

Abstract
Alzheimer's disease (AD), the most common neurodegenerative disorder, goes along with extracellular amyloid-β (Aβ) deposits. The cognitive decline observed during AD progression correlates with damaged spines, dendrites and synapses in hippocampus and cortex. Numerous studies have shown that Aβ oligomers, both synthetic and derived from cultures and AD brains, potently impair synaptic structure and functions. The cellular prion protein (PrPC) was proposed to mediate this effect. We report that ablation or overexpression of PrPC had no effect on the impairment of hippocampal synaptic plasticity in a transgenic model of AD. These findings challenge the role of PrPC as a mediator of Aβ toxicity.
Keywords: Alzheimer's disease, amyloid, prion protein, synaptic plasticity
→See accompanying Closeup by Benilova & de Strooper: http://dx.doi.org/10.1002/emmm.201000088
INTRODUCTION
Alzheimer's disease (AD) is an age-dependent neurodegenerative disorder that culminates in cognitive decline with limited treatment options. Oligomeric amyloid-β (Aβ), derived from the β and γ cleavage of β-amyloid precursor protein (APP), may drive AD pathogenesis by activating ill-defined signalling pathways (Walsh et al, 2005). Several molecules have been suggested to trigger the latter (De Felice et al, 2009; Shankar et al, 2007; Snyder et al, 2005). The cellular prion protein (PrPC) was reported to mediate the impairment of long-term potentiation (LTP) induced by synthetic Aβ oligomers in the hippocampal Schaffer collateral pathway (Lauren et al, 2009). Also, removal of PrPC from mice carrying APPswe and PSen1ΔE9 transgenes rescued early death and memory impairment (Gimbel et al, 2010).
PrPC is a membrane-anchored glycoprotein (Steele et al, 2007) crucial for axomyelinic integrity of peripheral nerves (Bremer et al, 2010). The remarkable finding that PrPC mediates Aβ-related synaptic toxicity was taken to suggest that interference with PrPC may represent a therapeutic option for AD (Lauren et al, 2009; Gimbel et al, 2010). However, upon intracerebral injection of synthetic Aβ oligomers, the absence of PrPC did not prevent deficits in hippocampal dependent behavioural tests (Balducci et al, 2010).
In view of these conflicting reports, we reasoned that a better understanding of the impact of PrPC onto AD may come from careful genetic analyses. Also, the utilization of a second, independent AD transgenic mouse model may help evaluating the universality of the observed phenomena. We therefore asked whether PrPC would modulate the degradation of LTP in an in vivo model of AD. We crossed mice lacking (Büeler et al, 1992) or overexpressing membrane-anchored (Fischer et al, 1996) or secreted PrP (Chesebro et al, 2005) with APPPS1+ mice coexpressing mutant APP (APPKM670/671NL) and mutant presenilin-1 (PS1L166P; Radde et al, 2006) which suffer from Aβ-dependent learning and memory deficits (Serneels et al, 2009; Table 1). We found that ablation or overexpression of PrPC had no effect on the impairment of hippocampal synaptic plasticity in a transgenic model of AD. These findings challenge the role of PrPC as a Aβ toxicity mediator.
Table 1.
Genetically modified mice used in this study
| Line | Description | Genetic modifications | Genetic background | References |
|---|---|---|---|---|
| APPPS1 | Alzheimer's disease mouse model displaying Aβ42 cerebral amyloidosis | APPKM670/671NL transgene | C57BL/6 | Radde et al (2006) |
| PS1L166P transgene (both on Mmu2) | ||||
| Prnpo/o | Mouse lacking cellular prion protein | Introduction of a neo cassette replacing PrP codon 4–187 in the Prnp locus in Mmu2 (Prnpo allele) | C57BL/6 and 129/Sv | Büeler et al (1992) |
| tga20 | Mouse overexpressing cellular prion protein | Introduction of a neo cassette replacing PrP codon 4–187 in the Prnp locus in Mmu2 (Prnpo allele) | C57BL/6 and 129/Sv | Fischer et al (1996) |
| Prnp minigene on Mmu17 | ||||
| tg44Prnp−/− | Mouse expressing GPI-anchorless prion protein | Introduction of a neo cassette into a KpnI site following residue 93 of PrP in the Prnp locus in Mmu2 (Prnp− allele) | C57BL/10 and 129/Ola | Chesebro et al (2005) |
| Anchorless PrP transgene |
Mmu2 and Mmu17: Mus musculus chromosome 2 and 17, respectively; neo: neomycin phosphotransferase; Prnpo and Prnp− denote by convention the ‘Zurich-I’ and ‘Edbg’ knockout alleles of Prnp, respectively.
