

Abstract
The representational difference analysis (RDA) and other subtraction techniques are used to enrich sample-specific sequences by elimination of ubiquitous sequences existing in both the sample of interest (tester) and the subtraction partner (driver). While applying the RDA to genomic DNA of cutaneous lymphoma cells in order to identify tumor relevant alterations, we predominantly isolated repetitive sequences and artificial repeat-mediated fusion products of otherwise independent PCR fragments (PCR hybrids). Since these products severely interfered with the isolation of tester-specific fragments, we developed a considerably more robust and efficient approach, termed ligation-mediated subtraction (Limes). In first applications of Limes, genomic sequences and/or transcripts of genes involved in the regulation of transcription, such as transforming growth factor β stimulated clone 22 related gene (TSC-22R), cell death and cytokine production (caspase-1) or antigen presentation (HLA class II sequences), were found to be completely absent in a cutaneous lymphoma line. On the assumption that mutations in tumor-relevant genes can affect their transcription pattern, a protocol was developed and successfully applied that allows the identification of such sequences. Due to these results, Limes may substitute/supplement other subtraction/comparison techniques such as RDA or DNA microarray techniques in a variety of different research fields.
INTRODUCTION
The analysis of variations or mutations in complex mammalian genomes is one of the major challenges for modern molecular biology. To approach this task, sample-specific sequences are identified by comparing one million or more individual ‘PCR-adjusted’ fragments from one source (tester) to appropriate counterpart DNA (driver). For example, viral sequences in the tester DNA or deletions in the driver DNA (∼10 000–200 000 bp) will generate only a few hundred or less tester-specific sequences among these millions of ubiquitous fragments.
As it was shown for the so-called differential display technique (1), tester-specific sequences can be isolated by ‘simple’ comparison of tester and driver DNA. However, only a limited number of several hundred fragments can be investigated in one differential display experiment and, accordingly, every effort was made to enhance this number. As a result, newly developed DNA microarray techniques allow immobilization of up to tens of thousands of distinct sequence clones on small solid matrices. Replicas of these sequences are hybridized with fluorescent-labeled tester and driver cDNA and software-supported confocal laser scanning microscope analysis of the hybrids displays quantitative differences between tester and driver. Different research groups have successfully applied such a technique and presented impressive patterns of gene expression revealing, for example, different entities of B-cell lymphoma with otherwise indistinguishable phenotypes (2). Due to the enormous complexity of mammalian genomic DNA, however, differential display and DNA microarray techniques are primarily used for monitoring differences in gene expression.
A first and elegant approach for the isolation of genomic alterations was the representational difference analysis (RDA) (3), which evolved from the subtractive hybridization technique (4). Subtractive hybridization stands for (physical) separation of tester-specific sequences from ubiquitous and/or driver-specific DNA after hybridization of tester to an excess of driver. The key idea of the RDA approach was to replace the physical separation by a PCR-aided enrichment of tester-specific fragments. In our hands, however, a successful application of RDA was hampered by the preferential amplification of repetitive sequences and by generation of artificial fusion sequences.
We came across these technical limitations as we attempted to isolate tumor-specific alterations in cutaneous lymphoma. Primary cutaneous lymphoma represent a heterogeneous group of T-cell and B-cell lymphoma (CTCL and CBCL, respectively), have a largely unknown etiology (5) and exhibit considerable variation in clinical presentation, histology, immunophenotype as well as prognosis. Retroviral (6), as well as herpes virus sequences (7 and references therein), were reported to be causative agents for cutaneous lymphoma. This is, however, under intense debate because other studies were not able to substantiate these observations (8,9). In addition, attempts to assign known tumor-associated genes to cutaneous lymphoma failed because no ‘good’ candidate gene could be detected so far.
In order to identify and isolate tumor-specifically mutated and/or expressed sequences, we developed and applied a novel technique, termed ligation-mediated subtraction (Limes) because a ligation step with a thermostable ligase was a crucial step for the successful application of our protocol presented in this report.
MATERIALS AND METHODS
Cell lines
A detailed description of the murine lymphoma line can be found at http://www.cabri.org/CABRI/cabri-srs-bin/wgetz?-id+BaV71Fjouq+e+[ECACC_CELL–Accession_number: ’ECACC_SP_8502310 5’]. The establishment of the tumor cell line My-La from a plaque biopsy specimen of a patient with mycosis fungoides as well as karyotypic changes of My-La during long-term culture has been described previously (10). The cytotoxic T cell line, named CTL here, recognizes epitopes presented by autologous My-La cells (11).
Generation of PCR libraries
A detailed protocol for the Limes approach is available at http://www.charite.d e/ ch/derm/Limes. Genomic DNA was prepared as described elsewhere (12). For cDNA preparation, the cells were dissolved in Trizol and total RNA was isolated as recommended by the manufacturer (Life Technologies, Karlsruhe, Germany). Total RNA (1 µg) was transcribed into cDNA and amplified by applying the SMART™ cDNA Synthesis kit (Clontech, Heidelberg, Germany).
Genomic DNA or amplified cDNA was digested with TspRI (New England Biolabs, Schwalbach, Germany). The digested tester DNA (1 µg) was incubated with 300 pmol of oligonucleotides N1 (5′-gga aca ccc tat gaa cta gtg nnc a(c/g)t gnn-3′), 5′-phosphorylated A1 (5′-cac tag ttc ata ggg tgt tcc-3′) and 1× Taq ligase buffer (New England Biolabs) at 55°C for 1 min. The mixture was gradually cooled to 45°C (temperature gradient: –0.5°C/min) and incubated overnight at 45°C with 40 U Taq DNA ligase (New England Biolabs).
