

Peptide ligands often suffer from poor bioavailability, in part because of the proteolytic degradation in vivo. Previous work has shown that homo-oligomers of N-methyl Phenyl alanine are highly resistant to proteolysis from either chymotrypsin or proteinase K.[1] This work also showed that oligopeptides made with a mixture of Phe and N-methyl Phe retain some protease resistance. Similarly, peptoids, polymers of N-alkyl glycine, show resistance to proteolysis[2] and may enhance affinity and specificity when substituted for proline.[3] Unfortunately, movement of the α-carbon side chain to the amide nitrogen atom often disrupts peptide function. We were interested in strategies that enhance proteolytic stability while preserving the function of the underlying peptide. Toward that end, we have explored the effect of inserting single N-methyl amino acids into functional peptides, a method we refer to as N-methyl scanning mutagenesis. Our hope was to find one or more substitutions that enhance protease resistance while retaining binding affinity.
We chose the G-protein-binding core-motif peptide DKLYWWEFL, which binds the heterotrimeric inhibitory G protein Gαi1*GDP with good affinity (Kd =200 nm), as our model system.[4] Our strategy was to systematically construct nine variants of this peptide bearing a single N-methyl analogue of the natural residue at each position. One challenge we faced was that previous synthetic methods for incorporating N-methyl residues involved long multistep reactions per coupling or resulted in low yields.[5,6] The synthetic difficulty is the result of coupling the secondary amine of an N-methyl peptide.[7] This coupling must go to near completion, in a short period of time to avoid diketopiperazine formation.[6] To couple N-methyl residues we followed a strategy similar to Giralt and co-workers,[6] but used the additive pair O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU)/1-hydroxy-7-azabenzotriazole (HOAt). Coupling each N-methyl amino acid goes to near completion in 20 min in one step with no preactivation for all residues we tested. As a result, we were able to construct the N-methyl peptides reliably with a final yield of 20–25% (Table 1).
Table 1.
N-methyl peptides examined. Peptides were synthesized with and without biotin conjugation at the N terminus. Additionally, five N-terminal Asn residues were added to enhance peptide solubility.
| Compound | Sequence |
|---|---|
| N-Me-D | NNNNN(N-MeD)KLYWWEFL |
| N-Me-K | NNNNND(N-MeK)LYWWEFL |
| N-Me-L8 | NNNNNDK(N-MeL)YWWEFL |
| N-Me-Y | NNNNNDKL(N-MeY)WWEFL |
| N-Me-W10 | NNNNNDKLY(N-MeW)WEFL |
| N-Me-W11 | NNNNNDKLYW(N-MeW)EFL |
| N-Me-E | NNNNNDKLYWW(N-MeE)FL |
| N-Me-F | NNNNNDKLYWWE(N-MeF)L |
| N-Me-L14 | NNNNNDKLYWWEF(N-MeL) |
To investigate the effect of N-methylation on protease activity, the half-life was measured for trypsin cleavage for N-methyl substitutions were examined at the P2, P1, P1′, and P2′ positions (N-Me-D, N-Me-K, N-Me-L8, and N-Me-Y, respectively; Figure 1). Inserting N-methyl residues at these positions results in a dramatic increase in proteolysis resistance, ranging from 72 to > 1000-fold (Figure 1C). To our surprise, the site of N-methyl incorporation does not need to be at the scissile bond or at the trypsin recognition site in order to confer this stability. Indeed, our observations indicate that inserting a single N-methyl residue substantially reduces proteolysis in a four-residue window.
Figure 1.
