

Abstract
For the human pathogen Leishmania major, a key metabolic function is the synthesis of thymidylate, which requires 5,10-methylenetetrahydrofolate (5,10-CH2-THF). 5,10-CH2-THF can be synthesized from glycine by the mitochondrial glycine cleavage complex (GCC). Bioinformatic analysis revealed the four subunits of the GCC in the L. major genome, and the role of the GCC in parasite metabolism and virulence was assessed through studies of the P subunit (glycine decarboxylase (GCVP)). First, a tagged GCVP protein was expressed and localized to the parasite mitochondrion. Second, a gcvP− mutant was generated and shown to lack significant GCC activity using an indirect in vivo assay after incorporation of label from [2-14C]glycine into DNA. The gcvP− mutant grew poorly in the presence of excess glycine or minimal serine; these studies also established that L. major promastigotes require serine for optimal growth. Although gcvP− promastigotes and amastigotes showed normal virulence in macrophage infections in vitro, both forms of the parasite showed substantially delayed replication and lesion pathology in infections of both genetically susceptible or resistant mice. These data suggest that, as the physiology of the infection site changes during the course of infection, so do the metabolic constraints on parasite replication. This conclusion has great significance to the interpretation of metabolic requirements for virulence. Last, these studies call attention in trypanosomatid protozoa to the key metabolic intermediate 5,10-CH2-THF, situated at the junction of serine, glycine, and thymidylate metabolism. Notably, genome-based predictions suggest the related parasite Trypanosoma brucei is totally dependent on the GCC for 5,10-CH2-THF synthesis.
Parasites of the trypanosomatid genus Leishmania are the cause of leishmaniasis, a group of diseases ranging from relatively mild cutaneous lesions to the disfiguring mucocutaneous manifestation and lethal visceral disease. In vertebrates, Leishmania reside within macrophages as non-motile amastigote forms and multiply within acidified phagolysosomes. They are transmitted by sand flies, where they grow as the flagellated promastigote stage, and ultimately differentiate into vertebrate-infective metacyclic forms. Current therapy is based on antimonials and is generally effective but suffers from toxicity, the requirement for parenteral administration, and prolonged courses of treatment (1). Furthermore, in some parts of India visceral leishmaniasis is commonly resistant to antimonials (2). Miltefosine is a promising orally effective treatment, but resistance to this drug appears to arise rapidly (3). New drug targets are, therefore, urgently required.
An established target for antimicrobial chemotherapy is dihydrofolate reductase (DHFR).2 DHFR inhibition kills cells by preventing recycling of dihydrofolate, a product of the conversion of deoxyuridylate to thymidylate by thymidylate synthase (TS). In protozoa and plants, DHFR and TS are present together in a bifunctional enzyme (4, 5) and, depending on the species, are located in several cellular compartments, including the cytoplasm, plastid, and mitochondrion (6–8). Leishmania mutants lacking DHFR-TS are thymidine auxotrophs and fail to survive after animal infection (9), providing a genetic validation of this pathway as a potential target for chemotherapy. However, in Leishmania, antifolate therapy is compromised by the presence of pteridine reductase 1, which is less sensitive to classical antifolates and can bypass DHFR inhibition (10, 11). Notably, compounds active against both pteridine reductase 1 and DHFR are highly toxic toward Leishmania, and some dual-target compounds also appear to inhibit a third target (11). Together these data suggest that other folate-dependent enzymes including ones participating in thymidylate synthesis may be logical targets for pharmacological intervention.
TS utilizes 5,10-methylene-tetrahydrofolate (5,10-CH2-THF), a key metabolite in 1-carbon metabolism. 5,10-CH2-THF additionally leads through methylene-tetrahydrofolate reductase to the formation of 5-methyl tetrahydrofolate, required for methionine synthesis and methylation reactions, and through the bifunctional methylene tetrahydrofolate dehydrogenase-methenyl-tetrahydrofolate cyclohydrolase to the synthesis of 10-formyl-tetrahydrofolate (Fig. 1A). In most organisms 10-formyl-tetrahydrofolate is required for purine as well as mitochondrial formyl-methionyl-tRNA synthesis. However, Leishmania is auxotrophic for purines (12), and the genomes for several trypanosomatids lack identified homologs of the 10-formyl-THF-utilizing enzymes involved in purine synthesis (13–15). 5,10-CH2-THF is made in the cytosol by serine hydroxymethyltransferase (SHMT) and in the mitochondrion primarily through the activity of SHMT and the glycine cleavage complex (GCC). In Leishmania and many eukaryotes, SHMT is encoded by two different genes specifying the cytosolic and mitochondrial forms (16). Glycine and in some cases serine are, therefore, important precursors to 1-carbon-THF metabolites in trypanosomatids.
FIGURE 1.

A, 5,10- CH2-THF formation by the glycine cleavage complex and related folate metabolism in Leishmania. Solid lines indicate direct interconversion of THF and 5,10-CH2-THF (and serine and glycine). The GCC (reaction 1) converts glycine and THF to 5,10-CH2-THF, NH3, and CO2 (NAD+ is also converted to NADH). 5,10-CH2-THF is also formed by SHMT (reaction 2), which is reversible. Dotted and dashed lines indicate indirect routes of THF to 5,10-CH2-THF conversion via dihydrofolate (DHF) and the activity of bifunctional DHFR-TS (reactions 3D and 3T, dotted line) or via 10-formyl-tetrahydrofolate (10-CHO-THF) and 5,10-methenyl-tetrahydrofolate (5,10-CH=THF) using the activity of reversible bifunctional methlenetetrahydrofolate dehydrogenase (4D)-5,10-methenyl-tetrahydrofolate cyclohydrolase (4C) and the activity of 10-formyl-tetrahydrofolate synthase (5) (dashed line). The pathway for incorporation of radiolabel from the 2 position of glycine into DNA is marked by asterisks. B, diagram of glycine cleavage complex. P protein decarboxylates glycine and transfers the amino-methylene moiety to lipoate arm of H protein. T protein releases ammonia and transfers the methylene group to THF, forming 5,10-CH2-THF. The H protein is re-oxidized by lipoate dehydrogenase (L) with concurrent production of NADH.
The GCC comprises four loosely associated subunits, the P, H, T, and L proteins, which typically occur in nonstoichiometric levels (17, 18). The mode of action of these subunits is outlined in Fig. 1B. The GCC has been found in bacteria, plants, yeast, and vertebrates (18–20). In photosynthetic tissues of plants, the GCC consumes large amounts of glycine produced during photorespiration (18), but the activity is still essential in Arabidopsis even under non-photorespiratory conditions (21). In Saccharomyces cerevisiae, mutants lacking any of the GCC-specific subunits can grow on minimal medium but cannot use glycine as a nitrogen source (20).