For the preparation of tester PCR libraries, ligation products were purified with the PCR Purification kit (Qiagen, Hilden, Germany) and aliquots of each DNA sample were amplified in a PTC 200 MultiCycler (MJ Research, Waltham, MA) as follows: 20 ng DNA was added to 150 pmol S1 (5′-gga aca ccc tat gaa cta gtg-3′), 1× Taq polymerase buffer, 200 µM of each deoxynucleotide triphosphate (dNTPs) and 2.5 U Taq DNA polymerase (buffer and enzyme: Roche Diagnostics, Mannheim, Germany) in a final volume of 100 µl, denatured for 45 s at 92°C, annealed for 90 s at 57°C and extended for 120 s at 72°C. Before the first of 25 cycles, template DNA was denatured for 3 min at 94°C and after the last cycle, the reaction mixture was incubated for 15 min at 72°C.
For preparation of driver DNA, oligonucleotides N2 (5′-tca agt gtt caa gtc gac ccn nca (g/c)tg nn-3′; the SalI-site is underlined) and 5′-phosphorylated A2 (5′−ggg-tcg-act-tga-aca-ctt-ga-3′; the SalI site is underlined) were ligated to the human DNA and amplified by using the primer S2 (5′-tca agt gtt caa gtc gac cc-3′; the SalI site is underlined) as described above.
Subtractive hybridization
Driver DNA was digested with SalI and purified with the PCR Purification kit (Qiagen). The tester hybridization partner was prepared by TspRI digestion of 2 µg of the tester PCR library, purification of the digested DNA with the PCR Purification kit (Qiagen) and ligation to 600 pmol oligonucleotide N3 (5′-ctg aat gat aac gga ccg agn nca (g/c)tg nn) as described above. Genomic DNA and cDNA (1 and 0.1 µg, respectively) copies of the purified tester were added to 40 µg of purified, SalI-digested driver, precipitated and washed with 70% ethanol (v/v). The dry precipitate was dissolved in 4 µl of N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid) (EPPS) buffer (30 mM EPPS pH 8.0, 3 mM EDTA). After incubation at 98°C for 4 min, 1 µl of 5 M NaCl was added to the dissolved DNA at 90°C and the solution was incubated for 12 h at 77°C, an additional 12 h at 72°C and, finally, for at least 36 h at 68°C. The DNA was precipitated, washed and dried.
Ligation-mediated enrichment of tester-specific sequences
The hybrids were ligated to 300 pmol of 5′-phosphorylated and 3′-biotinylated A3 (5′-ctc ggt ccg tta tca ttc ag-3′) by incubation in 1× Taq ligase buffer at 65°C for 1 min, gradual decrease of the temperature to 55°C (gradient: –0.5°C/min), addition of 40 U Taq DNA ligase and incubation for 4 h at 55°C. The ligation products were purified (Qiagen PCR Purification kit) and incubated with 1 mg of suspended streptavidin-coupled beads (Dynal, Hamburg, Germany) as the manufacturers have recommended. The beads were washed at room temperature or indicated temperature with 50 µl of any one of the following solutions: once with B&W buffer (Dynal), 1× TE (10 mM Tris–HCl pH 7.5, 0.1 mM EDTA), 1× H2O at 55°C for 10 min, 2× H2O, 1× 0.1 M NaOH for 7 min, 2× 0.1 M NaOH, 1× B&W buffer and, finally, 1× TE. Beads (10 µl) resuspended in 20 µl H2O were amplified as described except for the following modifications: the primer was S3 (5′-ctg aat gat aac gga ccg ag-3′), 1.5 U Taq DNA polymerase (Roche) were added and 35 PCR cycles were performed.
Subsequent rounds of enrichment
Enriched tester sequences were digested with TspRI, purified and ligated to N4 (5′-tcg tgg agc aat tta cta gtc nnc a(g/c)t gnn-3′). Genomic DNA and cDNA (200 and 20 ng, respectively) copies of the enriched tester were added to 40 µg of SalI-digested driver. Hybridization and subsequent enrichment were performed as described above. In additional rounds (genomic tester only) the amount of enriched tester was reduced to 40 ng and, subsequently, to 10 ng. The following N-oligonucleotides were used: N5 (5′-tcg aaa tga atc ggt ccg tcn nca (g/c)tg nn-3′) and N6 (5′-atc atc aca ttg gtc gac ggn nca (g/c)tg nn-3′). The sequences of A-oligonucleotides and S-primer were deduced from respective N-oligonucleotides.
Isolation of tester-specific sequences present in both enriched cDNA and genomic tester
Two-fold enriched tester cDNA (40 µg) was added to 500 ng of a 5-fold enriched, 5′-biotinylated genomic tester, which was prepared by amplification with a biotinylated S-primer (see above) and purified with the Qiagen PCR Purification kit. Precipitation, denaturation and hybridization were done as described above. The DNA was bound to 1 mg of streptavidin-coupled beads as recommended by the manufacturer (Dynal) and the washes were performed with 50 µl of any one of the following solutions at room temperature or indicated temperature: once with B&W buffer, 1× TE, 1× H2O at 55°C for 10 min, 2× H2O, 1× H2O at 55°C for 10 min and, finally, 2× H2O. Beads (10 µl) resuspended in 20 µl H2O were used for a 35-cycle amplification with cDNA-flanking S-primer as described above.
Screening for tester-specific clones
Series of 10-fold dilutions were prepared from HindIII- or TspRI-digested mixtures of bacteriophage λ DNA with mammalian genomic DNA. Dilution steps ranging from 100 ng to 1 pg DNA were amplified with primer Lam-5 (5′-tag agc gat tta tct tct gaa c-3′) and Lam-3 (5′-cgg cgc ata gct gat aac-3′) as described above except for the following modifications: the total number of cycles was 25 and the annealing temperature was 55°C.
