

Abstract
Because adolescent brains are undergoing extensive developmental changes, they may be uniquely sensitive to effects of addictive drugs like nicotine. We exposed adolescent and adult rats to nicotine infusion for two weeks, and then used whole genome microarray analysis to determine effects on gene expression in the ventral tegmental area. We examined brains immediately after two weeks of nicotine or saline, and also four weeks after termination of nicotine exposure. After identifying genes with a significant age X treatment interaction, we employed template matching to find specific patterns of expression across age and treatment. Of those genes that were transiently regulated (up- or down-regulated immediately following the end of nicotine treatment, but back to saline baseline 30 days later), two-thirds were specific to adult animals, while only 30% were specific to adolescents and 4% were shared across the two ages. In contrast, significant genes that were persistently regulated (altered following nicotine treatment and still altered 30 days later) were more likely (59%) to be adolescent, with only 32% in adults and 8% shared. The greatest number of significant genes was late-regulated (no change immediately after nicotine, but regulated 30 days later). Again, most were in adolescents (54%), compared to adults (10%) or shared (36%). Pathway analysis revealed that adolescent-specific genes were over-represented in several biological functions and canonical pathways, including nervous system development and function and long-term potentiation. Furthermore, adolescent-specific genes formed extensive interaction networks, unlike those specific for adults or shared. This age-specific expression pattern may relate to the heightened vulnerability of adolescents to the effects of addictive drugs. In particular, the propensity of adolescents to show persistent alterations in gene expression corresponds to the persistence of drug dependence among smokers who began their habit as adolescents. These findings support a model whereby adolescent brains are uniquely vulnerable to long-term changes in gene expression in the brain’s reward pathway caused by early exposure to nicotine.
Keywords: Nicotine addiction, Adolescent drug abuse, Long-term potentiation, Nervous system development, Nicotinic receptors
Recreational drug use is commonly initiated during adolescence, often beginning with tobacco products. Nicotine has been shown to have a number of adverse affects on the brain, particularly during early development. Epidemiological studies have demonstrated that adolescent smokers proceed to dependence quicker, are more likely to be dependent as adults, have higher average consumption, and exhibit increased co-morbidity with other drug use and more psychological problems compared to adults (Chen and Millar, 1998; Chambers et al., 2003; Adriani et al., 2006). Behavioral studies have shown distinct responses to nicotine in animals treated during adolescence relative to their adult counterparts. Pre-exposure to nicotine during, but not following, adolescence sensitizes rats to nicotine’s effects on locomotor response and conditioned place preference (CPP), and increases self-administration of nicotine (Adriani et al., 2006). Pre-exposure to nicotine during adolescence also sensitizes rats to the rewarding effects of other drugs, particularly psychostimulants such as cocaine (Collins and Izenwasser, 2004; McMillen et al., 2005; McQuown et al., 2007). These data strongly suggest an increased vulnerability to effects of nicotine during adolescence.
Adolescence is characterized by extensive physiological and psychological changes, and recent studies have shown the human brain continues to mature into the early 20’s (Sowell et al., 2003). This is characterized by extensive growth during early adolescence, followed by a decrease in grey matter, a gradual loss of synapses, and a strengthening of the remaining synapses. Neurotransmitter receptor populations peak during adolescence and decline thereafter. This has been shown for GABA, serotonin, norepinephrine, dopamine, and acetylcholine receptors (Lujan et al., 2005). These changes occur throughout the brain and coincide with changes in complex social behaviors characterized by increased impulsivity, risk-taking, and sensation-seeking (Sowell et al., 2003). Exposure to nicotine during this period may have systemic affects which persist into adulthood and contribute to the observed increase in consumption of tobacco and other drugs of abuse, and the subsequent difficulty of quitting in those individuals that began smoking during adolescence.
We have previously shown that chronic nicotine exposure during adolescence differentially regulates nAChR subtype number in adolescent compared to adult male Sprague-Dawley (SD) rats (Doura et al., 2008). The goal of the current study was to identify the short-term and long-term changes in gene expression unique to, and shared between, adolescent and adult male SD rats exposed to chronic nicotine. We examined whole genome expression in the ventral tegmental area (VTA) from nicotine- and saline-treated rats of both age groups. The VTA is the cell body region for the mesocorticolimbic dopamine reward pathway, and thus it is likely that gene expression in these cells may play important roles in mediating the addictive properties of drugs such as nicotine. We report striking differences in gene expression in response to chronic nicotine in a number of potentially relevant networks and pathways, including nervous system development and function, circadian rhythms, and LTP.
EXPERIMENTAL PROCEDURES
Treatment
Osmotic minipumps (Alzet model 2002; Durect Corporation, Cupertino, CA) were filled with sterile saline or with nicotine hydrogen tartrate dissolved in saline, at concentrations designed to achieve a dose of 6 mg/kg/day, calculated as nicotine free base (37 μmol/kg). Minipumps were implanted into male Sprague–Dawley rats (Hilltop Lab Animals, Scottdale, PA) of two ages, postnatal day (PN) 28-30 or PN 60–70; six animals were used for each treatment group. The period from PN 28–40 in rats is typically labeled periadolescence, that from PN 40–52 middle adolescence, and PN 52–60 late adolescence; “puberty” generally occurs during the last days of periadolescence (Spear, 2004). Thus, our treatment was performed at the early periadolescent and adult stages. Rats were anesthetized with isoflurane and the minipumps inserted into a subcutaneous pocket via a small incision made over the shoulders. While under anesthesia, animals were administered buprenorphine (0.1 mg/kg, s.c.) for post-operative pain. The wound was closed with clips and the area swabbed with antiseptic. After recovery from anesthetic (10–30 min), animals were returned to individual cages. Fourteen days after minipump implantation (PN 42-44 for adolescents PN 74–84 for adults), some animals were lightly anesthetized with isoflurane and sacrificed by decapitation. The remaining animals were left untreated for 30 days (minipumps empty 14 to 17 days after implant, so animals were nicotine-free for from 27-29 days), during which time the adolescents reach full adulthood. The remaining animals were then sacrificed as described. Altogether, there were 3 treatment groups, comprised of saline, nicotine, and nicotine + 30 days (nicotine withdrawal), for both the adolescent and adult animals.