In several recent global screens of intracellular pathogens during experimental infections, GCC mutants with attenuated virulence have been identified (22–24). Thus, given its importance in parasite metabolism and in the development of improved anti-parasitic chemotherapy and potential involvement in virulence, we investigated the role of the GCC in Leishmania major, focusing on the study of mutants lacking the GCC P subunit (glycine decarboxylase or GCVP; gcvP− mutants). Although dispensable for growth in vitro, gcvP− promastigotes were hypersensitive to the antifolate methotrexate and to excess glycine. Infectivity tests showed that, whereas gcvP− mutants grow normally within macrophages in vitro, they show an unusual pattern of attenuated virulence in experimental animals in vivo.
EXPERIMENTAL PROCEDURES
Parasites, Culture Media, and Drug Testing
L. major strain Friedlin clone V1 (MHOM/IL/81/Friedlin) was cultured as promastigotes in M199 medium as described (25) with the addition of 1 μg ml−1 biotin and 2 μg ml−1 biopterin. RPMI medium, used in nutritional and DNA-labeling studies, was prepared after a standard formulation (Invitrogen), except that L-serine was added to 95 μM (10 mg liter−1), MgCl2 (0.1 g liter−1) was substituted for MgSO4, and the medium was supplemented with 40 mM HEPES, pH 7.4, 2 μg ml−1 biopterin, 100 μM adenine, 5 μg ml−1 hemin, and 1% (v/v) heat-inactivated fetal bovine serum. All lines required several passages to adapt to normal growth in this medium after transfer from M199 medium. Cells were counted using a hemocytometer (RPMI medium experiments) or a model Z1 Coulter counter (M199 medium experiments). Growth inhibition was determined by seeding parasites at 5 × 105 cells ml−1 at various concentrations of drug and determining cell density when control cultures reached late-log phase. The EC50 value is the concentration of drug required to decrease growth by 50%. Methotrexate and O/129 (2,4-diamino-6,7-diisopropylpteridine) were purchased from Sigma; CB3717 (2-[(4-{[(2,4-diaminoquinazolin-6-yl)methyl](prop-2-yn-1-yl)amino}phenyl)formamido]-pentanedioic acid (26)) was a gift of Ann Jackman, Institute of Cancer Research, Surrey, UK; the folate analogues compound 34 (2,4-diamino-6-(3,4-dichlorophenoxy)-quinazoline) and compound 70 (2,4-diamino-6-benzyl-5-(3-phenylpropyl)-pyrimidine) were as described (11). RPMI low-serine medium for macrophages was prepared the same as the Leishmania RPMI medium, with the addition of 1 mM sodium pyruvate and 10% (v/v) heat-inactivated fetal bovine serum and the omission of serine, biopterin, adenine, and hemin.
Infectivity Studies
Before virulence tests, all lines were passed through mice once by injecting hind footpads of Balb/c mice (Charles River Laboratories, Wilmington, MA) with a large inoculum (1 × 106) of stationary-phase parasites and recovering parasites by needle aspiration of the footpad regardless of pathology 3–5 weeks afterward. Lines were passaged no more than 6 times in vitro before virulence tests, and parasite inocula were grown in the absence of selective drugs. Mouse and macrophage infections were performed as described (27). Mice were infected with 105 metacyclic promastigotes or 104 amastigotes. Macrophages were infected at a ratio of 5 metacyclics or 1 amastigote per macrophage. Metacyclics (for mouse infection) were isolated from stationary-phase cultures of promastigotes by negative selection with peanut agglutinin (28) or (for macrophage infections) by Ficoll gradient centrifugation (29). Amastigotes were isolated from infected mice showing lesions with thicknesses of 1–2 mm (27). Amastigotes in lesions were quantified using a limiting dilution assay (30), with dilutions made in liquid M199 medium.
DNA Manipulations
The open reading frame of the candidate L. major GCVP gene LmjF26.0030 was PCR-amplified in two pieces using KlentaqLA (DNA Polymerase Technology) and primers B2057 and B2062 and primers B2063 and B2058 (primer sequences are listed in supplemental Table S1). After digestion with SacI and BamHI, the fragments were ligated into the BamHI site of pBluescript SK− (yielding pBS-GCVP; laboratory strain B5086). Relevant portions of all constructs were confirmed by sequencing.
A gene disruption approach was used to inactivate the GCVP gene. An autonomous puromycin drug-resistance cassette was excised from pXGPAC (strain B3325) by digestion with XhoI and DdeI, filled in with Klenow fragment, and inserted into pBS-GCVP cut at a unique PmlI site in the GCVP open reading frame, yielding pBS-GCVP-PAC (strain B5121).
The disrupted gene fragment was excised from pBS-GCVP-PAC by digestion with BamHI and transfected into wild-type (WT) L. major promastigotes as described (31). Presumptive heterozygous clonal lines (GCVP/gcvP::PAC) were isolated by plating on semisolid M199 medium containing 1% Noble agar and 33 μg ml−1 puromycin. Lines bearing the planned PAC insertion were confirmed by PCR using a forward primer located 5′ of the GCVP open reading frame targeting fragment (B2137) with a reverse primer located within the PAC gene (B2013, Fig. 2A). Eight independent heterozygotes were obtained (formally gcvP::PAC/+), two of which were inoculated into mice and found to retain infectivity. For disruption of the second allele, pBS-GCVP-HYG (strain B5119) was created by excising an autonomous hygromycin B resistance cassette from pX63-HYG vector (strain B617) (9) by digestion with BamHI and PvuII. The resistance cassette was filled in and inserted into pBS-GCVP as described above. The disrupted gene was excised by digestion with BamHI and transfected as described above into heterozygote clone 8, and clonal lines were isolated on plates containing 20 μg ml−1 each puromycin and hygromycin. Gene disruption was evaluated first by PCR for retention of the PAC disruption as described above and by PCR for the HYG insertion using primer B2137 and a reverse primer within the HYG gene (B1675).
FIGURE 2. GCVP gene disruption.