RESULTS AND DISCUSSION
LTP impairment and APP processing are not altered in absence of the cellular prion protein
We crossed Prnpo/o mice lacking PrPC (Büeler et al, 1992) with APPPS1+ mice coexpressing mutant APP (APPKM670/671NL) and mutant presenilin-1 (PS1L166P; Radde et al, 2006). The resulting mice did not display any early death independently of the Prnp genotype (data not shown). High-frequency stimulation (HFS) of Schaffer collateral CA1 synapses induced an increase in field excitatory postsynaptic potentials (fEPSP) reflecting LTP in both 4-month-old Prnp+/+ and Prnpo/o mice (data not shown) as previously reported (Lledo et al, 1996). In contrast, age-matched APPPS1+Prnp+/+ (n = 6), APPPS1+Prnp+/o (n = 5) and APPPS1+Prnpo/o (n = 5) all exhibited defective LTP after HFS (114.23 ± 9.61; 111.72 ± 9.64 and 105.51 ± 12.23%, respectively; p < 0.001; Fig 1A). The fEPSP slopes during the first 2 min were similar in APPPS1+Prnp+/+ and wild-type mice (124.1 ± 7.0 and 184.8 ± 26.2%, respectively; p > 0.05), indicating that immediate post-tetanic potentiation was not affected. Basal synaptic transmission as assessed by input–output curve analysis was normal in all mice (Fig 1B and C), confirming that the APPPS1 transgene induces a selective impairment in synaptic plasticity. In contrast to 4-month-old animals, robust LTP was induced in 2-month-old APPPS1+Prnp+/+ (172.6 ± 14.6%; n = 5), APPPS1+Prnp+/o (168.9 ± 14%; n = 5) and APPPS1+Prnpo/o mice (204.4 ± 15.9%; n = 4) and was comparable to LTP in Prnp+/o (174.6 ± 7%; n = 5; Fig 1D). We conclude that the LTP impairment was age related, appeared only in mice carrying the APPPS1 transgene after >2 months, and was independent of Prnp gene dosage.
Figure 1. CA1 hippocampal LTP impairment in APPPS1+ mice occurs at 4 months of age and is not regulated by PrPC expression.

- CA1 hippocampal LTP was induced in acute slices from 4-month-old Prnp+/+ mice (black, n = 7), but was abolished in slices from age-matched APPPS1+Prnp+/+ (dark blue, n = 6), APPPS1+Prnp+/o (blue, n = 5) and APPPS1+Prnpo/o mice (light blue n = 5).
- fEPSP traces before (red) and after (black) LTP induction. Calibration: 1 mV; 10 ms.
- Input–output curves (stimulus intensity vs. fEPSP slope) indicative of normal basal synaptic transmission.
- Unaffected LTP in slices derived from 2-month-old APPPS1+Prnp+/+ (n = 5), APPPS1+Prnp+/o (n = 5), APPPS1+Prnpo/o (n = 4) and Prnp+/o mice (n = 5). These results indicate that LTP impairment in APPPS1+ mice was not a developmental defect, and occurred only after 2 months of age independently of Prnp gene dosage.
