | 118.6 (9) | C7—C8—H8 | 122.7 (9) |
| C2—C3—C4 | 119.26 (9) | C9—C8—H8 | 115.8 (9) |
| C2—C3—H3 | 121.6 (9) | O2—C9—C10 | 121.66 (9) |
| C4—C3—H3 | 119.1 (9) | O2—C9—C8 | 119.96 (9) |
| C5—C4—C3 | 122.53 (9) | C10—C9—C8 | 118.38 (9) |
| C5—C4—N1 | 121.19 (8) | C5—C10—C1 | 119.86 (9) |
| C3—C4—N1 | 116.27 (8) | C5—C10—C9 | 120.27 (8) |
| C4—C5—C10 | 118.10 (8) | C1—C10—C9 | 119.86 (9) |
| O1—C1—C2—C3 | −177.19 (9) | O3—C6—C7—C8 | 175.40 (10) |
| C10—C1—C2—C3 | 3.41 (15) | C5—C6—C7—C8 | −1.17 (15) |
| C1—C2—C3—C4 | −1.45 (15) | C6—C7—C8—C9 | −0.45 (16) |
| C2—C3—C4—C5 | −2.37 (15) | C7—C8—C9—O2 | 178.62 (10) |
| C2—C3—C4—N1 | 176.71 (9) | C7—C8—C9—C10 | −1.51 (15) |
| O4—N1—C4—C5 | 72.88 (12) | C4—C5—C10—C1 | −2.00 (14) |
| O5—N1—C4—C5 | −110.47 (10) | C6—C5—C10—C1 | 174.21 (8) |
| O4—N1—C4—C3 | −106.21 (10) | C4—C5—C10—C9 | 176.79 (8) |
| O5—N1—C4—C3 | 70.44 (11) | C6—C5—C10—C9 | −7.00 (14) |
| C3—C4—C5—C10 | 4.07 (14) | O1—C1—C10—C5 | 178.97 (9) |
| N1—C4—C5—C10 | −174.97 (8) | C2—C1—C10—C5 | −1.65 (14) |
| C3—C4—C5—C6 | −172.05 (9) | O1—C1—C10—C9 | 0.17 (14) |
| N1—C4—C5—C6 | 8.92 (14) | C2—C1—C10—C9 | 179.56 (9) |
| C4—C5—C6—O3 | 4.45 (15) | O2—C9—C10—C5 | −174.82 (9) |
| C10—C5—C6—O3 | −171.60 (9) | C8—C9—C10—C5 | 5.31 (14) |
| C4—C5—C6—C7 | −179.01 (9) | O2—C9—C10—C1 | 3.97 (15) |
| C10—C5—C6—C7 | 4.94 (13) | C8—C9—C10—C1 | −175.90 (9) |
Symmetry codes: (i) −x+2, −y, −z+1; (ii) −x+2, −y−1, −z+1.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1O1···O2 | 0.889 (18) | 1.769 (19) | 2.5695 (10) | 148.5 (16) |
| C2—H2···O3iii | 0.969 (15) | 2.547 (16) | 3.1853 (12) | 123.4 (12) |
| C3—H3···O5ii | 0.970 (15) | 2.577 (15) | 3.3827 (13) | 140.6 (11) |
| C7—H7···O1iv | 0.982 (16) | 2.561 (16) | 3.1851 (13) | 121.4 (12) |
| C8—H8···Cg1v | 0.950 (15) | 2.976 (14) | 3.6548 (11) | 129.5 (11) |
Symmetry codes: (iii) x−1/2, −y−1/2, z−1/2; (ii) −x+2, −y−1, −z+1; (iv) x+1/2, −y+1/2, z+1/2; (v) −x+3/2, y+1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: AT2591).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–S19.
- Anuradha, V., Srinivas, P. V., Aparna, P. & Rao, J. M. (2006). Tetrahedron Lett.47, 4933–4935.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Bruker (2005). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Cui, J. R., Jian, Y. J., Wu, Y. K. & Wang, S. M. (2006). Chin. J. Chem.24, 1163–1169.
- Guingant, A. & Barreto, M. M. (1987). Tetrahedron Lett.28, 3107–3110.
- Krohn, K., Sohrab, M. H. & Flörke, U. (2004). Tetrahedron Asymmetry15, 713–718.
- Larsen, D. S., O’Shea, M. D. & Brooker, S. (1996). Chem. Commun. pp. 203–204.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2003). J. Appl. Cryst.36, 7–13.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536808021594/at2591sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808021594/at2591Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report