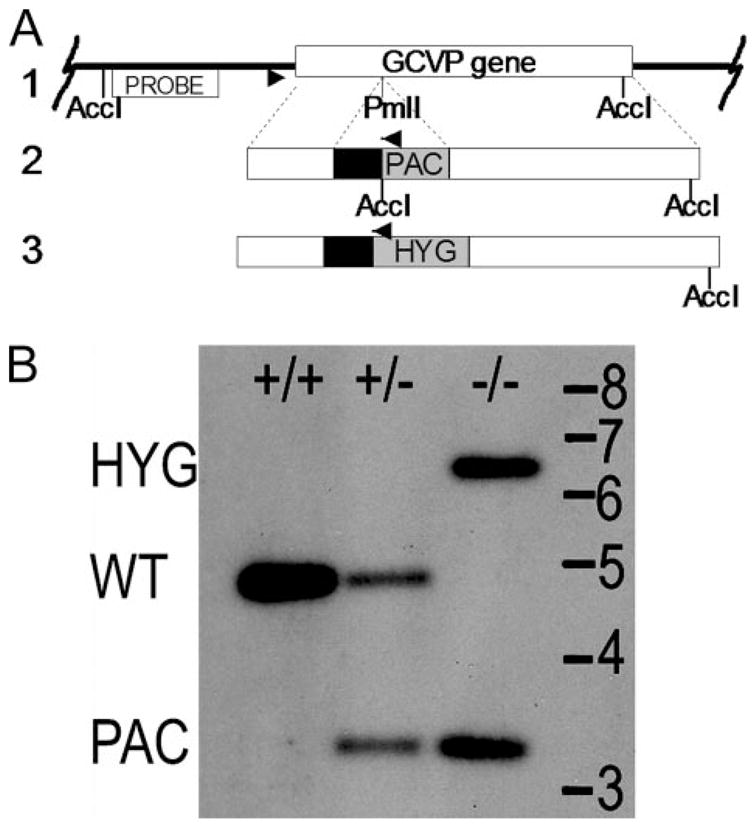
A, gene disruption scheme. Line 1 shows the GCVP gene (open box) in its chromosomal context. Lines 2 and 3 show GCVP disruption constructs. Disruption constructs consisted of the complete GCVP gene interrupted at a PmlI site 734 nucleotides from the 5′ end by autonomous drug resistance cassettes (PAC or HYG, 1.2 or 1.5 kb) consisting of the PAC or HYG open reading frame (gray boxes) whose expression was driven by a segment of 5′-flanking DNA bearing a functional splice acceptor (black boxes). Disruption cassettes were excised from their parental plasmids (pBS-GCVP-PAC and pBS-GCVP-HYG) and introduced into parasites successively. The locations of PCR primers used to confirm the homologous replacement of a WT allele with the PAC or HYG “disruptant” alleles are shown (arrowheads) as is the location of AccI sites and the hybridization probe used in the Southern blot shown in panel B. Dotted lines indicate areas of potential homologous recombination. B, Southern blot of GCVP disruption. DNA from parasite lines digested with AccI was blotted and hybridized with a probe matching the region of DNA indicated in panel A. Lane +/+, WT DNA; lane +/−, WT/gcvP::PAC heterozygote DNA; lane −/−, gcvP::PAC/gcvP::HYG double disruption (gcvP−). Positions of standards (in kb) and WT and HYG- and PAC-disrupted bands are shown.
Putative double disruptants were confirmed by Southern blot analysis using genomic DNA digested with AccI and a radiolabeled probe consisting of a 900-bp fragment located upstream of the GCVP-targeting fragment, derived by PCR using genomic DNA template and primers B2184 and B2185 (Fig. 2A). Genomic DNA was isolated using a LiCl-Triton X-100 method (32). Multiple double disruptants were obtained (formally gcvP::PAC/gcvP::HYG, referred to as gcvP− in this work); gcvP− clone #1 was used in all experiments reported here. Two other clones were used in mouse and macrophage infection experiments not shown with similar results.
To restore or overexpress the GCVP gene, the GCVP open reading frame was cut from pBS-GCVP with BamHI and inserted into the BamHI expression site of the pXG plasmid (strain B1288 (33)), creating pXG-GCVP (strain B5124). gcvP− or WT L. major were transfected with pXG-GCVP, and clones were isolated from plates containing 10 or 12.5 μg ml−1 G418; the presence of the plasmid was confirmed by re-isolating it from Leishmania, transforming Escherichia coli and verifying the plasmid by restriction digestion. These lines are referred to as gcvP−/+GCVP (formally gcvP::PAC/gcvP::HYG [pXG-GCVP]) or WT/+GCVP (formally +/+[pXG-GCVP]. gcvP−/+GCVP clone 1.1 was used for metabolic studies, and clone 1.6 was used in virulence tests. WT/+GCVP clone 1 was used in DNA labeling experiments.
For construction of a C-terminal GCVP-GFP fusion, we used pXG−/GFP+ (strain B2863) (33), which has a BamHI site upstream of a GFP cassette lacking an ATG, facilitating the creation of in-frame gene fusions. First, the 3′ 1.1 kb of GCVP was modified to replace the stop codon with a BglII site by PCR amplification with pBS-GCVP as template and primers B2086 and B2497 and inserted by TA cloning into pGEM-T (Promega) (to make strain B5745). pXG-GCVP-GFP was generated by a three-way ligation of the 0.7-kb NotI and BglII fragment of B5745 (bearing the modified GCVP 3′ region), a 2.2-kb BamHI-NotI fragment of pBS-GCVP (5′ region), and BamHI-cut and dephosphorylated pXG−/GFP+ to create pXG-GCVP-GFP (strain B5760). WT transfectants bearing this construct were generated as described above for pXG-GCVP and are termed WT/+pXG-GCVP-GFP (formally, +/+[pXG-GCVP-GFP]); two clonal lines were obtained with similar properties, and the results with clone 1 are shown.
Microscopy
WT/+pXG-GCVP-GFP promastigotes were stained with 5 μg/ml Hoechst 33342 (Vector Laboratories) for 30 min in phosphate-buffered saline at room temperature, washed in phosphate-buffered saline, fixed with 0.3% (w/v) formaldehyde, and observed by epifluorescence microscopy. In some studies, 40 nM Mitotracker Red 580 (Invitrogen) was added at the same time as the Hoechst dye.
Indirect GCC Assay
GCC activity was assessed by an indirect 14C incorporation assay in vivo. L. major promastigotes were grown in RPMI medium containing 27 μM (2 mg l−1) glycine. Labeling cultures were started at 3 × 105 cells ml−1, with each 10-ml culture containing 1 μCi of [2-14C]glycine (3.7 Ci mol−1; American Radiolabeled Chemicals). These were grown to a final density of 2–4 × 107 ml−1, harvested, washed twice in phosphate-buffered saline, and suspended in 0.5 ml lysis buffer consisting of 10 mM Tris-HCl pH 8.0, 25 mM EDTA, 150 mM sodium chloride, 0.5% (w/v) sodium dodecyl sulfate, and 20 μg ml−1 RNase. Lysates were incubated for 5 h at 37 °C before adding 0.1 mg ml−1 proteinase K and incubation for a further 16 h. Protein was extracted twice with phenol/chloroform/isoamyl alcohol (25:24:1), and DNA was precipitated with 2 volumes of ethanol and 0.1 volume of 3 M sodium acetate, pH 5.2. The pellet was washed twice with 70% ethanol, dried, and resuspended in 10 mM Tris-HCl, 500 μM EDTA, pH 8.0, containing 20 μg ml−1 RNase. DNA was quantified by UV absorption and 14C by scintillation counting. In WT cells less than 1% cellular labeling was found in purified DNA. Statistical significance was assessed using two-tailed t test.