Many genetic polymorphisms affect APP processing and Aβ levels (Lehman et al, 2003). The APPKM670/671NL and PS1L166P transgenes map to mouse chromosome 2 (Mmu2; Radde et al, 2006) along with Prnp, and are linked to a quantitative trait locus that modifies Aβ levels (Ryman et al, 2008). Furthermore, PrPC itself was reported to directly interfere with APP catabolism (Parkin et al, 2007). Each of these factors, alone or in combination, may modulate the production of soluble Aβ42, thereby indirectly affecting LTP impairment. However, we found that 2-month old gender-matched APPPS1+Prnp+/+ and APPPS1+Prnpo/o mice displayed similar levels of APP catabolites (Fig S1A) and soluble Aβ42 (Fig S1B). We conclude that the effects described here cannot be ascribed to any difference in APP generation or processing.
Evaluation of genetic confounders that might mask the impact of PrPC on LTP in 4-month-old APPPS1 mice
A genome-wide screen of 192 polymorphic microsatellites revealed that APPPS1+Prnpo/o mice contained significantly larger portions of 129/Sv-derived genome than APPPS1+Prnp+/+ mice (129/Sv-specific markers: average ± SEM: 60 ± 6.2 vs. 2 ± 0.4, respectively; p < 0.001). This genetic constellation may be taken to suggest that the above intercrosses have inadvertently introduced genetic biases affecting LTP independently of Aβ levels (Gerlai, 2002). However, in subsequent intercrosses, the content in genome-wide 129/Sv-specific markers was 55.3 ± 3.9 versus 41.7 ± 3.2 (n = 7 and 6, respectively; p < 0.05), yet this statistically significant difference disappeared upon exclusion of markers on Mmu2 (44.7 ± 3.8 vs. 38.0 ± 3.2, respectively; p > 0.05). This indicates that the latter mice, although not inbred, were genetically similar except for the Mmu2 genomic region that is closely linked to both Prnp and APPPS1 and does not desegregate easily from these loci by breeding. This genetic scenario may help explaining the differences in insoluble Aβ42 levels seen in F2 APPPS1+ mice with different Prnp genotypes generated by intercrosses of APPPS1+ and Prnpo/o mice (Fig S2; Ryman et al, 2008).
Transgenic PrPC overexpression disproves Mmu2 bias and does not aggravate APPPS1-induced LTP impairment
To formally discriminate between PrPC-dependent effect and potential confounders residing on Mmu2, we reintroduced PrPC into APPPS1+Prnpo/o mice via crosses to tga20 mice (Fischer et al, 1996) that carry a Prnp minigene on Mmu17 (Zabel et al, 2009) and overexpress PrPC about fourfold (Fig S3). LTP was again affected in 4-month-old APPPS1+tga20tg/−Prnpo/o (127.84 ± 12.61%; n = 4) and APPPS1+tga20−/−Prnpo/o littermates (106.56 ± 5.46%; n = 5; p = 0.137; Fig 2A). The genome-wide microsatellite patterns of these two groups of mice were indistinguishable even when Mmu2 markers were included (129/Sv-specific markers: 61.0 ± 2.1 vs. 61.7 ± 3.9, respectively; p > 0.05; Fig 2B), indicating that any contribution by genetic confounders to the phenotype is unlikely. To further explore the impact of supraphysiological levels on PrPC in LTP, we analyzed APPPS1+tga20tg/−Prnp+/o which overexpress ca. sevenfold PrPC (Fig S3) and APPPS1+tga20−/−Prnp+/o littermates. These two groups of mice shared similar genomic microsatellite patterns (Fig 3A). At 4 months of age, LTP was significantly reduced in both APPPS1+tga20tg/−Prnp+/o and APPPS1+tga20−/−Prnp+/o littermates (149.41 ± 11.81%, n = 6 vs. 121.56 ± 11.65%, respectively; n = 4; Fig 3B). Expression of the tga20 allele showed a tendency towards improved LTP that was not statistically significant, without altering APP catabolites and soluble and insoluble Aβ42 (Fig 3C and D). Therefore, PrPC overexpression did not enhance Aβ-mediated LTP impairment; if anything, it may have marginally antagonized it.
Figure 2. LTP in 4-month-old APPPS1+ mice expressing a PrPC transgene.