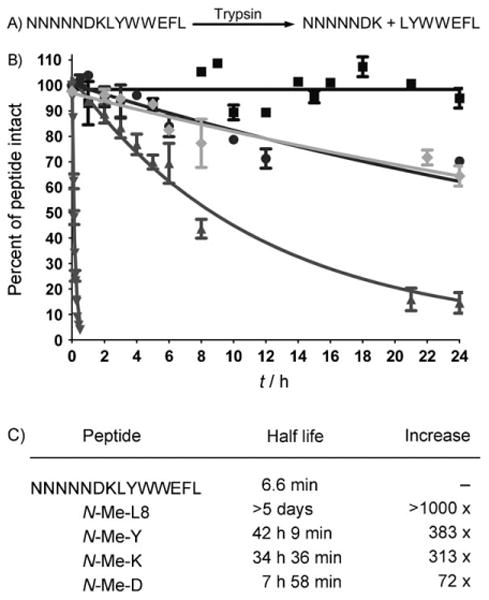
Peptide stability to proteolysis. A) Site of trypsin digestion. B) Proteolysis of unmodified peptide (▼), N-Me-L8 (■), N-Me-K (●), N-Me-Y (◆), and N-Me-D (▲) by trypsin. C) Experimentally determined half life data.
We next worked to determine if our N-methyl insertions altered the peptide binding affinity to Gαi1*GDP. While N-methyl amino acid incorporation generally reduced binding, we observed increased binding affinity for N-Me-K. We quantitated this increase trhough equilibrium competition with the R6A peptide (MSQTKRLDDQLYWWEYL; Kd = 60 nm).[8] This analysis indicated that N-Me-K enhanced binding by 2.5-fold, a ΔΔG of −0.50 kcal mol−1 (Figure 2).
Figure 2.
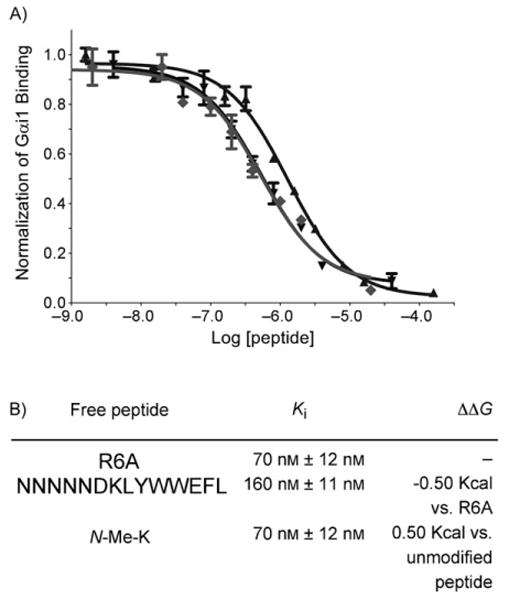
Relative peptide affinities. A) Radiolabeled Gαi1 was incubated with immobilized, biotinylated R6A and varying concentrations of either R6A (▼), unmodified peptide (▲), or N-Me-K (◆) for 3 h. at 4°C. The radiolabel bound was normalized to that which bound in the absence of competitor. The plotted values represent the mean of three experimental values and the error bars correspond to the standard deviation. B) Experimentally determined binding data is shown above. The error is calculated based on the standard error of the mean (SEM) for the log IC50 using GraphPad 5.0.
The crystal structure of R6A in complex with Gαi1*GDP (S. Sprang, A. Adhikari, personal communication) provides insight into our binding data. There, N-methyl substitutions would disrupt many of the peptide–protein interactions or the fold of the bound peptide. From the crystal structure of a similar sequence, we observe that a hydrogen bond is present from the backbone of V (equivalent to L in our peptide) to Gαi1*GDP.[9] This would indicate that N-methyl incorporation at this position would disrupt binding to Gαi1*GDP, which we observed. Additional insight may be inferred by the results of other peptides selected by mRNA display to bind to Gαi1*GDP, for which we have observed a significant conservation of the EFL residues.[10] Any mutations are subtle, E to D or F to Y, for example. Therefore, it would be suspected that N-methyl incorporation at any of these positions would be deleterious to peptide binding. This was experimentally confirmed here for our peptide binding to Gαi1*GDP.