Tissue Preparation
Rats were anesthetized with isoflurane and decapitatied; brains were quickly removed and frozen on dry ice. Brain slices were made with a cold stainless steel rat brain slicer matrix (Zivic Instruments, Pittsburgh, PA) with 1 mm coronal slice section intervals. Brain slices were immediately placed in cold saline and four punches (two from each side) were taken from the VTA of each animal using a 500 micron tissue biopsy punch (Zivic Instruments, Pittsburgh, PA). Identification of tissue was done by reference to a rat brain atlas (Paxinos and Watson, 2007); coronal sections were used from -4.6 mm to -5.0 mm from bregma. Tissue punches were stored in RNAlater (Qiagen, Valencia, CA) at -80°C.
Total RNA Isolation and Gene Expression Profiling
Each VTA sample (representing an independent biological replicate) consisted of a pool from two animals (a total of 8 punches). Total RNA was isolated and purified using Trizol reagent (InVitrogen, Carlsbad, CA) and the RNeasy micro kit (Qiagen), respectively, according to the manufacturers’ instructions. The purified RNA was amplified using the WT-Ovation Pico RNA Amplification System (Nugen, San Carlos, CA) according to the manufacturer’s instructions. cRNA target synthesis, hybridization onto the Rat 230 2.0 GeneChip, and posthybridization staining and scanning were performed using standard protocols as recommended by the manufacturer (Affymetrix, Santa Clara, CA). Chip data were scaled using GeneChip Operating Software (GCOS; Affymetrix) version 1.4 and expression values were log2-transformed. In total, 18 hybridizations were performed representing 3 independent replicates for each of 3 treatment groups in both adolescent and adult animals.
Statistical and Pathway Analyses
GCOS scaled log2-transformed expression data were analyzed using the Partek Genomic Suite (Partek, St. Louis, MO). We performed a one-way ANOVA followed by a 10% false discovery rate (FDR) to identify genes with a significant treatment effect in common between both age groups (Shared genes). To identify age-specific genes with a significant treatment effect (Age-specific genes), we performed a two-way ANOVA (age and treatment as the main factors) with 10% FDR and identified genes with a significant age X treatment interaction. The resulting significant genes were subjected to Template Matching/Feature Selection (Pavlidis and Noble, 2001) executed in MultiExperiment Viewer (Dana-Farber Cancer Institute, Boston, MA; www.tm4.org/mev) in order to identify genes exhibiting specified patterns of expression. This search is based on a Pearson Correlation and significance was determined at P < 0.05, allowing us to identify transient, persistent, and late response gene expression profiles that were unique to or shared between the two age groups. Further bioinformatics analysis was conducted on the significant genes to identify functional significance with respect to gene ontology, molecular networks and canonical pathways by means of Ingenuity Pathways Analysis (IPA 6.5 software; Ingenuity Systems, Redwood City, CA) and GeneSpring GX 11 (Agilent Technologies, Santa Clara, CA). Multigene interaction networks were produced using Pathway Studio 6 (Ariadne Genomics, Rockville, MD).
Quantitative real-time reverse transcriptase PCR
To validate expression data for selected genes, we performed quantitative real-time RT-PCR (qRT-PCR) using SYBR green on an ABI Prism 7700 Detection System (Applied Biosystems, Foster City, CA). Total RNA was reverse transcribed using random primers from TaqMan Reverse Transcription Reagents (Applied Biosystems) per manufacturer’s instructions. PCR primers were chosen for specificity by NCBI BLAST of the rat genome and amplicon specificity was verified by first-derivative melting curve analysis with software provided by Perkin Elmer and Applied Biosystems. Quantitation and normalization of relative gene expression was accomplished using the comparative threshold cycle method previously described (Joe et al., 2005). The house-keeping genes GLI1 and RPL36AL exhibited no differential expression in our array analysis and were used for normalization of target genes. Table 1 shows genes identified as differentially expressed across age and treatment groups and chosen for qRT-PCR validation.
Table 1.
List of primers used for real-time RTPCR validation. Shown are the NCBI accession numbers, primer sequences and product sizes.