RESULTS
GCVP Gene Comparison and GCVP Localization
Data base searches of the L. major genome (34) revealed strong candidates for all subunits of the GCC complex, including the T protein (two identical genes, LmjF36.3800 and LmjF36.3810), the H protein (LmjF35.4720), the L protein dihydrolipoamide dehydrogenase (LmjF32.3310), and the P protein (LmjF26.0030). In this work we focus on the P protein, predicted to encode a protein of 972 amino acids showing 48–50% identity to the GCVPs of Arabidopsis thaliana, bacteria, the slime mold Dictyostelium, yeast, and mammals. An alignment of the predicted L. major GCVP protein with those from other species is shown in supplemental Fig. S1. Structural studies of Thermus thermophilus GCVP identified 23 key active site residues, 20 of which were fully conserved in other GCVP proteins (35). These 20 residues are also present in L. major GCVP (supplemental Fig. S1), including the catalytic lysine (K721 in L. major). Seven conserved basic residues involved in H-protein interaction (35) are also present in the L. major sequence (supplemental Fig. S1).
In other organisms the GCC is located in the mitochondrion, and correspondingly, an N-terminal mitochondrial signal peptide of 21 amino acids was predicted for L. major GCVP by TargetP (36) (supplemental Fig. S1). Our analysis also predicted mitochondrial transit peptides for the other three GCC subunit open reading frames (data not shown). To confirm the mitochondrial localization prediction for the GCVP, we expressed a GCVP fusion protein bearing a C-terminal GFP reporter in L. major promastigotes. Leishmania and other trypanosomatids possess a single mitochondrion, which extends throughout the cell (37), and the fluorescence pattern of the GCVP-GFP-expressing cells showed a meshwork pattern typical of mitochondrially targeted proteins in Leishmania (Fig. 3, left panel). Hoechst 33342 staining was used to visualize the nucleus and kinetoplast (containing the parasite’s mitochondrial DNA network; Fig. 3, center panel). GFP fluorescence overlapped the kinetoplast but not the nucleus and did not show the perinuclear staining typical of ER proteins (Fig. 3, right panel). Mitochondrial localization was further confirmed by colocalization of GFP fluorescence with a red fluorescent mitochondrial dye, Mitotracker Red 580 (result not shown).
FIGURE 3. Localization of GCVP-GFP fusion protein.
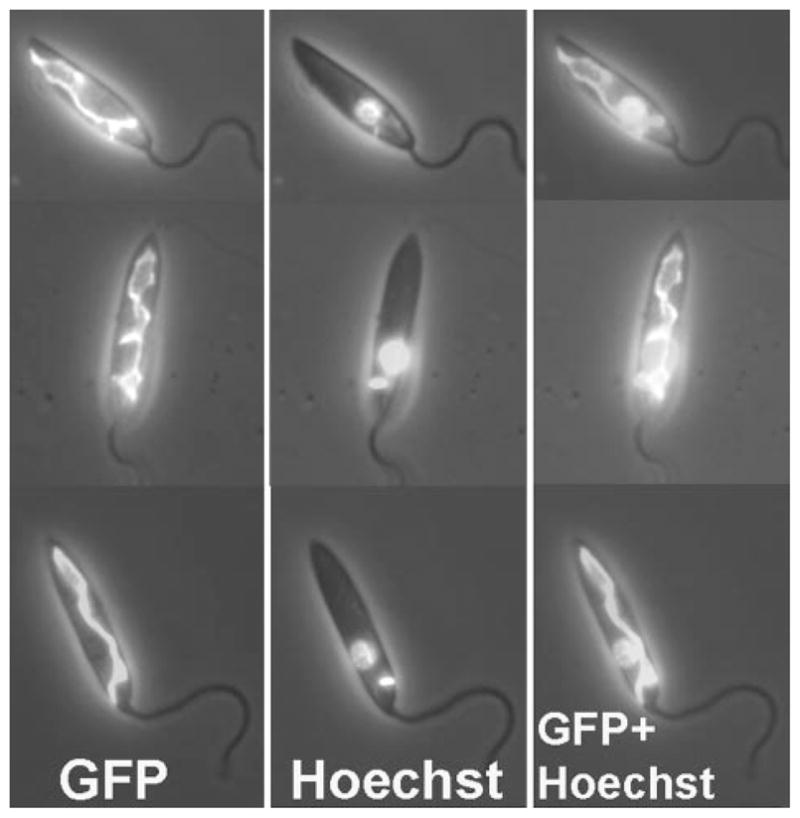
L. major WT-[pXG-GCVP-GFP] promastigotes expressing GCVP-GFP from the pXG-GCVP-GFP plasmid counter-stained with Hoechst 33342. Left panel, GFP fluorescence. Center panel, Hoechst fluorescence. Right panel, GFP and Hoechst fluorescence combined. All fluorescence images are shown overlaid on the phase-contrast image of the parasites.
GCVP Disruption
The Leishmania genome is predominantly diploid, and we used an insertional inactivation strategy to successively mutate both GCVP alleles in L. major, producing a gcvP− parasite (Fig. 2A). Successful disruption was demonstrated by PCR and Southern blot analysis (Fig. 2B or data not shown). To control for nonspecific effects arising from transfection and in vitro culture, we restored expression of GCVP in the gcvP− mutant by introduction of an episomal expression vector (pXG-GCVP), yielding the line gcvP−/+GCVP. Additionally, for effects arising from GCVP overexpression from multicopy episomal vectors, pXG-GCVP was also introduced into WT parasites, yielding a line termed WT/+GCVP.
The gcvP−, gcvP−/+GCVP, or WT/+GCVP lines showed no obvious phenotype during growth in M199 media and grew at the same rate and to the same stationary phase density (~5 × 107 cells/ml) as WT. Upon entry into stationary phase, gcvP− differentiated into peanut agglutinin-negative infectious metacyclic forms at a frequency similar to WT (5 ± 2 versus 5 ± 3%; n = 5).
Indirect Assay of GCC Activity in gcvP− Mutants
Attempts to measure GCC enzymatic activity directly using a conventional assay after the formation of 5,10-14CH2-THF (as [14C]formaldehyde) from 2-14C-labeled glycine were unsuccessful despite testing many different crude promastigote lysates and mitochondrial fractions under a range of conditions. Thus, we developed an indirect in vivo assay based upon the specific production of 5,10-14CH2-THF from 2-14C-labeled glycine via the action of the GCC followed by the incorporation of label into thymidylate (Fig. 1A) and DNA and, ultimately, quantification of the specific activity of labeled DNA. As anticipated, GCC activity was greatly decreased in the gcvP− line (20% WT). Although the residual activity might reflect a second GCC-like activity, the level of incorporation seen in the gcvP− mutant is very close to the experimental background of this assay (see legend to Table 1). GCC activity was restored partially (41%) upon re-expression of GCVP in the gcvP−/+GCVP cells (Table 1).