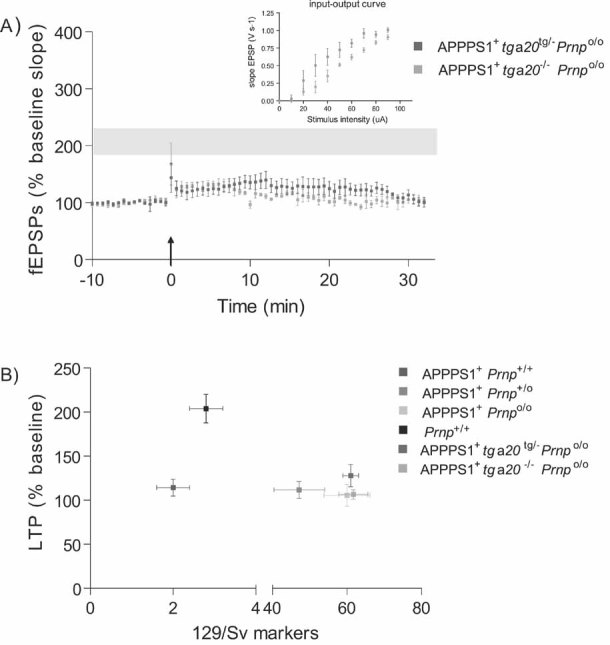
- At 4 months of age, LTP was impaired in slices from both APPPS1+tga20tg/−Prnpo/o (n = 4) and APPPS1+tga20−/−Prnpo/o (n = 5) but not in Prnp+/+ slices (n = 7; LTP mean ± SEM from Fig 1A represented as grey ribbon). Basal synaptic transmission was normal as indicated by normal input–output curve (stimulus intensity vs. fEPSP slope).
- Average fEPSP slopes (percentage of baseline) at 10–25 min post-LTP plotted against the average number of 129/Sv specific markers for mice depicted in panel A and Fig 1A. In all investigated paradigms, LTP suppression by the APPPS1 transgene was independent of the genetic background.
Figure 3. Analysis of 4-month-old APPPS1+ mice with supraphysiological levels of PrPC.

- Percentage of strain-specific microsatellites in APPPS1+tga20tg/−Prnp+/o (n = 6) and APPPS1+tga20−/−Prnp+/o (n = 4) mice is displayed by box plot. No significant difference in the genetic background of the two mouse strains was detected (Mann–Whitney U-test, two-tailed, p > 0.05).
- At 4 months of age, slices of both APPPS1+tga20tg/−Prnp+/o (n = 6) and APPPS1+tga20−/−Prnp+/o mice (n = 4) displayed reduced LTP when compared to Prnp+/+ mice (n = 7); LTP mean ± SEMfrom Fig 1A represented as grey ribbon. Basal synaptic transmission was normal as indicated by normal input–output curve (stimulus intensity vs. fEPSP slope). All error bars: standard errors of the mean.
- APP expression and processing by secretases were similar in 4-month-old APPPS1+ tga20tg/−Prnp+/o and APPPS1+tga20−/−Prnp+/o mice. Left panel: representative SDS–PAGE followed by immunoblotting using an APP C-terminal antibody detecting full-length APP and αβ-CTF; actin was used as loading control. Right panel: quantitation of chemiluminescence for APP, α-CTF and β-CTF.
- TRIS-soluble (left panel), detergent-soluble (middle panel) and insoluble (right panel) human Aβ42 levels as assessed by ELISA. Each symbol denotes one individual mouse.
Overexpression of a secreted PrPC variant reduced the impairment of LTP in 4-month-old APPPS1 mice
We next asked whether a soluble version of PrPC might intercept Aβ oligomers and interfere with synaptic toxicity. First we verified that interaction of PrPC with Aβ species (Balducci et al, 2010; Lauren et al, 2009) can occur in the absence of PrPC membrane anchoring. We therefore tested the binding properties of bacterially expressed recombinant full-length PrP (recPrP23–230). We found that recPrP23–230 bound low molecular weight Aβ42 species, and that binding was reduced by monoclonal anti-PrP antibodies (Polymenidou et al, 2008) raised against its N-proximal region (Fig S4). Also, we found that a shortened variant of recPrP lacking the amino-proximal residues 23–121 (recPrP121–230) did not bind Aβ42 (Fig S4). These results confirm that PrP, even when produced in bacteria and therefore, lacking all eukaryotic post-translational modifications including the addition of a glycolipid anchor, can efficiently bind Aβ species.