| Gene | Accession # | Forward Primer (5’-3’) | Reverse Primer (5’-3’) | Amplicon |
|---|---|---|---|---|
| CTNNB1 | NM_053357 | TCATGCGCCTTTGCGGGAAC | CGGACGCCCTCCACGAACTG | 126 |
| DDR1 | NM_001166022 | AGCCAAGTGCCGCTATGCCC | GGCACCAAGCCCCATCTCCA | 140 |
| GRINA | NM_153308 | CATGGGAACTACCAGGAGGA | GTCAGCACCAGGAACACCTT | 119 |
| KLF9 | NM_057211 | AAGCTCCGCAGCCACCCTCA | CGGCGCGGTTGGAGATGGAA | 125 |
| MRAS | NM_012981 | AGCCCAGTTCCCAGTCTCTT | TTGTCTGGTGCGTTGAATGT | 101 |
| NF1 | NM_012609 | GGCACTTCAAACCAAGCCGCA | GGAAGACCAGCGAGGAGCGAG | 101 |
| NINJ2 | NM_021595 | ATGCCCCTGCCTCTCACCCA | TGCAGTGCAACCAGAGTGCC | 150 |
| PPP1CA | NM_031527 | AACGAGATCCGTGGTCTTTG | CTCGAACAGCCGTAGAAGGT | 135 |
| PPP1R14A | NM_130403 | GGCAGACATGCCAGATGAG | CAGGCAAGCCTCCAAGAGT | 134 |
| PTPRF | NM_019249 | CAGCTCCTCTGAGCCATACC | GGTGTTCTGGGCTCTCTGAC | 103 |
| REST | NM_031788 | ATGGCGAGGCAGGAGCAGGT | TTGGCAGTGGTGACGGTGGG | 118 |
| VIP | NM_053991 | AGACCCAAGGAGGCACCGAGA | TAGAGAGGCCAGGCCAGCGA | 106 |
| GLI1 | XM_345832 | TTCCATATCAGAGCCCCAAG | ATGCAAAGCCAGATCCAAAC | 105 |
| RPL36AL | NM_031105 | GCCTCACAAAGTGACCCAGT | TGTGGTTTTGGCCTTCTTTC | 133 |
RESULTS
Whole genome expression was measured in the VTA of adolescent and adult male SD rats exposed to saline, chronic nicotine for 14 days, or 30 days withdrawal following chronic nicotine. A two-way ANOVA with 10% FDR revealed 4878 genes with a significant age X treatment interaction. Next, we applied template matching (also known as feature selection) (Pavlidis and Noble, 2001) to search for patterns of expression across age and treatment, identifying three distinct temporal expression patterns referred to as “transient response”, “persistent response” and “late response” genes. Transient response genes displayed an initial response (up- or down-regulated) to chronic nicotine compared to saline/baseline as measured at the end of the two week treatment period, but had returned to baseline expression levels at the end of the 30 day withdrawal period (Figs. 1A and 1B). There were 80 adolescent-specific genes (Fig. 1A) and 171 adult-specific genes (Fig. 1B) that exhibited a transient response to chronic nicotine. Persistent response genes displayed an initial response to chronic nicotine as measured at the end of the two week treatment period, and remained up- or down-regulated at the end of the 30 day withdrawal period (Figs. 1D and 1E). Sixty two adolescent-specific genes (Fig. 1D) and 33 adult-specific genes (Fig. 1E) exhibited a persistent pattern of response. Late response genes showed no initial response to chronic nicotine at the end of the two week treatment period, but showed significant up- or down-regulation at the end of the 30 day withdrawal period (Figs. 1G and 1H). A total of 532 adolescent-specific genes (Fig. 1G) and 101 adult-specific genes (Fig. 1H) exhibited a late response to chronic nicotine.
Figure 1.
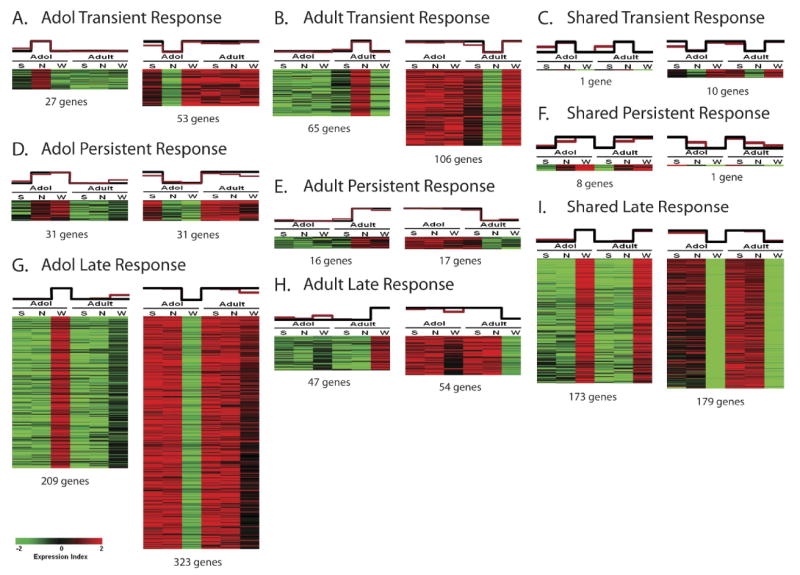
Gene expression correlation with chronic nicotine exposure. Heat maps of template-matched genes show positive and negative correlations with chronic nicotine treatment. The black graph above each heat map represents the specific template employed for matching, whereas the red graph represents the mean expression values of all matched genes within each column. Each pattern includes both a positive template (corresponding to gene up-regulation, left) and a negative template (corresponding to gene down-regulation, right). A. Genes showing a transient response to chronic nicotine in adolescent rats. B. Genes showing a transient response to chronic nicotine in adult rats. C. Genes showing a transient response to chronic nicotine shared between adolescent and adult rats. D. Genes showing a persistent response to chronic nicotine in adolescent rats. E. Genes showing a persistent response to chronic nicotine in adult rats. F. Genes showing a persistent response to chronic nicotine shared between adolescent and adult rats. G. Genes showing a late response to chronic nicotine in adolescent rats. H. Genes showing a late response to chronic nicotine in adult rats. I. Genes showing a late response to chronic nicotine shared between adolescent and adult rats.
A one-way ANOVA with 10% FDR revealed 973 adult genes and 3072 adolescent genes that were significant for chronic nicotine regulation. Of these, 425 genes were shared between the two age groups. Eleven shared genes exhibited a transient response (Fig. 1C), 9 shared genes exhibited a persistent response (Fig. 1F), and 352 shared genes exhibited a late response to chronic nicotine (Figs. 1I). It is noteworthy that the majority of shared gene regulation (≈83%) occurred after the end of chronic nicotine treatment (late response shared genes). By comparison, the number of transient and persistent response shared genes accounted for less than 6% of all shared genes (Fig. 2).