TABLE 1. Incorporation of label from [2-14C]glycine into DNA by transgenic L. major bearing modifications at the GCVP locus.
Values reflect the average of eight determinations in two independent experiments.
| Line | cpm/μg of DNAa (n = 8) | Percent (vs. WT) | p value |
|---|---|---|---|
| WT | 41 ± 13 | 100 | |
| gcvP− | 8 ± 2 | 20 | 0.0002 (vs. WT) |
| gcvP−/+GCVP | 17 ± 3 | 41 | 0.01 (vs. WT) 0.00002 (vs. gcvP−) |
| WT/+GCVP | 38 ± 4 | 93 | 0.59 (vs. WT) |
Machine background (44–49 cpm) was subtracted from counts. Counts (including background) for gcvP− samples ranged from 105 to 150 cpm.
Transfection of WT cells with pXG-GCVP did not increase labeling of DNA by [2-14C]glycine (Table 1). Because GCC activity is mediated by four proteins in other organisms that occur at nonstoichiometric levels (18), this suggests that the glycine decarboxylation reaction was not rate-limiting. Other explanations include various forms of post-transcriptional regulation, as often seen in Leishmania (38). Because the GCC is located in the mitochondrion, potentially only the mitochondrial (kinetoplast) DNA would be labeled after the cleavage of [2-14C]glycine. However, preliminary studies indicated that the specific activity of labeled DNA in WT cells was similar in total and mitochondrial minicircle DNA fractions (data not shown). This suggests that as in other organisms there is exchange of one carbon units (in the form of serine, glycine, or formate) (39) and/or thymidine/thymidylate (40) across the mitochondrial membrane.
Phenotype of gcvP− Mutants
From knowledge of the metabolic pathways involving GCC in Leishmania and yeast (41), we predicted the gcvP− mutant would be sensitive to elevated glycine or to decreased serine in the culture medium (Fig. 1A). To test these predictions, promastigotes were grown in a semi-defined RPMI-based media. Although WT L. major could grow without glycine, removal of this amino acid reduced the rate of growth by more than 50% (not shown). Unexpectedly, we also found that WT L. major requires exogenous serine for optimal growth (supplemental Fig. S2). Promastigotes grew poorly in medium lacking serine, even when tested in the presence of 10-fold elevated glycine (1.3 mM) in an effort to drive serine production through SHMT (Fig. 1A; data not shown). The effect of serine deprivation was more pronounced in the WT L. major Friedlin V1 line studied here than in another virulent L. major line (LV39; supplemental Fig. S2B). A requirement for exogenous serine is consistent with the absence of genes required for several pathways of de novo serine synthesis in trypanosomatids (15) (see “Discussion”).
When grown in RPMI medium containing standard levels of glycine and serine (133 and 95 μM, respectively), the gcvP− mutant grew as well as WT (Table 2; full growth curves are shown in supplemental Fig. S3). In media containing elevated glycine (13.3 mM), growth of gcvP− was strongly inhibited, an effect substantially reversed by restoration of GCVP expression (Table 2). In serine-deficient media (9.5 μM), growth of all lines was impaired somewhat, but the growth of gcvP− was 58% less than WT in the same medium (Table 2). From the metabolic pathway shown in Fig. 1A and previous studies in yeast (41), we predicted that reduced growth rate under low-serine conditions might be reversed by formate, which can be converted by formyl-tetrahydrofolate ligase and the bifunctional methylene-tetrahydrofolate dehydrogenase-methenyl-tetrahydrofolate cyclohydrolase into 5,10-CH2-THF (Fig. 1A). As anticipated, the addition of 10 mM formate to the serine-deficient medium restored the growth of gcvP− to that of WT (Table 2). In total, these data suggest that the metabolic consequences of GCC inactivation in Leishmania were similar to those seen in other organisms.
TABLE 2. Relative growth of L. major gcvP− mutants in semi-defined RPMI media.
Standard RPMI growth medium contained 133 μM glycine and 95 μM L-serine. Data indicate growth as percent relative to growth of WT in the same medium after 72 h (or 103 h in low serine media where the growth rate of all lines was slower). Data are the mean values ± S.D. for three independent cultures.
| Line | Standard medium | High glycine (13 mM) | Low serine (9.5 μM) | Low serine (9.5 μM) + formate (10 mM)a |
|---|---|---|---|---|
| WT | 100 t2 = 9.8 ha |
100 t2 = 11.5 h |
100 t2 = 13.6 h |
100 t2 = 12.6 h |
| gcvP− | 111 ± 4% | 13 ± 1% | 42 ± 3% | 122 ± 16% |
| gcvP−/+GCVP | 67 ± 4% | 88 ± 5% | 57 ± 1% | 110 ± 14% |
Values for the doubling time (t2) were calculated from logarithmic growth of WT cells in the respective media.
gcvP− Leishmania Are Hypersensitive to Methotrexate but Not Other DHFR or TS Inhibitors
We anticipated that inhibitors of THF synthesis would deplete the one-carbon folate pool and would act synergistically with disruption of GCVP in inhibiting growth of Leishmania (see Fig. 1A). Consistent with this, the gcvP− mutant showed a 4-fold increase in sensitivity to the DHFR inhibitor methotrexate, whereas the sensitivity of the gcvP−/+GCVP line was similar to WT (Table 3). WT and gcvP− parasites were equally sensitive to the TS inhibitor CB3717 (42), which does not act through modulation of THF levels. However, the gcvP− line showed no change in sensitivity toward several other compounds shown previously to inhibit both DHFR and pteridine reductase 1 activities, including 6,7-diisopropyl-4-amino pteridine (O/129), or compounds 70 or 34 (Table 3; see the legend for their chemical names (11)). These data agree with the previous work establishing that the cytotoxicity of these compounds arises from targets beyond DHFR and/or pteridine reductase 1 (11) and suggests that the susceptibility of Leishmania and/or these other targets is not dependent upon GCC activity.
TABLE 3. Effects of antifolate compounds on growth of FV1 promastigote lines grown in M199 culture medium.