Enriched tester DNA was ligated to oligonucleotides N5 and A5 (see above) and amplified with S5 as described. SalI-digested S5-amplification products were ligated into SalI-digested, dephosphorylated pTZ19R plasmids. TOP10 bacteria were transformed with the ligation products as recommended by the manufacturers (Invitrogen, Groningen, The Netherlands) and bacteria from blue/white-selected single colonies were transferred into 100 µl reaction mixtures containing 1× AmpliTaq PCR buffer II, 2 mM MgCl2, 30 pmol F-primer (5′-agg cga tta agt tgg gta ac-3′), 30 pmol R-primer (5′-cag cta tga cca tga tta cg-3′), 200 µM of each dNTP and 1 U AmpliTaq (Perkin Elmer, Weiterstadt, Germany). Thirty-five PCR cycles were performed as described above (‘Miniprep-PCR’). The PCR products derived from the CTL/My-La experiments (2 µl), were spotted on Nytran membranes (Schleicher & Schuell, Dassel, Germany). Denaturation, UV crosslinking and hybridization were performed as recommended by the manufacturer of the DIG High Prime DNA Labeling and Detection kit (Roche). Positively selected fragments as well as arbitrarily chosen λ-Miniprep-PCR clones served as templates for labeling reactions with the DNA Sequencing Kit (Applied Biosystems, Weiterstadt, Germany) by extension of primer SeqF (5′-ttg taa aac gac ggc cag tg-3′) or SeqR (5′-aat acg act cac tat agg gaa ag-3′). The sequence analysis was performed on an ABI Prism 373A and the obtained data were compared to the GenBank, Est and Htgs databases with the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/blast.cgi).
The Genetics Computer Group (GCG, Madison, WI) program package, version 10.0 was used to design pairs of primer specific for the following sequences: TSC-22R (I: 5′-tag aca aca aga tcg aac ag-3′; II: 5′-tgt gct agg tgt aaa gtt ctc-3′), TSPY (I: 5′-gaa gaa tcg tcc att tcc aga atc-3′; II: 5′-aag tct gat ggg gca aca gc-3′), TAP2 (I: 5′-ggt gaa caa caa agt ctt gat gtg-3′; II: 5′-tgt ctt agt ctc ctg gaa gaa ac-3′), HLA-DQA (I: 5′-gag gaa gga gac tgt ctg g-3′; II: 5′-agg gag gaa ggt gag gta ac-3′), HLA-DRB (I: 5′-tga tgc tgg aaa cag ttc ctc-3′; II: 5′-gtc atc tgc act tca gct c-3′), HLA-DRA (I: 5′-tcc cag aga cta cag aga ac-3′; II: 5′-ctc tct aag aaa cac cat cac-3′) and Notch4 (I: 5′-ctg tga gga gaa cct gga tg-3′; II: 5′-tga cac agg cag agt gtg g-3′). For amplification of the indicated sequences, 30 PCR cycles were performed as described above.
RESULTS
Setup and control experiments
In initial experiments, bacteriophage λ DNA was mixed to genomic DNA of the murine lymphoma cell line EL4 at a molar ratio of 1:1 and subtracted with pure EL4 driver DNA. Both driver and tester were digested with HindIII and subtracted by RDA as recommended by Lisitsyn and Wigler (13). Four successive rounds of RDA were performed. After two rounds, the pattern of enriched DNA changed from a smear to a set of about 10 discrete bands in a size range of 300–900 bp (data not shown). In order to obtain a rough estimation of the efficacy of each RDA round, dilution series of starting tester, the initial PCR library and enriched tester sequences were amplified with a pair of λ-specific primers. These primers annealed to the 564 bp λ–HindIII fragment and gave rise to a 307 bp amplification product. For amplification of the starting mixture ∼100-fold more target DNA (100 ng) was necessary to generate a visible 307 bp band than for the PCR library (1 ng) (data not shown). This reduction of complexity can be explained by the fact that most HindIII fragments are too large for being an optimal target for PCR amplification.
Another 3-fold DNA (∼300 pg) was required for obtaining a visible 307 bp band of λ-DNA after the first RDA round. During the subsequent three RDA rounds, however, the amount of required target DNA increased to 1 ng again (data not shown).
These rough estimations indicated that DNA fragments other than the 564 bp λ–HindIII were enriched during RDA. In order to determine those sequences, 4-fold enriched tester DNA was cloned and nine independent and arbitrarily chosen clones were sequenced (Table 1).
Table 1. Sequence analysis of nine arbitrarily chosen clones derived from a pool of tester sequences, which were 4-fold enriched by RDA.
| Clone | Fusion partner one | Fusion partner two | Fusing region | Hybrid |
| 2 | AC013350 | Unknown | GA-motif | Yes |
| 13 | AC013350 | AC084286 | G-rich | Yes |
| 29 | R74960 | Partially identical with clone 6 and 14 | CA-motif | Yes |
| Clone | Partial identity with | Remaining sequence | Putative fusion region | Hybrid |
| 6 | 14 and 29 | Identical with 11 | G-rich | Probable |
| 14 | 6 and 29 | Unknown | GA-motif | Probable |
| 11 | 6 | Unknown | G-rich | Probable |
| Clone | Identified sequence | Repeat | Hybrid | |
| 10 | Multiple | LINE/L1 | No | |
| 12 | No | No | ? | |
| 20 | No | No | ? |
As indicated by the respective accession number, clones 2, 13 and 29 were partly identical to already sequenced HindIII fragments of the murine genome (fusion partner one). The second moiety of each of those clones, however, represented a completely different sequence (fusion partner two). One half of clones 2 and 13, for instance, share the same sequence as indicated by the same GenBank accession number. Their second moiety did not show any similarity to sequences present in the GenBank database (clone 2) or revealed to be derived from an independent HindIII fragment (clone 13). The second part of clone 29 was also unknown. Surprisingly, this unknown sequence was identical to regions of clones 6 and 14. The remaining part of clone 6, in turn, was identical to a region of clone 11. Accordingly, it was very likely that at least two of clones 6, 14 and 11 represented hybrid sequences as well.
One of the final three clones was revealed to be a member of the highly repetitive family of long interspersed elements (LINE) (clone 10). This finding prompted us to inspect the putative fusion region between the fusion partner for sequences with reduced complexity. We have found either a G-rich region (>30 bases), GA-motifs (>50 bp) or a CA-repeat (>50 bp) in the six clones that were suspected of being hybrids.