We then crossed APPPS1+Prnpo/o mice to mice expressing GPI-anchorless PrP (secPrP) which is secreted into body fluids of tg44Prnp−/− transgenic mice (Chesebro et al, 2005). The Prnpo and Prnp− alleles refer to the ‘Zurich-I’ (Büeler et al, 1992) and ‘Edbg’ (Manson et al, 1994) gene ablation events. We measured LTP in hippocampal slices derived from 4-month-old APPPS1+tg44tg/−Prnp−/o (n = 7) and APPPS1+tg44−/−Prnp−/o (n = 6) littermates with comparable genomic microsatellite patterns (Fig 4A). Remarkably, secPrP significantly suppressed the APPPS1-related LTP impairment (151.5 ± 11 and 108.5 ± 7.5%, respectively; p < 0.05, ANOVA and Tukey's multiple comparison test, see Fig 4B). The metabolism of APP and the levels of soluble and insoluble Aβ42 did not appear to be altered by the tg44 transgene (Fig 4C and D), suggesting that secPrP exerted its beneficial effects interfering with the effectors of Aβ toxicity.
Figure 4. Anchorless soluble PrPC reduces hippocampal LTP impairment in APPPS1+ mice.

- Percentage of strain-specific microsatellites in APPPS1+tg44tg/−Prnp−/o (n = 5) and APPPS1+tg44−/−Prnp−/o (n = 5) mice is displayed by box plot. No significant difference in the genetic background was detected (Mann–Whitney U-test, two-tailed, p > 0.05).
- LTP was induced in slices prepared from 4-month-old tg44tg/−Prnp−/o (n = 5) and tg44−/−Prnp−/o (n = 7) mice, but was impaired in slices from APPPS1+tg44−/−Prnp−/o mice (n = 6) and partially rescued in APPPS1+tg44tg/−Prnp−/o (n = 7) mice. Basal synaptic transmission was normal as indicated by normal input–output curve (stimulus intensity vs. fEPSP slope). All mice were compound heterozygotes for the ‘Zurich-I’ (Prnpo) and the ‘Edbg’ (Prnp−) knockout alleles of Prnp.
- APP expression and processing by secretases were similar in APPPS1+tg44tg/−Prnp−/o and APPPS1+tg44−/−Prnp−/o mice at 4 months of age. Left panel: representative SDS–PAGE followed by immunoblotting using an APP C-terminal antibody detecting full-length APP and C-terminal fragments (αβ-CTF); actin was used as loading control. Right panel: quantitation of chemiluminescence revealed no difference in APP, α-CTF and β-CTF between the two groups.
- TRIS-soluble (left panel), detergent-soluble (middle panel) and insoluble (right panel) human Aβ42 levels as assessed by ELISA. Each symbol denotes one individual mouse.
Despite decades of research, the cascade of events that originates with the aggregation of Aβ and leads up to cognitive impairment continues to be poorly understood. Many observations point to a crucial role of transmembrane signaling events triggered by aggregated Aβ. Several membrane proteins have been reported to bind soluble Aβ oligomers—thereby candidating as potential transducers of toxicity (Deane et al, 2004; De Felice et al, 2009; Shankar et al, 2007; Snyder et al, 2005; Yan et al, 1996). A great deal of excitement was generated by the recovery of PrPC from an expression screen for soluble Aβ oligomer binders, particularly as synthetic soluble Aβ oligomers were found to damage hippocampal LTP in a PrPC-dependent manner (Lauren et al, 2009) and impairment of spatial memory was rescued by genetic ablation of PrP in a mouse model of AD (Gimbel et al, 2010). However, the report that removal of PrPC did not prevent the behavioural deficits caused by intracerebral injection of synthetic Aβ oligomers (Balducci et al, 2010) challenged the role of PrPC as a crucial mediator of Aβ synaptotoxicity.