The Q residue in R6A (equivalent to K in our peptide) adopts phi and psi angles of −60 and 128, respectively. Previous work indicated that peptoids have a limited number of energetically favorable conformations.[11] It is likely that the incorporation of an N-methyl amino acids results in a conformation similar to an optimal orientation for Gαi1 binding. This leads us to a conformational hypothesis for N-methylation. Assuming no unfavorable steric interactions, we would predict that N-methyl amino acid incorporation will be favorable or neutral when Phi, Psi angles are similar to those found in peptoids.[11] This heuristic should provide a good starting point for N-methyl amino acid modifications that improve peptide stability without impairing function. Further structural analysis could be beneficial and will be carried out in future work.
Finally, we wanted to see if N-methyl incorporation had altered the specificity of our peptides. Members of the peptide library were tested for binding against nine members of the G-protein family known to have various affinities to R6A. Five of the nine N-methyl peptide variants showed a dramatic increase in specificity (see the Supporting Information). One variant showed specific affinity for Gα12 over Gαi1 which the original selection targeted (Figure 3). This results in the first peptide ligand able to specifically bind to Gα12.We have shown that the incorporation of N-methyl amino acids can dramatically increase the function and stability of a peptide selected by mRNA display. Selective incorporation of N-methyl amino acids into peptides with a Phi, Psi angle similar to those stated for N-Me-K would likely have the same benefits from their incorporation. This relative synthetic ease and predictability makes incorporating of N-methyl amino acids into peptides derived from selections a practical first step in increasing bioavailability.
Figure 3.
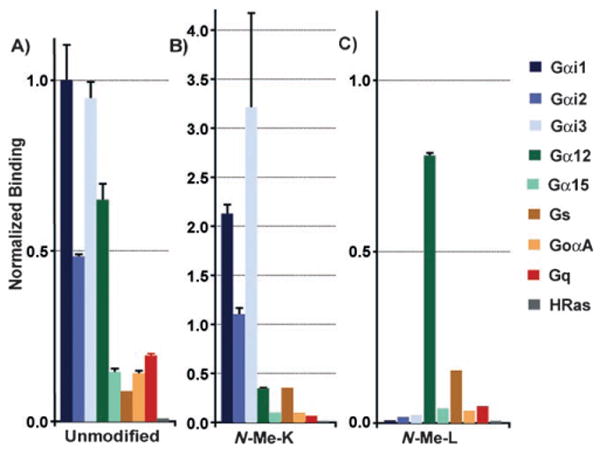
Binding of radiolabeled Gα subunits to immobilized peptides. Gαi1, Gαi2, Gαi3, Gq, Gs, GoαA, Gα12, and Gα15 pcDNA were translated into 35S-Met-labeled proteins using TnT-coupled transcription/translation system (Promega). Translation reactions were desalted and applied to A) Unmodified peptide, B) N-Me-K, and C) N-Me-L8 immobilized to neutravidin-agarose. Values represent the mean value from three experiments, and the error bars represent the standard deviation.
Experimental Section
Peptide synthesis
All solvents were purchased from Sigma. All peptides except N-Me-L14 were synthesized by manual solid-phase peptide synthesis with preloaded Leu-2-chlorotrityl resin (250 mg, 0.15 mmol; Anaspec). N-Me-L14 was synthesized after loading 2-chlorotrityl resin (Anaspec) with Fmoc-N-methyl leucine (Fmoc = 9-fluorenylmethoxycarbonyl; Anaspec). Five N-terminal Asn residues were added to enhance peptide solubility. Loading was accomplished by adding Fmoc-N-Me-L14 to 2-chlorotrityl resin (5 equiv; 250 mg, 0.35 mmol) in DMF with N,N-diisopropylethylamine (DIEA; 5 equiv) for 2 h. Standard couplings were carried out with monomer (5 equiv; Novabiochem) in HATU (2 mL, 0.6 mmol; Novabiochem), HOAt (1.2 mmol; Genescript) in DMF with DIEA (1.8 mmol) at room temperature for 15 min. Coupling to an N-methyl amino acid followed the same procedure with a 30 min coupling time. Fmoc deprotection was carried out with 20% piperidine (Anaspec) at room temperature for 15 min. Following, deprotection, cleavage with 95% TFA, filtration and ether extraction, the crude product was purified on a Vydac C-18 reversed-phase column using gradient elution (0% B for 5 min, 10–50% B in 40 min. Solvent A: H2O with 0.1% TFA, Solvent B: CH3CN with 0.035% TFA). Lyophilized solid was reconstituted in DMSO and quantitated by absorbance at 280 nm (ε280 = 12490 L mol−1 cm−1). Yield = 20–25%.