Figure 2.
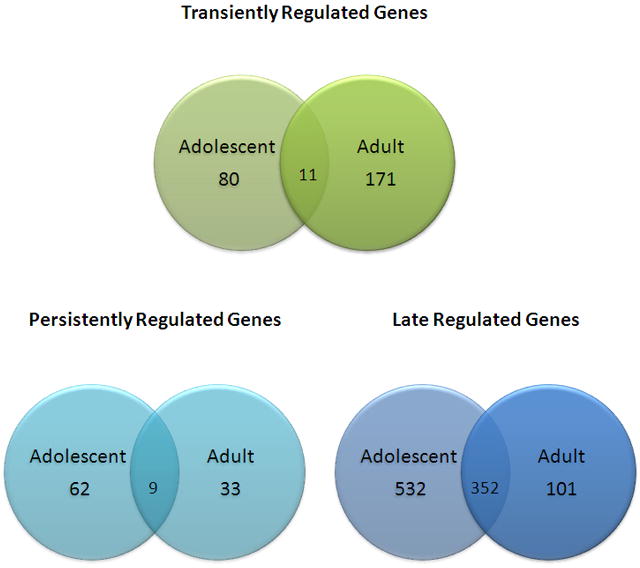
Venn diagram of template-matched genes significantly regulated by chronic nicotine treatment in adolescent and adult rats.
Adolescent- and adult-specific genes (transient, persistent and late response) were further analyzed for biological significance. Ingenuity Pathway Analysis (IPA) was employed for the identification of top ranked biological functions, significant networks, and canonical pathways associated with these age-specific genes. Figure 3 shows high level biological functions identified by IPA that were significantly overrepresented in our adolescent-specific, adult-specific, and shared data sets, including genetic disorders, psychological disorders, neurological disease and behavior. Further analysis focused on genes that contribute to nervous system development and function. This category consisted of 42 adolescent-specific, 14 adult-specific, and 23 shared genes involved in a number of biological processes (Table 2). Figure 4 depicts an interaction network of adolescent- (Fig. 4A) and adult-specific genes (Fig. 4B) involved in nervous system development and function. The adolescent-specific genes form an extensive interaction network in contrast to adult-specific genes.
Figure 3.

Over-represented biological functions with possible biological relevance to chronic nicotine treatment. These data were generated in Ingenuity Pathways Analysis and significance is expressed as the inverse log of the p-value.
Table 2.
Number and function of genes identified by Ingenuity Pathways Analysis involved in nervous system development and function in adolescent and adult animals including age-specific and shared genes.
| Function | Adolescent | Adult | ||
|---|---|---|---|---|
| P-Value | # of Genes | P-Value | # of Genes | |
| Development of brain | 1.33E-02 | 16 | – | – |
| Development of forebrain | 1.95E-02 | 8 | – | – |
| Development of neurites | 2.65E-03 | 18 | – | – |
| Growth of neurites | 7.78E-03 | 24 | – | – |
| Learning of mice | 4.23E-02 | 5 | 1.10E-02 | 5 |
| Length of dendrites | 2.00E-02 | 3 | – | – |
| Length of neurites | 7.54E-03 | 6 | – | – |
| Long term depression | 9.52E-03 | 6 | – | – |
| Morphogenesis of dendrites | 2.72E-02 | 6 | – | – |
| Morphogenesis of neurites | 4.89E-03 | 11 | – | – |
| Neurological process of axons | 3.03E-02 | 11 | – | – |
| Neurological process of cerebral cortex | 4.23E-02 | 5 | – | – |
| Neurological process of mice | 2.59E-02 | 19 | – | – |
| Outgrowth of neurites | 1.38E-02 | 21 | – | – |
| Quantity of presynaptic terminals | – | – | 1.06E-04 | 3 |
Figure 4.
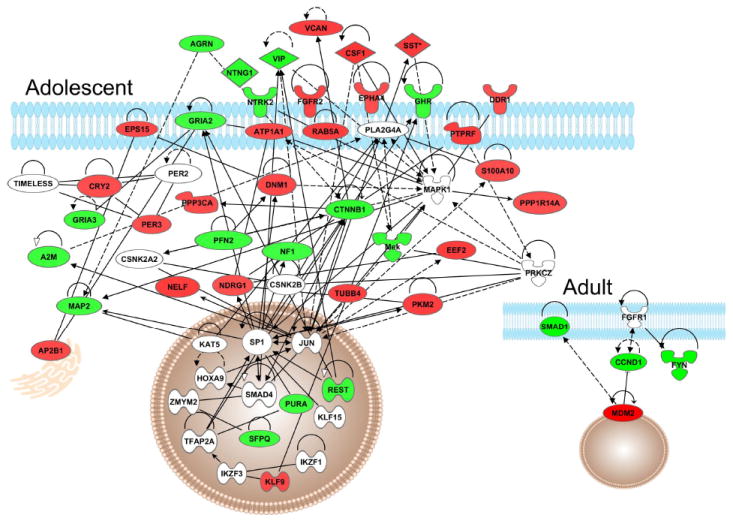
Interaction network of genes significant for regulation by chronic nicotine falling within Nervous System Development and Function, showing Adolescent network (main figure) and corresponding Adult network (inset).
Figure 5 depicts the canonical pathways significantly over-represented by adolescent-specific, adult-specific or shared genes regulated by chronic nicotine treatment. Five of the seven canonical pathways shown are unique to either adolescents or adult animals. Of note is the synaptic long-term potentiation (LTP) canonical pathway, where 8 adolescent-specific genes were identified as part of the LTP canonical pathway (Fig. 6). By comparison, the LTP canonical pathway was not significantly overrepresented by adult-specific genes. In fact, no adult-specific genes were placed in this pathway, and two genes were shared between the age groups (not significant).