We crossed mice expressing human Aβ to mice lacking or overexpressing PrPC or a soluble variant thereof to evaluate if the impact of PrP is persistent also in another AD mouse model which suffer from Aβ-dependent learning and memory deficits (Serneels et al, 2009). The latter experimental paradigm may more closely approximate the human disease than the previously published models (Balducci et al, 2010; Lauren et al, 2009) as exposure to Aβ species is chronic and uninterrupted over a protracted period, which is arguably more realistic than hyperacute exposure of brain tissue to Aβ. Furthermore, Aβ exists in AD brains as a vastly heterodisperse spectrum of assemblies ranging from monomers and dimers to oligomers and extremely large fibrillary aggregates, each one of which may partly contribute to the AD phenotype (Lesne et al, 2006; Shankar et al, 2008, 2009; Walsh et al, 2002). As the relative affinity of the various Aβ assemblies for PrPC is not known in detail, transgenic mice expressing many such assemblies may reveal phenomena that might go unrecognized in simpler systems, such as application of defined synthetic Aβ oligomers.
On the other hand, the genetic crosses described in our study and in previous work (Gimbel et al, 2010) may suffer from limitations. PrPC was reported to regulate β-secretase cleavage (Parkin et al, 2007), and overexpression may interfere with APP metabolism and Aβ levels, thereby indirectly affecting LTP impairment. Indeed, careful genetic quality control revealed a mouse-strain dependent effect on insoluble Aβ42 levels—a phenomenon that should be taken into account while interpreting results from mouse AD models. However, all mice analyzed in this study displayed similar levels of APP catabolites and Aβ42 independently of Prnp gene dosage.
We also considered the possibility that potential confounders residing on Mmu2 might have introduced alterations of the experimental evaluation (Steele et al, 2007), a problem which remains unsolved in the study by Gimbel et al. However, in our paradigm, genome-wide microsatellite analyses and expression of PrPC from the tga20 minigene on chromosome Mmu17 disproved any Mmu2 bias.
Additionally, one might argue that the exceedingly rapid amyloid pathology of APPPS1 mice used in our study leads to irreversible synaptic damage that is independent of Aβ oligomers and, consequently, of PrPC. However, the original report (Radde et al, 2006) and our observations indicate that immunohistochemically and biophysically recognizable amyloid deposition does not occur in APPPS1 hippocampi before 4–5 months of age (Fig S5). Therefore, at the time of our analysis, there was no massive amyloid deposition in the hippocampus. Furthermore, the rescue of LTP impairment by secPrP negates the possibility that an overly aggressive amyloid pathology precludes the evaluation of the role of PrPC in these mice.
The combined weight of all these results favours the conclusion that, however enticing, the hypothesis of PrPC being a crucial mediator of Aβ synaptotoxicity might be not universal.
MATERIALS AND METHODS
Mice
To remove the prion protein locus (Prnp), Prnpo/o mice (Büeler et al, 1992) were crossed with APPPS1 mice (Radde et al, 2006). APPPS1+Prnpo/o or APPPS1 mice were then crossed with tga20tg/−Prnpo/o (Fischer et al, 1996) or tg44tg/−Prnp−/− mice (Chesebro et al, 2005) to generate the different APPPS1+ and APPPS1-littermate control mice (Table 1 and Fig S6). The genetic pattern of mouse strains was determined with a panel of 192 polymorphic microsatellites as described (Bremer et al, 2010). All mice were maintained under specific pathogen-free conditions. Housing and experimental protocols were in accordance with the Swiss Animal Welfare Law and in compliance with the regulations of the Cantonal Veterinary Office, Zurich.
The paper explained
PROBLEM
Alzheimer's disease (AD), the most common neurodegenerative disorder, culminates in cognitive decline with limited treatment options. Aggregated Aβ, possibly in the form of oligomers, accumulates in the brain of affected individuals and may drive AD pathogenesis by activating ill-defined signaling pathways. The PrPC was reported to mediate the impairment of LTP induced by synthetic Aβ oligomers and removal of PrPC from an AD mouse model rescued early death and memory deficit. In another study, however, the absence of PrPC did not prevent deficits in hippocampal dependent behavioural tests caused by intracerebral injection of Aβ oligomers. To investigate the universality of the observed phenomena, we asked whether PrPC modulates LTP in a second independent AD mouse model.