The experimental design
According to the RDA concept, three different types of double-stranded DNA will be generated during hybridization of tester strands to an excess of driver DNA: driver/driver homodimers, driver/tester heterodimers and tester/tester homodimers. The last population contains tester-specific fragments because these sequences do not find any hybridization partners in the driver pool. The essential idea of RDA was to convert these tester/tester hybrids into PCR-competent sequences by an enzymatic filling of tester-specific 5′-protruding oligonucleotide flanks.
As described above, we tried to isolate bacteriophage λ sequences out of an equimolar mixture of λ and murine genomic DNA. We concluded from these preliminary experiments that, along with perfectly matched tester/tester homodimers, D-, P- or Y-shaped as well as other partial hybrids were generated during hybridization (Fig. 1). Due to the high copy number of repetitive sequences in mammalian genomes, some of these partial hybrids contained sufficiently well matched 3′-ends, and were elongated during the RDA-characteristic fill-in reaction. Finally, the newly synthesized fusion products dominated our pool of PCR-enriched tester sequences after repeated rounds of RDA. Attempts to avoid such kinds of artefacts resulted in the development of Limes (Fig. 2).
Figure 1.

Scheme of reaction pathways leading to ‘PCR hybrids’. During hybridization of tester and driver, members of a repeat family (open box) annealed to each other (partial hybrids) regardless of whether they are located on the reverse complementary or different strands (dotted and broken lines). In case of matched 3′-ends of tester/tester hybrids, the respective strand was elongated during an RDA-characteristic, enzymatic fill-in reaction leading to artificial PCR hybrids after amplification with a tester-specific primer (closed box).
Figure 2.
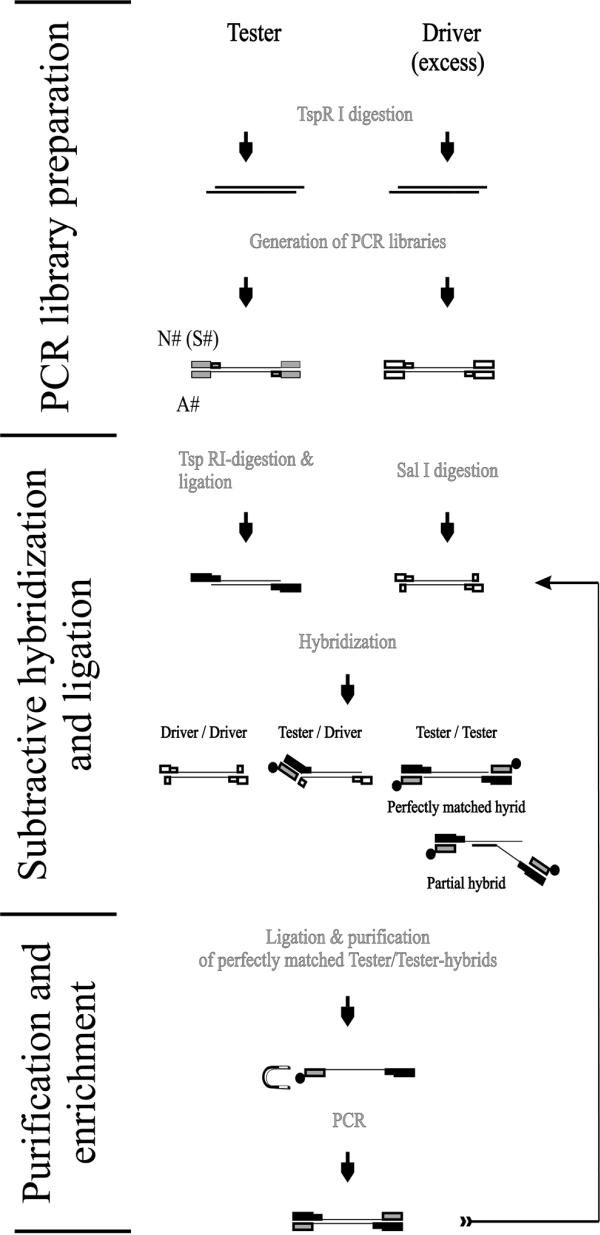
Scheme of Limes pathway. One round of Limes can be divided into three different parts: (i) preparation of PCR libraries, (ii) subtractive hybridization and ligation and (iii) purification and amplification of enriched tester sequences. The enriched material can be used for additional rounds of Limes. Initial PCR libraries were prepared by digestion of the subtraction partners with TspRI, by subsequent ligation of PCR-competent flanking sequences (N# and A#) to their ends with the Taq DNA ligase and amplification of the ligation products. The primers (S#) for the library amplification were identical to the respective N# primer apart from the lack of a TspRI recognition site at their 3′-end. In preparation for the subtractive hybridization, the tester library was digested with TspRI again and ligated with a new 5′ flanking N-primer. The driver was digested with SalI. After denaturation and hybridization of tester to an excess of driver, the pool of perfectly matched tester/tester homohybrids containing the tester-specific sequences was ligated with 3′-biotinylated (closed circle) oligonucleotides and purified by separation with streptavidin-coupled beads (horseshoe-shaped magnet) from driver/driver as well as tester/driver hybrids and all kinds of partial hybrids. The purified sequences were amplified with tester-specific S-primers.
Analogous to RDA, the Limes protocol starts with the preparation of tester and driver PCR libraries. For this purpose the sample DNA was digested with the restriction endonuclease TspRI, which was chosen for two main reasons. (i) Prevention of extensive loss of complexity in the resulting PCR libraries. TspRI recognizes the CA(C/G)TG-motif and, thus, the sizes of most of the generated fragments are well adapted for PCR because, from an arithmetical point of view, this motif is present in the genome every ∼512 bp. (ii) Usage of a highly specific thermophilic DNA ligase (Taq DNA ligase). TspRI generates an extraordinarily long 3′-overhang of nine bases (NNCA(G/C)TGNN), which allowed ligation of pre-annealed oligonucleotides (tester, N1 and A1; driver, N2 and A2) to TspRI-digested DNA (Fig. 2). The exceedingly selective Taq DNA ligase joins only perfectly matched ends without gaps between bases to be ligated (14).