Figure 5.

Canonical pathways regulated by chronic nicotine. These data were generated in IPA and are expressed as the inverse log of the p-value.
Figure 6.

Synaptic long term potentiation canonical pathway in adolescent rats treated with chronic nicotine. Genes shown in red are positively correlated and genes shown in green are negatively correlated.
Shared genes (transient, persistent, and late response) analyzed for biological significance with IPA were similarly over-represented in the same biological functions first seen in the adolescent- and adult-specific data sets. High level biological functions identified as overrepresented by the shared genes (Fig. 3) include nervous system development and function, cell morphology, cellular development, genetic disorder, neurological disease and psychological disorders. On the other hand, two overrepresented canonical pathways identified by IPA were unique to the shared data set. Top canonical pathways identified as overrepresented by the shared genes include semaphorin signaling in neurons, shown to function in synaptogenesis, axon pruning, and the density and maturation of dendritic spines (Pasterkamp and Giger, 2009), and ephrin receptor signaling, shown to function in dendritic spine morphology (Lai and Ip, 2009) (Fig. 5).
Quantitative real-time RT-PCR was performed to validate microarray results of 12 adolescent-specific genes differentially regulated by nicotine (1 transient, 1 persistent and 10 late response genes; see Fig. 7). These include four genes in the LTP canonical pathway and eight genes involved in nervous system development and function. All twelve genes were validated by RT-PCR as significant by ANOVA and post-hoc Tukey test (P < 0.05).
Figure 7.

Quantitative real time RT-PCR validation of microarray data. Comparison of individual template matched genes measured by microarray analysis (white bars) to real time RT-PCR (black bars). Expression values for adolescents treated with saline (Sal), nicotine (Nic), or nicotine followed by 30 days withdrawal (NicWD) were compared by one-way ANOVA followed by Tukey’s post-hoc test. *Significantly different from Saline; +significantly different from NicWD at P<0.05. Data are represented as the mean ± S.E.M. of 3-5 independent determinations.
DISCUSSION
A number of studies have been conducted examining the gene expression profiles of animals exposed to drugs of abuse in an attempt to identify the genetic pathways involved in the etiology of addiction. Thus far, few studies have examined the differences in the response of adolescent and adult animals. Epidemiological data clearly suggest that adolescents exposed to drugs of abuse have unique responses which persist into adulthood. We have conducted whole genome mRNA analysis of the VTA from adolescent and adult SD rats treated with chronic nicotine and find that adolescent rats also show unique genomic responses that persist into adulthood.
A two-way ANOVA identifying age specific genes revealed that adolescent rats have a strikingly distinct response to nicotine’s genetic regulatory effects relative to adults. Adolescent animals had only half as many age-specific genes affected transiently, whereas they had twice as many age-specific genes regulated persistently, and five times as many showing delayed regulation relative to adult animals. This suggests the developing adolescent brain may be significantly more sensitive to long-lasting effects of nicotine exposure at the transcriptional level. This sensitivity may relate to documented sensitivities of adolescents to the addictive effects of nicotine (and other drugs of abuse). Taken together, this suggests the age of onset of tobacco use significantly affects the response to exposure. Further, chronic nicotine exposure causes downstream effects that are seen long after cessation of treatment. This may cause changes in the developing adolescent brain which persist into adulthood and augment response to later drug exposure.
Over-represented classes of genes specific to adolescent chronic nicotine exposure included those associated with genetic and psychological disorders, developmental disorders, and nervous system development and function. Network analysis of genes from the latter category reveals that these adolescent-specific nicotine-regulated genes may form extensive networks in adolescent VTA, but that is not the case for adult-specific genes in the VTA. Also, a number of genes unique to adolescents were found to regulate neurite structure. Groups of genes were identified that regulate development of neurites (11 genes), neurite morphogenesis (7 genes), growth of neurites (15 genes), length (4 genes) and branching of neurites (5 genes). These genes are of particular interest because of their possible roles in structural and behavioral plasticity. Pathway analysis showed several canonical pathways over-represented by adolescent-specific genes in adolescent VTA, notably long-term potentiation, circadian rhythm signaling and CDK5 signaling. Animal studies have found that the circadian genes are important regulators of drug responsiveness, sensitization, and reward (Falcón and McClung, 2009). CDK5 has been shown to modulate dopamine signaling in neurons (Bibb et al., 1999). Pathways over-represented by adult-specific genes included protein ubiquitination, PI3K/AKT signaling and HIF1α signaling. PI3K/AKT signaling is activated by other drugs of abuse, and is known to function in neuronal branching, dendritic spine density, and cell body size (Russo et al., 2009).
Nervous System Development and Function
Drugs of abuse are known to produce widespread effects on neuronal structure and function. This is hypothesized to be the basis for the long lasting changes in behavior seen in addiction. Long-term changes that occur within the brain’s reward circuitry are central to reward related behaviors and drug response. In particular, the changes in dopaminergic neurons of the VTA and nucleus accumbens (Acb) are thought to alter reward responses leading to tolerance, increased intake, reward dysfunction, and addiction (Russo et al., 2009).