RESULTS
We crossed mice lacking or over-expressing PrPC with APPPS1+ mice coexpressing mutant APP and mutant presenilin-1, which suffer from Aβ-dependent learning and memory deficits. We found defective LTP in APPPS1+ mice at 4 months of age. Ablation or overexpression of PrPC had no effect on this impairment of hippocampal synaptic plasticity.
IMPACT
The results reported here suggest that PrPC may not be a universal mediator of Aβ synaptotoxicity. Additional work is required to refine our understanding of the interaction between PrPC and Aβ and establish whether PrPC is a viable target for pharmaceutical interventions in AD.
Electrophysiology
Hippocampal slice preparation from male mice and fEPSPs recordings in the CA1 region were as described (Knobloch et al, 2007). The LTP induction protocol was considered successful, and entered in the analysis, only if a stable baseline for at least 10 min was achieved. To generate input–output curves, slices were prepared as above and stimulated every 20 s with increasing intensity (from 0.0 to 0.1 mA in 0.01 mA increments) using a total of 10 stimuli. For comparing groups, potentiation of fEPSP slopes during the interval 10–25 min post-tetanus was evaluated. Data points were normalized to the mean baseline value and expressed as mean ± SEM All numbers in brackets indicate analyzed mice; 2–3 slices were typically analyzed for each mouse.
Tissue preparation
Brain fractionation was performed as described (Shankar et al, 2008) with modifications. Briefly, snap frozen forebrains were homogenized in ice-cold tris buffered saline (TBS), after centrifugation at 100,000 × g for 1 h the supernatant (called soluble fraction) was used to determine soluble Aβ42. The pellet was homogenized in phosphate buffered saline plus 0.5% 4-nonylphenyl-polyethylene glycol (NP40S), 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and spun at 16,000 × g for 30 min. The resultant supernatant was used to quantify APP, α-C terminal fragment (CTF) and β-CTF and the remaining pellet was solubilized in 70% formic acid and insoluble Aβ42 was measured after tris(hydroxymethyl)aminomethane (TRIS)-base neutralization.
Quantification of Aβ42 and PrPC
Levels of Aβ42 were assessed by sandwich enzyme-linked immunosorbent assay (ELISA; hAmyloid Aβ42, The Genetics Company) according to manufacturer's instructions. PrPC concentration was determined by sandwich ELISA as described (Polymenidou et al, 2008).
Immunoblotting
To determine APP and CTFs levels, 20 µg of proteins were separated by electrophoresis on a 4–12% polyacrylamide gel. Primary antibodies were: anti-APP C-terminal (Sigma) recognizing both mouse and human APP and CTFs; anti-actin (Chemicon). Protein bands were detected by adding SuperSignal West Pico Chemiluminescent Substrate (Pierce) and exposing the blot in a Stella detector (Raytest). Chemiluminescence quantification was performed by TINA software.
++++3.6++++In vitro binding assay
Binding of synthetic human Aβ42 (Bachem AG) to immobilized recombinant PrP (Zahn et al, 1997) was analyzed by ELISA. Recombinant PrP (recPrP23–231 or recPrP121–231) was immobilized overnight at 4°C on 96-well microtiter plates. Varying concentrations of synthetic human Aβ42 were added to wells and incubated for 1 h. Bound proteins were detected by incubation with 6E10 antibody (Covance) followed by horseradish peroxidase-conjugated antimouse IgG1. Absorbance was measured at 450 nm. For Western blot analysis various concentrations of Aβ42 were incubated in the same conditions, followed by Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and blotting with 6E10 antibody. Binding of human Aβ42 (25 nM) to recPrP23–231 was assessed also in presence of decadic dilutions (100, 10 and 1 nM) of anti-PrP antibodies (Polymenidou et al, 2008).