Peptides were characterized by HPLC and MALDI-TOF. Peptides were run on a Vydac C-18 column using gradient elution (5–95% B). Buffers were the same as described above. Absorbance at 280 nm was recorded for biotin-labeled peptides excluding Bio-N-Me-D, Bio-R6A and Bio-unmodified peptide. Absorbance at 215 nm was recorded for the remaining peptides. The peak around 4 min for peptides at 215 nm was the solvent peak. The next major peak (or first major peak in the case of 280 nm recording) was collected and analyzed by MALDI-TOF.
Binding affinity of the N-methyl peptide library
TnT pulldown experiments (Promega) were carried out as described in ref. [4] with some modifications. Human cDNA clones encoding G proteins with the pcDNA3.1+ vector (Invitrogen) were obtained from the UMR cDNA Resource Center (http://www.cdna.org). The DNA clones for Gαi1, and HRas were translated into 35S-Met-labeled proteins using a TnT-coupled transcription/translation system (Promega). Translation reactions were desalted with G-25 spin columns (GE Healthcare), and the translation yield was calculated by trichloroacetic acid (TCA) precipitation. Equal amounts of each labeled subunit were added to neutravidin-agarose (5 μL) containing prebound biotin-labeled peptide (2 nmol) in HBS-EP buffer (1 mL). Control neutravidin-agarose beads were loaded with (d)-biotin (Anaspec). Binding reactions proceeded for 2 h at 4°C followed by filtration and washing with HBS-EP. The matrix was analyzed by scintillation counting and the percent bound was determined by the matrix counts divided by the total counts as determined by TCA precipitation. Values were normalized to the percent bound of unmodified peptide.
R6A-1 competition/equilibrium binding
R6A (MSQTKRLDDQLYWWEYL) and Bio-R6A (Bio-MSQTKRLDDQLYWWEYL) were synthesized as described above. This peptide was immobilized to NeutrAvidin beads (Promega) using the manufacturer's instructions. Gαi1 was expressed using the TnT reticulocyte lysate system (T7 promoter, Promega). (250 ng) plasmid DNA and (25 μCi) l-35S-Met (MP Biomedicals) were used in an (25 μL) expression. Gαi1 translations were desalted using MicroSpin G-25 columns (GE healthcare) into modified HBS-EP buffer: HEPES (10 mm, pH 7.4), NaCl (150 mm), EDTA (3 mm), polysorbate 20 (0.005%; Tween 20), MgCl2 (8 mm), GDP (30 μm), and BSA (0.05% (w/v)). Specific activity was measured by TCA precipitation.
Immobilized R6A (400 pmol) and Gαi1 (100000 cpm) were suspended in HBS-EP buffer (1 mL). Varying quantities of free peptide (R6A, unmodified peptide, or N-Me-K) were added. The final DMSO concentration was adjusted to 0.5%. Samples were rotated for 3 h at 4°C followed by brief centrifugation and supernatant removal. Samples were washed four times by resuspension in modified HBS-EP buffer at 4°C, centrifugation, and supernatant removal. After the final wash, the immobilized sample was transferred to scintillation vials and analyzed by scintillation counting. CPM values were normalized by dividing by the average value obtained in the absence of added peptide. Binding analysis was performed by GraphPad Prism 4.0.