Changes in spine morphology are critical for synaptic function (Sorra and Harris, 2000; Hering and Sheng, 2001; Yuste and Bonhoeffer, 2001). For instance, larger spines support stronger synaptic transmission (El-Husseini et al., 2000; Murthy et al., 2001). Spine morphology also plays an important role in synaptic plasticity. The autonomous space created by the spine neck facilitates activity-triggered calcium increases (Wang et al., 2000; Sabatini et al., 2002), which activates calcium-activated kinases and phosphatases that underlies synaptic plasticity (Morishita et al., 2001; Thiels and Klann, 2001; Lisman et al., 2002). Chronic administration of cocaine and amphetamine produce increased spine density in the Acb (Robinson and Kolb, 1997, 1999a). Similarly, chronic nicotine exposure increases spine density within the Acb and medial prefrontal cortex (Brown and Kolb, 2001; Robinson and Kolb, 2004). Opioids have the opposite effect: morphine decreases spine density and dendritic branching in the accumbens shell and medial prefrontal cortex (Robinson and Kolb, 1999b; Robinson et al., 2002; Robinson and Kolb, 2004). These structural changes are seen long after cessation of drug treatment, up to 3.5 months (Kolb et al., 2003; Robinson and Kolb, 2004).
Circadian Rhythms
Drosophila studies first showed that mutations in the circadian rhythm genes Per, Clock, Cycle, or Doubletime had an effect (fail to sensitize after repeated exposure) on cocaine response (Andretic et al., 1999). Several subsequent studies have shown that cocaine can induce or repress the expression of circadian rhythm genes in several brain regions (Yuferov et al., 2003; McClung and Nestler, 2003; Uz et al., 2005). Mice lacking a functional mPer1 gene were demonstrated to have no CPP response, and failed to sensitize to cocaine (Abarca et al., 2002). Mice with a mutant Clock gene, however, showed robust cocaine sensitization, increased cocaine preference, and increased reward as measured by intracranial self-stimulation following cocaine treatment (McClung et al., 2005; Roybal et al., 2007). A recent microarray study identified 29 circadian rhythm genes differentially regulated in animals self-administering cocaine (Lynch et al., 2008). Studies have also suggested that circadian rhythm genes play a role in alcohol (Spanagel et al., 2005) and morphine (Liu et al., 2005) response. These data and our own suggest that drugs of abuse alter expression of circadian rhythm genes. Other studies show altering circadian rhythm gene expression alters drug sensitivity and reward. This suggests a direct role for circadian rhythm genes in substance abuse. Further study of this area may prove useful in understanding the differential response to drugs of abuse, and identifying therapeutic targets for the treatment of various forms of drug addiction.
Long Term Potentiation (LTP)
There is a large body of data supporting the involvement of LTP in drug addiction. In fact, addiction has been described as “a pathological usurpation of learning and memory” (Hyman, 2005). Behavioral sensitization, conditioned locomotor response, and CPP are drug-associated phenomena that are context- and NMDA-dependent, suggesting the involvement of LTP (Hyman and Malenka, 2001). Evidence suggests that drug-induced synaptic plasticity is mediated by a process similar to long term potentiation (Ungless et al., 2001; Saal et al., 2003). A single dose of a drug may be enough to change the AMPA/NMDA receptor balance that blocks LTP induction (Ungless et al., 2001; Saal et al., 2003). Psychostimulants may facilitate synaptic potentiation by interfering with long term depression (LTD) in the same cells (Jones et al., 2000; Gutlerner et al., 2002). Neuronal nicotinic receptors are known to play a role in cognition; recent studies have detailed the effects of nicotine on learning and LTP (Tang and Dani, 2009).
Synaptic plasticity in the VTA has been implicated in the acquisition of nicotine dependence (Dani et al., 2001). It has been demonstrated that a single dose of nicotine is sufficient to induce synaptic plasticity in VTA DA neurons (Gray et al., 1996; Mansvelder et al., 2000; Saal et al., 2003; Pidoplichko et al., 2004). These synaptic alterations correlate with behavioral sensitization (Borgland et al., 2004) and self-administration (Chen et al., 2008) in animal studies. Prolonged nicotine exposure causes desensitization of β2-containing nicotinic receptors, reducing GABAergic inhibitory tone and increasing DA neuron excitability and the probability of glutamatergic LTP (Dani et al., 2001).
While epidemiological data clearly show that adolescents are more vulnerable to drugs of abuse, and a number of studies have examined the effects of addictive drugs in adolescent rodent models, little is known about the developmental factors affecting changes in plasticity caused by nicotine. The hippocampus is vital for encoding and recalling episodic memory (Squire et al., 2004). It is now clear that DA affects LTP, and that the hippocampus receives significant DA innervation from the VTA (Gasbarri et al., 1997). DA cells increase burst firing in response to unexpected reward and reduce burst firing when expected reward is omitted (Schultz and Dickinson, 2000). Both conscious and unconscious memory mechanisms contribute to the rewarding effects of nicotine and other drugs of abuse (Kauer and Malenka, 2007; Kelley, 2004; Dani and Montague, 2007). Nicotine increases neuronal excitability and strengthens synapses in several brain regions (Kauer and Malenka, 2007; Mansvelder and McGehee, 2002). Recent evidence suggests rather than directly encoding reward, DA signals contribute to associative learning in response to behaviorally important cues (Schultz, 2007). Nicotine-induced DA signaling in hippocampus has been shown to be necessary for LTP induction and nicotine reinforcement as measured by CPP (Tang and Dani, 2009).
Exposure to nicotine and other drugs of abuse induces long term changes similar to LTP in the motivational/reward pathway (Jones and Bonci, 2005; Kauer and Malenka, 2007). Identifying the mechanism by which nicotine induces conscious and unconscious memory is important for understanding addiction. Further, the differential response to chronic nicotine exposure seen between our adolescent and adult animals may play a role in smoking acquisition and the disparate outcomes observed between adolescent and adult smokers.