Histological analyses
Brains were removed and fixed in 4% formaldehyde in phosphate buffered saline, pH 7.5, paraffin embedded and cut into 2–4 µm sections. Sections were stained with hematoxylin–eosin (HE) or antibodies against glial fibrillary acidic protein (GFAP) (DAKO), ionized calcium binding adapter molecule 1 (Iba1; WAKO) and Aβ (4G8; Signet).
Statistical analyses
Statistical significance was determined according to one-way ANOVA followed by Tukey's post-test for multiple comparison, unpaired Student's t-test and Mann–Whitney test using Prism software (GraphPad Software). Error bars in the graphs and numbers following the ± sign denote standard errors of the mean unless otherwise indicated.
Acknowledgments
We thank Dr M. Jucker for APPPS1 mice, Dr B. Chesebro and Dr M.B.A. Oldstone for tg44 mice, Dr S. Hornemann for providing advice on protein purification, P. Schwarz and M. Delic for technical assistance, M. Bieri and N. Wey for software development and Dr F.D. Heitz for helpful comments on the manuscript. This work was supported by grants of the European Union, the Swiss National Research Foundation, the Novartis Foundation, the National Center for Competence in Research ‘Neural Plasticity and Repair’ and an Advanced Investigator Grant of the European Research Council to A.A. A.M.C. is partly supported by the ‘Alzheimer und Depression Fonds der SAMW’. M.N. is partly supported by an investigator fellowship of Collegio Ghislieri, Pavia, Italy.
Supporting information is available at EMBO Molecular Medicine Online.
The authors declare that they have no conflict of interest.
Author contributions
A.M.C. designed the study, organized and maintained the mouse colony, performed biochemical and histologic analyses, analyzed the data and cowrote the paper; M.F. performed electrophysiology experiments, analyzed the data and cowrote the paper; M.N. helped in organizing and maintaining the mouse colony, performed genetic analyses, analyzed the data and cowrote the paper; O.M. performed electrophysiology experiments and analyzed the data; R.M. performed biochemical experiments; J.F. performed biochemical experiments; I.M.M. supervised electrophysiology experiments, analyzed the data and wrote the paper; A.A. designed and coordinated the study, supervised biochemical, genetic and histologic analyses, analyzed the data and wrote the paper.
For more information
Accompanying Closeup:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, et al. Synthetic amyloid-{beta} oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci USA. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- Büeler HR, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci USA. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Gerlai R. Hippocampal LTP and memory in mouse strains: is there evidence for a causal relationship. Hippocampus. 2002;12:657–666. doi: 10.1002/hipo.10101. [DOI] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Farinelli M, Konietzko U, Nitsch RM, Mansuy IM. Abeta oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J Neurosci. 2007;27:7648–7653. doi: 10.1523/JNEUROSCI.0395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman EJ, Kulnane LS, Gao Y, Petriello MC, Pimpis KM, Younkin L, Dolios G, Wang R, Younkin SG, Lamb BT. Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse. Hum Mol Genet. 2003;12:2949–2956. doi: 10.1093/hmg/ddg322. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Tremblay P, Dearmond SJ, Prusiner SB, Nicoll RA. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci USA. 1996;93:2403–2407. doi: 10.1073/pnas.93.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci USA. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, Hafner-Bratkovic I, Jerala R, Hornemann S, Wuthrich K, et al. The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS ONE. 2008;3:e3872. doi: 10.1371/journal.pone.0003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman D, Gao Y, Lamb BT. Genetic loci modulating amyloid-beta levels in a mouse model of Alzheimer's disease. Neurobiol Aging. 2008;29:1190–1198. doi: 10.1016/j.neurobiolaging.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, Van Houtvin T, Esselmann H, Paul S, Schafer MK, Berezovska O, et al. Gamma-secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, Selkoe DJ, Lemere CA, Walsh DM. Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion. 2007;1:83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, et al. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Zabel M, Greenwood C, Thackray AM, Pulford B, Rens W, Bujdoso R. Perturbation of T-cell development by insertional mutation of a PrP transgene. Immunology. 2009;127:226–236. doi: 10.1111/j.1365-2567.2008.02944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn R, von Schroetter C, Wüthrich K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 1997;417:400–404. doi: 10.1016/s0014-5793(97)01330-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.