Compound 34 is (2,4-diamino-6-(3,4-dichlorophenoxy)-quinazoline); Compound 70 is (2,4-diamino-6-benzyl-5-(3-phenylpropyl)-pyrimidine). The density of uninhibited cultures was in the range 1–3 × 107 cells ml−1 when EC50 values were evaluated and did not differ significantly between WT, gcvP−, and gcvP−/+GCVP cultures.
| Compound | WT FV1 EC50 | gcvP− EC50 | -Fold hypersensitivity gcvP−vs. WT | gcvP−/+GCVP EC50 |
|---|---|---|---|---|
| nM | nM | nM | ||
| Methotrexate | 42 ± 2 | 10 ± 1 | 4 | 48 ± 7 |
| O/129 | 200 ± 20 | 220 ± 70 | 0.9 | 270 ± 40 |
| Compound 34 | 890 ± 50 | 720 ± 50 | 1.2 | 900 ± 100 |
| Compound 70 | 140 ± 5 | 210 ± 80 | 0.7 | 260 ± 30 |
| CB3717 | 21 ± 2 | 20 ± 1 |
gcvP− Leishmania Have Attenuated Virulence in Animal Infections
Because the gcvP− mutant grew and differentiated to infective forms normally in standard culture media, we asked whether the ability of the parasite to survive as the amastigote form in macrophages and/or induce disease in a mouse infection model was altered. Infective metacyclic promastigotes from different lines were inoculated into susceptible BALB/c mice, and lesion formation followed over time. Inoculation of WT or gcvP−/+GCVP metacyclics produced lesions that appeared after 14 days and progressed rapidly (Fig. 4A). In contrast, lesions in mice infected with gcvP− parasites did not appear until 24 days and progressed slightly less rapidly than lesions produced by WT parasites. In a separate experiment a limiting dilution assay was used to assess parasite numbers after mouse infection. After 22 days post-inoculation, WT-infected mice displayed lesions averaging 1.05 mm in thickness and containing 3–8 × 107 parasites, whereas gcvP− infected mice showed only slight inflammation without any swelling and contained 1000-fold fewer parasites (2–4 × 104). Thus, gcvP− parasites displayed less pathology and replicated less well in BALB/c mice.
FIGURE 4. Lack of GCVP delays lesion development in mouse infections.

Lesion development after inoculation of BALB/c (A and C) or C57/BL6 (B) mouse footpads with 1 × 105 metacyclic (peanut agglutinin minus) promastigotes (A and B) or 1 × 104 amastigotes (C). ●, WT; □, gcvP−; ▲, gcvP−/+GCVP parasites. Error bars indicate S.E. of mean of measurements of four infected mice. Similar results were seen in five other experiments using BALB/c mice (three with metacyclic parasites and two with amastigotes).
Similar studies were performed in genetically-resistant C57/Bl6 mice, which ultimately control L. major infections and heal. In this background WT infections developed pathology at about 18 days, which progressed to a maximum around 30 – 40 days and then healed over the next ~90 days (Fig. 4B). As in the susceptible BALB/c mouse, the gcvP− mutant showed a delay in lesion formation, with lesions appearing around 33 days. Lesions grew somewhat more slowly and peaked at a lower thickness (0.9 versus 1.4 mm), after which they healed with similar kinetics (Fig. 4B). Interestingly, the gcvP−/+GCVP parasite appeared to form somewhat larger lesions, but this was not statistically significant, and the timing of their appearance and healing was similar to that seen with WT infection (Fig. 4B).
After resolution of the lesion pathology, WT L. major persist indefinitely at low levels in both macrophage and non-macrophage cells (43), a phenomenon that may contribute to maintenance of protective immunity (44). We used the limiting dilution assay to assess parasite numbers 9 months after infection, long after lesion pathology had subsided (Fig. 4B). WT and gcvP−/+GCVP parasites persisted at similar levels (3,300 ± 7,100 WT parasites per lesion, n = 5; 2,200 ± 2,700 gcvP−/+GCVP parasites per lesion, n = 8), whereas gcvP− were found at somewhat higher levels (9,300 ± 14,000 parasites per lesion, n = 9), although this difference was not statistically significant.
gcvP− Amastigotes Show Attenuated Virulence
In previous studies of other mutant L. major, we showed that a “delayed lesion” phenotype similar to that seen with gcvP− metacyclic promastigote infections arose from a defect in the virulence of metacyclic promastigotes but not of amastigotes (27, 45– 47). This model was tested here by purifying amastigotes from gcvP−-infected mice showing significant pathology and inoculating them directly back into mice as amastigotes (Fig. 4C). As seen in promastigote infections, WT and gcvP−/+GCVP amastigote infections yielded lesions that appeared and progressed rapidly, whereas gcvP− amastigote infections produced lesions only after a further ~21-day delay and progressed more slowly (Fig. 4C). Thus, both gcvP− metacyclic promastigotes and amastigotes show a similar delay in disease induction, arguing against a stage-specific effect in parasite survival.
gcvP− Metacyclic Promastigotes Show No Attenuation in Macrophage Infections
A further test of the virulence of gcvP− parasites was made by infection of primary peritoneal macrophages in vitro. In these studies infections were performed with metacyclic promastigotes in the presence of high (400 μM) or low (with only ~10 μM present in 10% serum) levels of serine or with amastigotes (high serine medium only) (Fig. 5). In all these experiments WT and gcvP− parasites entered macrophages, survived, and replicated to similar levels. These data argue that despite the attenuation of virulence in the mouse models, the gcvP− parasite shows no intrinsic defect in its ability to survive in this macrophage system.
FIGURE 5. Macrophage infections with gcvP− metacyclic promastigotes or amastigotes.

Infection of macrophages in vitro with metacyclic promastigotes (A, high serine medium; B, low serine medium) or amastigotes (C, high-serine medium). ●, WT; □, gcvP−. Error bars indicate S.E. from triplicate macrophage coverslip cultures.
DISCUSSION
We used bioinformatic approaches to identify the presumptive Leishmania orthologs of the genes encoding the four subunits of the glycine cleavage complex (GCVP, GCVT, GCVH, and GCVL/dihydrolipoamide dehydrogenase; Fig. 1B). We focused on the GCVP protein, which mediates the first catalytic step of the GCC, decarboxylation of glycine, and generated mutants and relevant control lines to explore its role and that of the GCC in Leishmania metabolism. In other species the GCC is localized to the mitochondrion, and correspondingly, presumptive mitochondrial targeting pre-sequences were identified for all of the Leishmania GCC subunit orthologs (supplemental Fig. S1 or not shown). We confirmed experimentally that a GFP-tagged GCVP protein was targeted to the parasite mitochondrion (Fig. 3).