As schematically shown in Figure 2, the resulting ligation products were amplified with respective S-primers giving rise to PCR libraries. An excess of SalI-digested driver PCR libraries (40 µg) served as subtraction partner for genomic (1 µg) or cDNA (100 ng) tester, which was ligated to N3 but not to oligonucleotide A3. The post-hybridizational ligation of A3 to perfectly matched tester/tester hybrids is one of the important differences between Limes and RDA. Albeit annealed to N3 overhangs of tester/driver heterohybrids as well as partial tester hybrids (Fig. 2), only ligated A-oligonucleotides transformed perfect tester/tester homodimers into PCR-competent sequences.
The annealing and extension steps of PCR are short-term hybridizations that are followed by sequence elongations as described for RDA. Accordingly, above-mentioned artefacts could also be generated during PCR-aided enrichment of tester/tester hybrids. To avoid this, 3′-biotinylated A3 ligation products were physically separated from other hybrids with streptavidin-coupled magnetic beads (Fig. 2).
The efficacy of our approach was essentially examined as described above. As a consequence of the enormous increase of sequence data that was provided by the Human Genome Project we decided, however, to add bacteriophage λ DNA to human genomic My-La DNA instead of murine EL4.
As shown at http://www.charite.de/ch/derm/Limes (figure 1 therein), a set of discrete bands in a range of 250–600 bp appeared after the third round of Limes and the same pattern remained constant until the fifth subtraction. The efficacy of each round of Limes was monitored by using the same set of primers as described above because both oligonucleotides are located on the same 519 bp TspRI fragment. Also shown at http://www.charite.de/ch/derm/Limes (figure 2 therein), 10 times less initial (unsubtracted) PCR library than untreated mixture of λ and genomic DNA was required to yield comparable amounts of the 307 bp λ-PCR fragment. This result implied a 10-fold reduction of complexity in favor of the investigated fragment. During the first three rounds of Limes an extra 100-fold enrichment of the λ fragment could be observed. This overall enrichment factor of 1:1000, however, decreased during the last two rounds to 1:10, although no obvious differences could be seen in the ‘macroscopic’ pattern of the enriched sequences. This observation can be explained by a growing domination of other sequences with an increasing number of rounds of Limes.
To analyze this pool of dominant fragments, the 4-fold enriched tester DNA was cloned and 10 arbitrarily chosen clones were sequenced (Table 2). Two of the 10 clones (clones 7 and 12) were in fact derived from different TspRI fragments of the bacteriophage λ genome. At least seven of the other eight fragments were revealed to be complete TspRI fragments of the sequenced human genome. Even for the exceptional clone (clone 11) whose sequence is still partially unknown, we did not see any evidence for the generation of fusion artefacts, although at least one of the 10 clones contained parts of a repetitive element (clone 3; Alu-repeat).
Table 2. Sequence analysis of 10 arbitrarily chosen clones derived from a pool of 4-fold enriched Limes tester sequences.
| Clone |
Bacteriophage λ (position
on λ) |
Human DNA, accession no. (position) |
Repeat |
Hybrid |
| 3 | No | AC020683 (1074–1451) | ∼80 bp Alu | No |
| 4 | No | AC002476 (106923–107213) | No | No |
| 7 | Yes (31793–32261) | No | No | No |
| 8 | No | AC018769 (149243–149568) | No | No |
| 11 | No | NM_003247.1(92 bp—thrombospondin exon—197 bp) | No | ? |
| 12 | Yes (43835–44425) | No | No | No |
| 14 | No | AC004782 (116637–117093) | No | No |
| 18 | No | AC025310 (37584–37943) | No | No |
| 22 | No | AC022490 (139531–140049) | No | No |
| 27 | No | AL358412 (65265–65550) | <25 bp MIR | No |
Isolation of tester-specific cDNA sequences by Limes
After completion of the described setup experiments, two cell lines were used for first subtraction experiments, the cutaneous lymphoma derived My-La (driver) and a CTL (tester) specifically recognizing and killing My-La cells (11). This tester/driver combination was chosen because My-La cells are known to be genetically unstable. These cells have most likely lost genetic information before establishing as a line.
In order to be prepared for the handling of limited amounts of material, reverse transcription was performed with the SMART™ cDNA Synthesis kit (Clontech) permitting the synthesis of PCR-based cDNA libraries from mRNA that is present in minute amounts of total RNA.
After two rounds of subtraction, the resulting enriched tester libraries contained approximately 10 dominant and an unknown number of less frequent PCR products (data not shown). One thousand cloned PCR products were investigated in parallel by dot blot hybridization with labeled probes derived from both initial tester and driver libraries. Most arrayed clones were positive for tester and negative for driver cDNA (see figure 3 at http://www.charite.de/ch/derm/Limes). All positively selected PCR fragments were clustered by their restriction fragment length pattern and 10 independent clones were sequenced. Comparison of these sequences to the GenBank database revealed that all 10 clones, except two (GenBank accession nos of clone 1: AF153603, AF183393 or HSM800841; clone 2: e.g. NM_001223.1 or U13697) were either unknown or identical/similar to expressed sequences tags (EST) with unknown functions. Clone 1 turned out to be the ‘related to transforming growth factor β stimulated clone 22’ (TSC-22R) and clone 2 was caspase-1 or interleukin-1β converting enzyme (ICE). As a final control, genomic DNA and cDNA from both initial libraries was amplified with primers derived from selected sequences. As was explicitly shown for clone 1 (Fig. 3A), it was found for both clones that amplification products of calculated sizes could be detected in genomic DNA (lane 1) as well as cDNA (lane 3) of the CTL sample. Amplification of genomic My-La DNA (lane 2) but not cDNA (lane 4) resulted in TSC-22R indicating PCR products. Caspase-1 amplification products could be obtained for My-La DNA but not for My-La cDNA (data not shown).
Figure 3.