We have demonstrated striking differences in gene expression response of adolescent and adult SD rats to chronic nicotine exposure. VTA gene expression profiles reveal a response pattern suggesting adolescents are more severely affected in the long term relative to adult animals. Further, chronic nicotine was shown to affect genes associated with neuronal structure and function, LTP, circadian rhythm, and a number of psychological disorder related pathways more robustly in our adolescent animals. Analysis of the significant genes shared between age groups revealed convergence in some pathways (nervous system and development, circadian rhythms) relative to age specific genes. These effects were much more robust, however, in our adolescent animals. Our data suggest that adolescent rats have a greater sensitivity to chronic nicotine’s effects and show a prolonged response in a number of genetic pathways lasting well into adulthood. Further study of these areas may provide promising targets for the treatment of drug abuse.
Acknowledgments
This work was supported by NIH grants DA015767 (DCP) and DA015087 (NHL), and is in part from a dissertation by Mr. Doura to be presented to the Pharmacology Program of the George Washington University Institute for Biomedical Sciences in partial fulfillment of the requirements for the PhD degree.
Abbreviations
- Acb
nucleus accumbens
- AMPA
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
- CPP
conditioned place preference
- DA
dopamine
- FDR
false discovery rate
- GABA
gamma amino butyric acid
- GCOS
Gene Chip Operating Software
- IPA
Ingenuity Pathway Analysis
- LTP
long-term potentiation
- NMDA
N-methyl D-aspartic acid
- PN
post-natal day
- RT-PCR
reverse transcriptase polymerase chain reaction
- SD
Sprague-Dawley
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abarca C, Albrecht U, Spanagel R. Cocaine sensitization and reward are under the influence of circadian genes and rhythm. Proc Natl Acad Sci U S A. 2002;99:9026–9030. doi: 10.1073/pnas.142039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adriani W, Deroche-Gamonet V, Le MM, Laviola G, Piazza PV. Preexposure during or following adolescence differently affects nicotine-rewarding properties in adult rats. Psychopharmacology (Berl) 2006;184:382–390. doi: 10.1007/s00213-005-0125-1. [DOI] [PubMed] [Google Scholar]
- 3.Andretic R, Chaney S, Hirsh J. Requirement of circadian genes for cocaine sensitization in Drosophila. Science. 1999;285:1066–1068. doi: 10.1126/science.285.5430.1066. [DOI] [PubMed] [Google Scholar]
- 4.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signaling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 5.Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown RW, Kolb B. Nicotine sensitization increases dendritic length and spine density in the nucleus accumbens and cingulate cortex. Brain Res. 2001;899:94–100. doi: 10.1016/s0006-8993(01)02201-6. [DOI] [PubMed] [Google Scholar]
- 7.Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry. 2003;160:1041–1052. doi: 10.1176/appi.ajp.160.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Millar WJ. Age of smoking initiation: implications for quitting. Health Rep. 1998;9:39–46. [PubMed] [Google Scholar]
- 10.Collins SL, Izenwasser S. Chronic nicotine differentially alters cocaine-induced locomotor activity in adolescent vs. adult male and female rats. Neuropharmacology. 2004;46:349–362. doi: 10.1016/j.neuropharm.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 11.Crews F, He J, Hodge C. Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav. 2007;86:189–199. doi: 10.1016/j.pbb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–352. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- 13.Dani JA, Montague PR. Disrupting addiction through the loss of drug-associated internal states. Nat Neurosci. 2007;10:403–404. doi: 10.1038/nn0407-403. [DOI] [PubMed] [Google Scholar]
- 14.Doura MB, Gold AB, Keller AB, Perry DC. Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res. 2008;1215:40–52. doi: 10.1016/j.brainres.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- 16.Falcon E, McClung CA. A role for the circadian genes in drug addiction. Neuropharmacology. 2009;56(Suppl 1):91–96. doi: 10.1016/j.neuropharm.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasbarri A, Sulli A, Packard MG. The dopaminergic mesencephalic projections to the hippocampal formation in the rat. Prog Neuropsychopharmacol Biol Psychiatry. 1997;21:1–22. doi: 10.1016/s0278-5846(96)00157-1. [DOI] [PubMed] [Google Scholar]
- 18.Gold AB, Keller AB, Perry DC. Prenatal exposure of rats to nicotine causes persistent alterations of nicotinic cholinergic receptors. Brain Res. 2009;1250:88–100. doi: 10.1016/j.brainres.2008.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 20.Gutlerner JL, Penick EC, Snyder EM, Kauer JA. Novel protein kinase A-dependent long-term depression of excitatory synapses. Neuron. 2002;36:921–931. doi: 10.1016/s0896-6273(02)01051-6. [DOI] [PubMed] [Google Scholar]
- 21.Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- 22.Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci. 2001;2:695–703. doi: 10.1038/35094560. [DOI] [PubMed] [Google Scholar]
- 23.Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- 24.Joe B, Letwin NE, Garrett MR, Dhindaw S, Frank B, Sultana R, Verratti K, Rapp JP, Lee NH. Transcriptional profiling with a blood pressure QTL interval-specific oligonucleotide array. Physiol Genomics. 2005;23:318–326. doi: 10.1152/physiolgenomics.00164.2004. [DOI] [PubMed] [Google Scholar]