When grown as the promastigote stage in vitro culture, an L. major gcvP− mutant grew and differentiated normally to the infective metacyclic form. We attempted to assay GCC activity enzymatically in crude and fractionated parasite extracts but were unsuccessful. In other non-photosynthetic organisms where glycine cleavage activity has been measured, it is very low; 0.1– 0.2 nmol/min/mg of protein for yeast and E. coli (48). Thus, we devised an alternative indirect in vivo assay for GCC activity after incorporation of label from [2-14C]glycine through 5,10-CH2-THF into thymidylate and then DNA (Table 1). Preliminary studies indicated that nuclear and mitochondrial DNA were similarly labeled by [2-14C]glycine, implying that either or both 5,10-CH2-THF and thymidine/thymidylate are able to pass out of the mitochondrion. In rodent liver mitochondria, one-carbon THF derivatives are trapped inside by polyglutamylation (49, 50), but the 14C label in thymidylate (or thymidine) can be exported to the cytosol/nucleus (40). Importantly, incorporation was greatly decreased in the gcvP− mutant and was partially restored after re-expression of the GCVP in the mutant background. These data in combination with the metabolic data discussed below argue that the GCVP protein identified here participates in GCC activity as expected. In these studies some residual labeling very close to the experimental background was found in the gcvP− mutant (Table 1). This could arise from contamination of the DNA preparations with other abundant [2-14C]glycine-labeled molecules (proteins being the most likely candidate), and we cannot absolutely exclude the possibility of an alternative novel glycine cleavage activity and/or novel metabolic pathways for incorporation. Several studies proposed that GCVP may function in nitric oxide synthesis in plants (51). However, consistent with the later withdrawal of this claim (52), nitricoxide synthase activity was not found in preparations of recombinant Leishmania GCVP protein (data not shown).
The metabolic role of the GCC can be seen as somewhat redundant with that of SHMT, which occurs in both cytoplasmic and mitochondrial compartments in forms encoded by distinct genes (16). This may explain why in two standard Leishmania culture media the gcvP− mutant grew and differentiated normally. However, under conditions of excess glycine, growth of the gcvP− mutant was greatly inhibited (Table 2), as seen previously in plants or yeast (41, 48, 53). This may arise from the observation in other organisms that excess glycine contributes to decreased 5,10-CH2-THF production by SHMT (54), which would further reduce 5,10-CH2-THF levels in GCC-deficient mutants. Removal of glycine is a major role for GCC in normal plant metabolism, which produces large amounts of glycine in the oxidative photosynthetic carbon cycle (18). Potentially, Leishmania GCC may also function to metabolize excess glycine produced in vivo either within the parasite or by mammalian or insect hosts.
The interplay of GCC and SHMT in the production of 5,10-methylene-THF prompted us to examine serine metabolism in these lines. Unexpectedly we found that WT L. major grew poorly in semi-defined RPMI-based media in the absence of glycine or serine (Fig. S2). In other organisms serine may arise de novo from glycine through the action of SHMT or through two pathways leading from the glycolytic intermediate 3-phosphoglycerate (the phosphorylated and non-phosphorylated pathways) (55). Genome-based analyses of Leishmania metabolic pathways have yielded disparate conclusions about the ability of Leishmania to synthesize serine de novo (15, 56, 57). Our genome analysis indicated that Leishmania lacks the pathways for serine synthesis from 3-phosphoglycerate. This suggests that the only sources of serine may be through glycine and SHMT and/or salvage from the external environment. Thus, the GCC may contribute to Leishmania serine synthesis through SHMT by driving 5,10-methylene-THF production. Consistent with this model, growth of the gcvP− mutant was poorer in conditions of serine deficiency (Table 2 and Fig. S3). In yeast, mitochondrial SHMT, but not the cytosolic isoform, can synthesize enough serine in combination with the GCC to sustain normal growth rates (41, 58). However, in WT L. major this pathway is apparently unable to provide sufficient serine to sustain maximal growth, even in the presence of 10-fold more glycine, which would be expected to further drive SHMT-mediated serine production. These findings suggest that L. major may be susceptible to inhibition by drugs that interfere with the uptake and utilization of serine.
Role of the GCC in Leishmania Virulence
Infections of either genetically susceptible Balb/C or resistant C57BL6 mice with gcvP− L. major resulted in a significant delay in the emergence of lesion pathology relative to that seen in WT infections (Fig. 4, A and B). This effect was specifically attributable to the lack of gcvP−, as restoration of GCVP expression (gcvP−/+GCVP cells) returned the lesion progression profile to that of WT (Fig. 4, A and B). Measurements of parasite numbers in Balb/C mouse infections showed that the decrease in lesion size correlated to decreased numbers of parasites, confirming that loss of gcvP− reduced both parasite replication and pathology induction during this early phase. Interestingly, infections of other intracellular pathogens, Francisella tularensis, Brucella abortus, or Mycobacterium tuberculosis are likewise dependent on the GCVP or other GCC subunit genes (22–24), possibly indicating similar constraints impacting the glycine/serine/5,10-CH2-THF pathways.
Once lesions ultimately appeared in gcvP−-infected mice, they progressed and grew at rates only slightly slower than seen in WT parasite infections (Figs. 4, A and B). In previous studies of other mutant L. major, we showed that the delayed lesion phenotype similar to that seen with gcvP− metacyclic promastigotes infections arose from stage-specific effects (27, 45–47). In such mutants, the ability of the metacyclic promastigote to establish infections in macrophages and/or mice was compromised but not its ability to replicate and induce pathology as the amastigote stage. As seen in other organisms, virulence factors in Leishmania can operate at one or more stages of the infectious cycle. This form of the delayed lesion phenotype thus reflects the initial destruction of a high proportion of metacyclics and the eventual outgrowth as amastigotes of those managing to survive.
However, although gcvP− promastigotes showed delayed lesion formation, when amastigotes were isolated directly from progressing gcvP− infections and used immediately to re-infect naïve animals, the delayed lesion phenotype remained (Fig. 4C). This effect was specifically attributable to loss of GCVP as it was reversed by restoration of GCVP expression (Fig. 4C). This suggests there is no process of “adaptation” of gcvP− promastigotes to mammalian growth conditions, as gcvP− amastigotes should already have undergone adaptation. It also argues against the possibility that the delayed lesions arising from promastigote infections arise through genetic reversion.
A second unexpected finding given the mouse infectivity studies was that in infections of peritoneal macrophages in vitro we were unable to detect any difference in the entry, survival, and replication of the gcvP− L. major relative to WT when tested as either metacyclic promastigotes or as amastigotes (Fig. 5). This was seen even in the absence of added serine in the macrophage culture media in an experiment designed to exploit the reduced serine growth deficiency seen in vitro with promastigotes (Table 2).
These data suggest that GCVP and/or the GCC carries out some unique role in animal infections that is not adequately probed by growth tests in vitro or in macrophages. Potentially this is unrelated to its known role in GCC activity, although this is a theoretical possibility we think it unlikely. A second explanation invokes differences in the immune response to WT and gcvP− parasites. Again we consider this unlikely, as the delayed lesion phenotype was seen in highly susceptible Balb/c mice that lack an effective anti-leishmanial immune response as well as C57BL6, which effectively contain Leishmania pathology and replication (Fig. 4).