N-methyl selectivity
Gαi1, Gαi2, Gαi3, Gq, Gs, GoαA, Gα12, and Gα15 pcDNA were translated into 35S-Met-labeled proteins using the TnT-coupled transcription/translation system (Promega). Translation reactions were desalted using G-25 spin columns (GE Healthcare) and applied to peptide immobilized to neutravidin-agarose as previously described. The immobilized peptides were N-Me-D, N-Me-K, N-Me-L8, N-Me-Y, N-Me-W10, N-Me-W11, N-Me-E, N-Me-F, or N-Me-L14 as illustrated. Binding was allowed to proceed for 3 h at 4°C after which the resin was filtered and washed. Scintillation counting of the resin yielded the cpm bound, which was normalized as detailed in the Supporting Information. Values represent the mean value from three experiments and the error bars represent the standard deviation. Binding analysis and normalization performed as outlined in R6A-1 competition/equilibrium binding.
Protease resistance experiment
Unmodified peptide, N-Me-D, N-Me-K, and N-Me-L8 (7.5 mm) in DMSO (2 μL) were added to buffer (98 μL) containing Tris (50 mm), CaCl2 (20 mm), pH 7.75. This was used to reconstitute modified trypsin (20 μg; New England Bio-labs). The reaction proceeded at 37 °C for either 30 min (unmodified peptide) or 24 h (N-Me-D, N-Me-K, N-Me-L8, N-Me-Y). An aliquot (8 μL) was removed for each time point and diluted in ACN (25 μL). Samples were flash frozen until analysis was to be performed. At this time 0.1 % TFA in H2O (67 μL) was used to dilute the samples to 100 μL. The sample was injected onto a C-18 reversed-phase column and separated by gradient elution (5–90% B in 10 min. Solvent A: H2O with 0.1% TFA, Solvent B: CH3CN with 0.035% TFA). The area under the starting material peak was quantitated using the 32 KaratGold Software package (Beckman). The plotted values represent the mean of three experimental value and the error bars represent the standard error of the mean. The graph was generated by fitting the data to a one-phase exponential decay equation (GraphPad Prism).
Supplementary Material
Acknowledgments
The authors would like to thank Steven W. Millward from The California Institute of Technology and Ryan Austin from The University of Southern California for consultation on this project. This work was supported by NIH grant NIH GM R01 60416 and NIH GM R21 76678.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Frankel A, Millward SW, Roberts RW. Chem Biol. 2003;10:1043–1050. doi: 10.1016/j.chembiol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg Med Chem Lett. 1994;4:2657–2662. [Google Scholar]
- 3.Nguyen JT, Turck CW, Cohen FE, Zuckermann RN, Lim WA. Science. 1998;282:2088–2092. doi: 10.1126/science.282.5396.2088. [DOI] [PubMed] [Google Scholar]
- 4.Ja WW, Adhikari A, Austin RJ, Sprang SR, Roberts RW. J Biol Chem. 2005;280:32057–32060. doi: 10.1074/jbc.C500319200. [DOI] [PubMed] [Google Scholar]
- 5.Biron E, Kessler H. J Org Chem. 2005;70:5183–5189. doi: 10.1021/jo050477z. [DOI] [PubMed] [Google Scholar]
- 6.Teixido M, Albericio F, Giralt E. J Pept Res. 2005;65:153–166. doi: 10.1111/j.1399-3011.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- 7.DiGioia ML, Leggio A, Liguori A. J Org Chem. 2005;70:3892–3897. doi: 10.1021/jo0478959. [DOI] [PubMed] [Google Scholar]
- 8.Ja WW, Roberts RW. Biochemistry. 2004;43:9265–9275. doi: 10.1021/bi0498398. [DOI] [PubMed] [Google Scholar]
- 9.Johnston CA, Siderovski DP. Proc Natl Acad Sci USA. 2007;104:2001–2006. doi: 10.1073/pnas.0608599104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Millward SW, Fiacco S, Austin RJ, Roberts RW. ACS Chem Biol. 2007;2:625–634. doi: 10.1021/cb7001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Spellmeyer DC, Tan R, Frankel AD, Santi DV, Cohen FE, Bartlett PA. Proc Natl Acad Sci USA. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.