- 25.Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci. 2000;20:5575–5580. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones S, Bonci A. Synaptic plasticity and drug addiction. Curr Opin Pharmacol. 2005;5:20–25. doi: 10.1016/j.coph.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 28.Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 29.Kolb B, Gorny G, Li Y, Samaha AN, Robinson TE. Amphetamine or cocaine limits the ability of later experience to promote structural plasticity in the neocortex and nucleus accumbens. Proc Natl Acad Sci U S A. 2003;100:10523–10528. doi: 10.1073/pnas.1834271100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai KO, Ip NY. Synapse development and plasticity: roles of ephrin/Eph receptor signaling. Curr Opin Neurobiol. 2009;19:275–283. doi: 10.1016/j.conb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Wang Y, Wan C, Zhou W, Peng T, Liu Y, Wang Z, Li G, Cornelisson G, Halberg F. The role of mPer1 in morphine dependence in mice. Neuroscience. 2005;130:383–388. doi: 10.1016/j.neuroscience.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lujan R, Shigemoto R, Lopez-Bendito G. Glutamate and GABA receptor signaling in the developing brain. Neuroscience. 2005;130:567–580. doi: 10.1016/j.neuroscience.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 34.Lynch WJ, Girgenti MJ, Breslin FJ, Newton SS, Taylor JR. Gene profiling the response to repeated cocaine self-administration in dorsal striatum: a focus on circadian genes. Brain Res. 2008;1213:166–177. doi: 10.1016/j.brainres.2008.02.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 36.Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- 37.McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- 38.McClung CA, Sidiropoulou K, Vitaterna M, Takahashi JS, White FJ, Cooper DC, Nestler EJ. Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc Natl Acad Sci U S A. 2005;102:9377–9381. doi: 10.1073/pnas.0503584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McMillen BA, Davis BJ, Williams HL, Soderstrom K. Periadolescent nicotine exposure causes heterologous sensitization to cocaine reinforcement. Eur J Pharmacol. 2005;509:161–164. doi: 10.1016/j.ejphar.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 40.McQuown SC, Belluzzi JD, Leslie FM. Low dose nicotine treatment during early adolescence increases subsequent cocaine reward. Neurotoxicol Teratol. 2007;29:66–73. doi: 10.1016/j.ntt.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC. Regulation of synaptic strength by protein phosphatase 1. Neuron. 2001;32:1133–1148. doi: 10.1016/s0896-6273(01)00554-2. [DOI] [PubMed] [Google Scholar]
- 42.Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- 43.Pasterkamp RJ, Giger RJ. Semaphorin function in neural plasticity and disease. Curr Opin Neurobiol. 2009;19:263–274. doi: 10.1016/j.conb.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavlidis P, Noble WS. Analysis of strain and regional variation in gene expression in mouse brain. Genome Biol. 2001;2:RESEARCH0042. doi: 10.1186/gb-2001-2-10-research0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Amsterdam: Academic Press/Elsevier; 2007. [Google Scholar]
- 46.Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, Dani JA. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem. 2004;11:60–69. doi: 10.1101/lm.70004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 1997;17:8491–8497. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- 49.Robinson TE, Kolb B. Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse. 1999;33:160–162. doi: 10.1002/(SICI)1098-2396(199908)33:2<160::AID-SYN6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 50.Robinson TE, Gorny G, Savage VR, Kolb B. Widespread but regionally specific effects of experimenter- versus self-administered morphine on dendritic spines in the nucleus accumbens, hippocampus, and neocortex of adult rats. Synapse. 2002;46:271–279. doi: 10.1002/syn.10146. [DOI] [PubMed] [Google Scholar]
- 51.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 52.Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, Chakravarty S, Peevey J, Oehrlein N, Birnbaum S, Vitaterna MH, Orsulak P, Takahashi JS, Nestler EJ, Carlezon WA, Jr, McClung CA. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci U S A. 2007;104:6406–6411. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56(Suppl 1):73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 55.Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- 56.Schultz W, Dickinson A. Neuronal coding of prediction errors. Annu Rev Neurosci. 2000;23:473–500. doi: 10.1146/annurev.neuro.23.1.473. [DOI] [PubMed] [Google Scholar]
- 57.Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 58.Sorra KE, Harris KM. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus. 2000;10:501–511. doi: 10.1002/1098-1063(2000)10:5<501::AID-HIPO1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 59.Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human life span. Nat Neurosci. 2003;6:309–315. doi: 10.1038/nn1008. [DOI] [PubMed] [Google Scholar]
- 60.Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med. 2005;11:35–42. doi: 10.1038/nm1163. [DOI] [PubMed] [Google Scholar]
- 61.Spear LP. Adolescent brain development and animal models. Ann N Y Acad Sci. 2004;1021:23–26. doi: 10.1196/annals.1308.002. [DOI] [PubMed] [Google Scholar]
- 62.Squire LR. Memory systems of the brain: a brief history and current perspective. Neurobiol Learn Mem. 2004;82:171–177. doi: 10.1016/j.nlm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 63.Suarez AP, Staffa JA, Fletcher P, Jones JK. Reason for discontinuation of newly prescribed antihypertensive medications: methods of a pilot study using computerized patient records. Pharmacoepidemiol Drug Saf. 2000;9:405–416. doi: 10.1002/1099-1557(200009/10)9:5<405::AID-PDS521>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 64.Tang J, Dani JA. Dopamine enables in vivo synaptic plasticity associated with the addictive drug nicotine. Neuron. 2009;63:673–682. doi: 10.1016/j.neuron.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thiels E, Klann E. Extracellular signal-regulated kinase, synaptic plasticity, and memory. Rev Neurosci. 2001;12:327–345. doi: 10.1515/revneuro.2001.12.4.327. [DOI] [PubMed] [Google Scholar]
- 66.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 67.Uz T, Ahmed R, Akhisaroglu M, Kurtuncu M, Imbesi M, Dirim AA, Manev H. Effect of fluoxetine and cocaine on the expression of clock genes in the mouse hippocampus and striatum. Neuroscience. 2005;134:1309–1316. doi: 10.1016/j.neuroscience.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 68.Wang SS, Denk W, Hausser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- 69.Yuferov V, Kroslak T, LaForge KS, Zhou Y, Ho A, Kreek MJ. Differential gene expression in the rat caudate putamen after “binge” cocaine administration: advantage of triplicate microarray analysis. Synapse. 2003;48:157–169. doi: 10.1002/syn.10198. [DOI] [PubMed] [Google Scholar]
- 70.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]