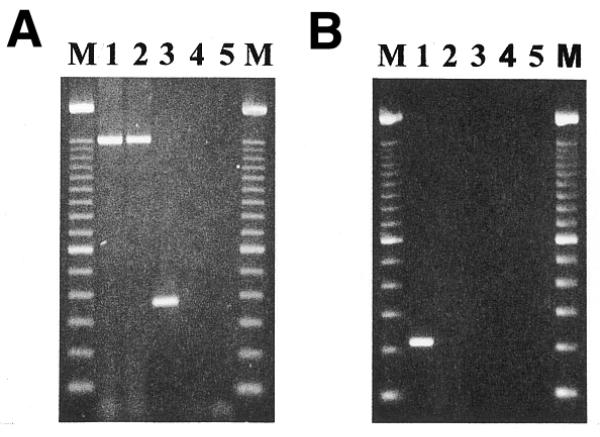
Amplification of tester-specific TSC-22R (A) and TSPY intron sequences (B). Genomic DNA of CTL (lane 1) and My-La (lane 2), cDNA of CTL (lane 3) and My-La (lane 4) and non-template DNA (lane 5) were amplified with a pair of primers that was either derived from a TspRI fragment of TSC-22R cDNA (A) or a TSPY intron (B) sequence and electrophoretically separated on an ethidium bromide-stained 1.5% agarose gel. M, 100 bp ladder size marker. The calculated size of genomic and cDNA TSC-22R amplification products was 1520 and 370 bp, respectively. The calculated size of the TSPY intron fragment was 220 bp.
Isolation of tester-specific genomic sequences by Limes
Analogous to the protocol described above, genomic DNA from the two cell lines was used to enrich tester-specific CTL sequences.
We started with 1 µg tester DNA and, for the following three of five Limes rounds, the enriched tester was diluted 4–5-fold after each round (200, 40, 10 and 10 ng). After the second round, the smear-like pattern of enriched tester was gradually replaced by distinct fragments. The enriched tester sequences were finally cloned and screened as described above. Cross-hybridization of repetitive sequences present on both the probe and spotted DNA was suppressed by blocking of repetitive sequences on the probe with an excess of unlabeled human DNA (15). Putative tester positive clones were grouped into clusters and representatives of each group were sequenced. So far, only one clone could be assigned to a gene with known function, the so-called testis specific protein Y-encoded (TSPY) (GenBank accession nos: M98524 or AF106331). The sequence analysis revealed the isolation of TSPY intron sequences and PCR with primers derived from this intron confirmed its tester specificity (Fig. 3B). The TSPY intron could be detected in the genome of CTL (lane 1) but was transcriptionally silent (lane 3). In My-La, the fragment was deleted at the chromosomal level (lane 2) and, accordingly, was not expressed either (lane 4). Cytogenetic studies confirmed that the examined My-La subline had lost its Y chromosome (data not shown). This is in contrast to the karyotype of the original My-La cells showing a duplication of the Y chromosome (10). The loss of these chromosomes indicated that neither gain nor loss of chromosome Y had any effect on cell growth.
‘Extract’ of genomically and cDNA-enriched tester
As shown in the upper experiment, large deletions such as loss of whole chromosomes may facilitate the isolation of tester-specific fragments but complicate the analysis of relevant sequences. TSPY, for instance, was genomically deleted in the driver but was most likely irrelevant for tumor presentation or progression because this gene was not expressed in the tester (data not shown). Moreover, the λ control experiment mentioned above revealed that some of the tester-specific λ sequences were two or three orders of magnitude less abundant in the finally enriched tester than other λ fragments. Accordingly, mutated genes of interest may have been lost because they were dominated by other less informative genomic or cDNA fragments.
In order to extract those fragments out of enriched libraries that were genomically mutated and showed an altered expression pattern, a 5′-biotinylated enriched library of genomic DNA was hybridized to an excess of an enriched cDNA library derived from the same specimen. Genomic sequences were separated from other sequences with streptavidin-coupled magnetic beads and attached cDNA sequences were amplified with a primer specific for the cDNA library. Again, the obtained fragments were cloned and replicated arrays were hybridized to either initial library of both tester and driver. Approximately 10% of the tested clones were specific for genomic as well as cDNA tester. Representatives of different clustered clones were sequenced and three sequences were allocated to genes with known function so far. All three, the DQA1, DRB1 and DRA genes belong to the HLA class II cluster (16). As confirmed by PCR (Fig. 4), these genes were expressed in CTL and deleted in My-La cells. In order to estimate the extent of the bi-allelic deletion in the HLA region on chromosome 6, HLA class II flanking Notch4 and TAP2 genes were analyzed. TAP2 was found to be expressed in My-La and CTL and, albeit not expressed, genomic Notch4 sequences could be detected in both cell lines (Fig. 4). These results confirmed data obtained after phenotype analyses of primary My-La cells with monoclonal antibodies against HLA class II antigens (10).
Figure 4.
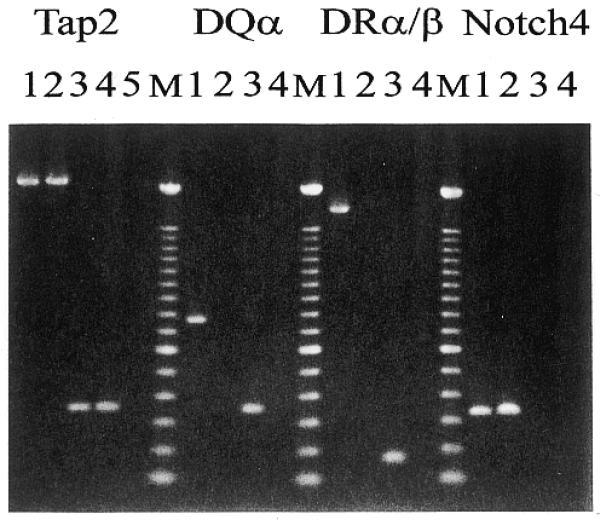
Amplification of HLA class II and flanking region genes. Genomic DNA of CTL (lane 1) and My-La (lane 2), cDNA of CTL (lane 3) and My-La (lane 4) and non-template DNA (lane 5) were amplified with pairs of primers that were derived from the indicated genes Tap2 (calculated sizes: 2197 and 345 bp for genomic DNA and cDNA, respectively), HLA-DQα (763 and 351 bp for genomic DNA and cDNA, respectively), HLA-DRB1 (1728 bp for genomic DNA), HLA-DRα (165 bp for cDNA) and Notch4 (329 and 209 bp for genomic DNA and cDNA, respectively). Amplification of genomic DNA and cDNA with primers from HLA-DRα (calculated size: 904 bp) and HLA-DRB1 (250 bp), respectively, were not shown but confirmed the shown results. The PCR products were electrophoretically separated on a 1.5% agarose gel and stained with ethidium bromide. M, 100 bp ladder size marker.
DISCUSSION
During our initial RDA experiments we came across two phenomena. On one hand, seven of nine sequences isolated by RDA contained repetitive sequences or elements with reduced complexity (e.g. G-stretches). This observation is in good agreement with a panel of other publications that also have described a preferential amplification of repetitive sequences by RDA of genomic DNA (e.g. 13 and 17). The second observation was the unusually high incidence of fusion artefacts (six of nine clones analyzed). To our knowledge, such a phenomenon was not reported so far, although the tendency of RDA to be prone to the production of false positives was noted by other groups as well. O’Neill and Sinclair (18), for instance, reported that the occurrence of some RDA artefacts is due to illegitimate ligation of partially degraded oligonucleotides to the ends of tester DNA.
We speculated that there is a correlation between the preferential amplification of repetitive sequences and the creation of fusion artefacts and hypothesized that the RDA-characteristic, enzymatic extension of hybrids may facilitate the production of fusion artefacts starting from repeat-mediated partial hybrids.
The aims of the key steps of Limes were (i) minimal loss of complexity during library preparation by use of TspRI-flanking PCR libraries; (ii) minimal artefact production by exclusive conversion of perfectly matched tester/tester homodimers into PCR competent sequences with a highly specific ligation reaction; and (iii) supplement prevention of artefacts by purification of such ligation products via streptavidin-coupled magnetic beads. The efficacy of this novel protocol was investigated by an experimental setting that was as close as possible to that used in the initial RDA control experiments. We have chosen the same DNA as tester (λ-DNA; size of the monitoring fragment: 519 instead of 564 bp) and mammalian genomic DNA as driver (human instead of murine DNA). The efficacy of Limes was proven by the successful isolation of two tester-specific λ sequences out of 10 enriched fragments. In addition, fusion artefacts were efficiently avoided.
We have found by using the same settings for serial dilutions of RDA-enriched as well as Limes-enriched DNA that our Limes libraries were ∼10-fold more complex than HindIII-, BamHI- or BglII-digested genomic libraries. RDA or other methods, such as a very recently published differential subtraction chain (DSC) technique (19), typically investigate the latter. Our results were in good agreement with results of Luo et al. (19) who also reported an 80–100-fold reduction of complexity at this step of the RDA protocol.
In the case of DSC, perfectly matched tester/tester homodimers are discriminated from other sequences by removal of flanking single-stranded overhangs with Mung Bean Nuclease. The authors suggest that the ratio between PCR-competent specific sequences and ubiquitous fragments is considerably improved by repeated cycles of hybridization and digestion. It is also conceivable, however, that additional benefit resulted from the destruction of partial hybrids by the Mung Bean Nuclease. It is not trivial, however, to adjust the optimal enzyme concentration for an almost complete digestion of single-stranded DNA without destruction of heteroduplexes.
In the case of less complex cDNA libraries, two rounds of Limes subtraction were sufficient to obtain a highly enriched pool of tester-specifically expressed sequences containing transcripts such as TSC-22R, a candidate tumor suppressor gene or the protease caspase-1 involved in processing of inflammatory cytokines, such as IL-1β or IL-18, and in apoptosis (20). Homo- and heterodimers of leucine zipper-containing members of the TSC-22 family most likely seem to mediate transcriptional repressor activity (21) and the absence of TSC-22R expression in the cutaneous lymphoma line, My-La, corresponded well to the suggested suppressive function of the gene. Other sequences obtained during this run remain either unknown or still not analyzed because the abundance of enriched tester sequences spanned several orders of magnitude.
The screening for genomic mutations was stopped at the point when we realized that the examined My-La subline had lost its complete Y chromosome, because large genomic deletions impeded the identification of candidate oncogenes or tumor suppressor genes. It is worth noting, however, that the majority of the analyzed genomic clones were single copy sequences indicating that the frequency of repetitive elements decreased considerably in the enriched library.
Instead of continued screening of genomic DNA, the concept of searching for mutated genomic sequence with altered expression patterns was developed and different genes of the HLA class II region were found by this approach. There seems to be no unequivocal correlation between HLA class II expression on tumor cells and prognosis of the disease and, accordingly, this issue is controversially discussed. Recently, a caspase-independent pathway of MHC class II-mediated apoptosis, induced by an antibody-mediated HLA-DR stimulation, was detected in human B cells (22).
In summary, the Limes approach is a technique for the isolation of tester-specific sequences from genomic DNA, cDNA and a combination of both. Limes is resistant to artefact production. In first experiments, we were able to isolate tester-specific sequences belonging to genes involved in regulation of transcription, in processing of cytokines, in antigen presentation and in apoptosis. The experimental protocol of this method was designed to begin experiments with minute amounts of RNA or DNA and we are currently applying Limes to small numbers of tumor cells derived from patients with cutaneous lymphoma. We hope that this novel technical tool will help to facilitate the identification of differences in a variety of DNA sources.
Acknowledgments
ACKNOWLEDGEMENTS
We thank P. Walden and other members of the Klinische Forschergruppe ‘Tumoren der Haut’ at the Charité/Department of Dermatology and Allergy for fruitful discussions and manifold support. We are also grateful to Petra Schneider and Anke Herrmann for their excellent technical assistance. This study is supported by a grant from the Deutsche Forschungsgemeinschaft to W.S.
References
- 1.Liang P. and Pardee,A.B. (1992) Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science, 257, 967–971. [DOI] [PubMed] [Google Scholar]
- 2.Alizadeh A.A., Eisen,M.B., Davis,R.E., Ma,C., Lossos,I.S., Rosenwald,A., Boldrick,J.C., Sabet,H., Tran,T., Yu,X. et al. (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 403, 503–511. [DOI] [PubMed] [Google Scholar]
- 3.Lisitsyn N., Lisitsyn,N. and Wigler,M. (1993) Cloning the differences between two complex genomes. Science, 259, 946–951. [DOI] [PubMed] [Google Scholar]
- 4.Hedrick S.M., Cohen,D.I., Nielsen,E.A. and Davis,M.M. (1984) Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature, 308, 149–153. [DOI] [PubMed] [Google Scholar]
- 5.Willemze R., Kerl,H., Sterry,W., Berti,E., Cerroni,L., Chimenti,S., Diaz-Perez,J.L., Geerts,M.L., Goos,M., Knobler,R., Ralfkiaer,E., Santucci,M., Smith,N., Wechsler,J., van Vloten,W.A. and Meijer,C.J. (1997) EORTC classification for primary cutaneous lymphomas: a proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood, 90, 354–371. [PubMed] [Google Scholar]
- 6.Pancake B.A., Zucker-Franklin,D. and Coutavas,E.E. (1995) The cutaneous T cell lymphoma, mycosis fungoides, is a human T cell lymphotropic virus-associated disease. A study of 50 patients. J. Clin. Invest., 95, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivadeneira E.D., Ferrari,M.G., Jarrett,R.F., Armstrong,A.A., Markham,P., Birkebak,T., Takemoto,S., Johnson-Delaney,C., Pecon-Slattery,J., Clark,E.A. and Franchini,G. (1999) A novel Epstein-Barr Virus-like virus, HV(MNE), in a Macaca nemestrina with mycosis fungoides. Blood, 94, 2090–2101. [PubMed] [Google Scholar]
- 8.Boni R., Davis-Daneshfar,A., Burg,G., Fuchs,D. and Wood,G.S. (1996) No detection of HTLV-I proviral DNA in lesional skin biopsies from Swiss and German patients with cutaneous T-cell lymphoma. Br. J. Dermatol., 134, 282–284. [PubMed] [Google Scholar]
- 9.Henghold W.B.,II, Purvis,S.F., Schaffer,J., Giam,C.Z. and Wood,G.S. (1997) No evidence of KSHV/HHV-8 in mycosis fungoides or associated disorders. J. Invest. Dermatol., 108, 920–922. [DOI] [PubMed] [Google Scholar]
- 10.Kaltoft K., Bisballe,S., Dyrberg,T., Boel,E., Rasmussen,P.B. and Thestrup-Pedersen,K. (1992) Establishment of two continuous T-cell strains from a single plaque of a patient with mycosis fungoides. In Vitro Cell. Dev. Biol., 28A, 161–167. [DOI] [PubMed] [Google Scholar]
- 11.Linnemann T., Wiesmüller,K.H., Gellrich,S., Kaltoft,K., Sterry,W. and Walden,P. (2000) A T-cell epitope determined with random peptide libraries and combinatorial peptide chemistry stimulates T cells specific for cutaneous T-cell lymphoma. Ann. Oncol., 11, 95–99. [PubMed] [Google Scholar]
- 12.Yokota S., Hansen-Hagge,T.E., Ludwig,W.D., Reiter,A., Raghavachar,A., Kleihauer,E. and Bartram,C.R. (1991) Use of polymerase chain reactions to monitor minimal residual disease in acute lymphoblastic leukemia patients. Blood, 77, 331–339. [PubMed] [Google Scholar]
- 13.Lisitsyn N. and Wigler,M. (1995) Representational difference analysis in detection of genetic lesions in cancer. Methods Enzymol., 254, 291–304. [DOI] [PubMed] [Google Scholar]
- 14.Housby J.N. and Southern,E.M. (1998) Fidelity of DNA ligation: a novel experimental approach based on the polymerisation of libraries of oligonucleotides. Nucleic Acids Res., 26, 4259–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sealey P.G., Whittaker,P.A. and Southern,E.M. (1985) Removal of repeated sequences from hybridisation probes. Nucleic Acids Res., 13, 1905–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The MHC sequencing consortium (1999) Complete sequence and gene map of a human major histocompatibility complex. Nature, 401, 921–923. [DOI] [PubMed] [Google Scholar]
- 17.Nekrutenko A., Makova,K.D. and Baker,R.J. (2000) Isolation of binary species-specific PCR-based markers and their value for diagnostic applications. Gene, 249, 47–51. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill M.J. and Sinclair,A.H. (1997) Isolation of rare transcripts by representational difference analysis. Nucleic Acids Res., 25, 2681–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo J.H., Puc,J.A., Slosberg,E.D., Yao,Y., Bruce,J.N., Wright,T.C., Becich,M.J. and Parsons,R. (1999) Differential subtraction chain, a method for identifying differences in genomic DNA and mRNA. Nucleic Acids Res., 27, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatsuta T., Shiraishi,A. and Mountz,J.D. (2000) The prodomain of caspase-1 enhances Fas-mediated apoptosis through facilitation of caspase-8 activation. J. Biol. Chem., 275, 14248–14254. [DOI] [PubMed] [Google Scholar]
- 21.Kester H.A., Blanchetot,C., den Hertog,J., van der Saag,P.T. and van der Burg,B. (1999) Transforming growth factor-beta-stimulated clone-22 is a member of a family of leucine zipper proteins that can homo- and heterodimerize and has transcriptional repressor activity. J. Biol. Chem., 274, 27439–27447. [DOI] [PubMed] [Google Scholar]
- 22.Drenou B., Blancheteau,V., Burgess,D.H., Fauchet,R., Charron,D.J. and Mooney,N.A. (1999) A caspase-independent pathway of MHC class II antigen-mediated apoptosis of human B lymphocytes. J. Immunol ., 163, 4115–4124. [PubMed] [Google Scholar]