A third explanation invokes some differences in physiological environment experienced by parasites in the first month or so but not later in the infection. In careful studies of C57BL6 mice it has been shown that for the first 4 – 6 weeks after infection, parasite replication is substantial but occurs in the absence of significant tissue pathology (59). After that time, parasite replication continues but is accompanied by significant inflammatory pathology, which in C57BL6 is also a period marked by the appearance of an active immune response capable of clearing the parasite. Although little is known about host tissue metabolic changes accompanying Leishmania infection during these pre- versus post-inflammatory periods, it is well known that significant metabolic changes occur during inflammation in other circumstances (60, 61). These considerations prompt the suggestion that that in vivo gcvP− parasites are less able to survive (or, at least, replicate more slowly) under nutritional conditions imposed by a non-inflammatory setting, accounting for the delay in parasite replication and lesion pathology. However, upon commencement of the inflammatory response and accompanying metabolic perturbations, the parasites now find themselves under metabolic conditions more conducive for their growth. It has been proposed recently that the tropism of Leishmania for the macrophage phagolysosome is dependent on that environment being rich in amino acids (62), and our data suggest the possibility that the “richness” of the phagolysosome may vary as the host infection proceeds. Our model also suggests that there may be other Leishmania genes required for growth or survival early versus late in the infection, as the environment experienced by Leishmania within the host macrophage evolves.
This model could be explored further by experimental manipulations of macrophage metabolism during Leishmania infection and aided by better knowledge of host tissue metabolism via metabolic profiling at various stages after animal infection. The fact that WT and gcvP− parasites survive equally well in the starch-elicited peritoneal macrophage preparations used here suggests that these tests took place under “nutritionally permissive” conditions, even under conditions of serine limitation in the macrophage culture media (Fig. 5). Potentially, macrophage subtypes and/or conditions more representative of those occurring in natural lesions (as opposed to the artificial setting of peritoneal macrophages grown in rich media) may be required to critically assess this hypothesis. An improved understanding of how lesions develop and the role of host tissues in supporting parasite metabolism and growth under various circumstances could aid in the design of novel therapies. In this respect infections of mice by the gcvP− mutant may provide a marker to study the physiology of cutaneous leishmaniasis lesions.
Perspective
These studies on the role of glycine cleavage in Leishmania folate metabolism and virulence may aid the development of novel treatments for leishmaniasis in two main ways. The results from in vitro culture of the gcvP− mutant confirm predictions from L. major genomics linking folate metabolism to the use of glycine, serine, and formate as carbon donors. However, the finding that a nutritional requirement for serine cannot be replaced by glycine underlines the importance of experimental verification of genomic-based metabolic predictions. Although the GCC itself may not be a singular drug target, as evidenced by the rapidly progressing lesions arising late after gcvP− infection, this broader understanding of Leishmania folate metabolism will assist the study of other folate-utilizing enzymes with unique functions that may provide critical drug targets. Interestingly, a comparison of Leishmania folate metabolism with that of its relative T. brucei (13) (the African sleeping sickness parasite) implies that T. brucei has a reduced folate metabolism and in particular lacks SHMT and 10-formyl-tetrahydrofolate synthetase, which could make it critically dependent on the activity of the GCC.
Supplementary Material
Acknowledgments
We thank Ann Jackman for providing the TS inhibitor CB3717.
Footnotes
This work was supported by National Institutes of Health Grant AI21903 (to S. M. B.) and European Molecular Biology Organization Long Term Fellowship ALTF 106-2005 (to T. V.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S3.
The abbreviations used are: DHFR, dihydrofolate reductase; TS, thymidylate synthase; 5,10-CH2-THF, 5,10-methylenetetrahydrofolate; GCVP, glycine cleavage complex P subunit; SHMT, serine hydroxymethyltransferase; GCC, glycine cleavage complex; WT, wild type; kb, kilobase(s); GFP, green fluorescent protein.
References
- 1.Herwaldt BL. Lancet. 1999;354:1191–1199. doi: 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- 2.Lira R, Sundar S, Makharia A, Kenney R, Gam A, Saraiva E, Sacks D. J Infect Dis. 1999;180:564–567. doi: 10.1086/314896. [DOI] [PubMed] [Google Scholar]
- 3.Berman J, Bryceson AD, Croft S, Engel J, Gutteridge W, Karbwang J, Sindermann H, Soto J, Sundar S, Urbina JA. Trans R Soc Trop Med Hyg. 2006;100(Suppl 1):41–44. doi: 10.1016/j.trstmh.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Garrett CE, Coderre JA, Meek TD, Garvey EP, Claman DM, Beverley SM, Santi DV. Mol Biochem Parasitol. 1984;11:257–265. doi: 10.1016/0166-6851(84)90070-7. [DOI] [PubMed] [Google Scholar]
- 5.Stechmann A, Cavalier-Smith T. Science. 2002;297:89–91. doi: 10.1126/science.1071196. [DOI] [PubMed] [Google Scholar]
- 6.Swafford JR, Beverley SM, Kan CC, Caulfield JP. XIIth International Congress of Electron Microscopy; San Francisco, CA: San Francisco Press, Inc; 1990. pp. 610–611. [Google Scholar]
- 7.Luo M, Orsi R, Patrucco E, Pancaldi S, Cella R. Plant Mol Biol. 1997;33:709–722. doi: 10.1023/a:1005798207693. [DOI] [PubMed] [Google Scholar]
- 8.Neuburger M, Rebeille F, Jourdain A, Nakamura S, Douce R. J Biol Chem. 1996;271:9466–9472. doi: 10.1074/jbc.271.16.9466. [DOI] [PubMed] [Google Scholar]
- 9.Cruz A, Coburn CM, Beverley SM. Proc Natl Acad Sci U S A. 1991;88:7170–7174. doi: 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nare B, Hardy LW, Beverley SM. J Biol Chem. 1997;272:13883–13891. doi: 10.1074/jbc.272.21.13883. [DOI] [PubMed] [Google Scholar]
- 11.Hardy LW, Matthews W, Nare B, Beverley SM. Exp Parasitol. 1997;87:157–169. [PubMed] [Google Scholar]
- 12.Hammond DJ, Gutteridge WE. Mol Biochem Parasitol. 1984;13:243–261. doi: 10.1016/0166-6851(84)90117-8. [DOI] [PubMed] [Google Scholar]
- 13.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Bohme U, Hannick L, Aslett MA, Shallom J, Marcello L, Hou L, Wickstead B, Alsmark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K, Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN, Corton CH, Cronin A, Davies RM, Doggett J, Djikeng A, Feldblyum T, Field MC, Fraser A, Goodhead I, Hance Z, Harper D, Harris BR, Hauser H, Hostetler J, Ivens A, Jagels K, Johnson D, Johnson J, Jones K, Kerhornou AX, Koo H, Larke N, Landfear S, Larkin C, Leech V, Line A, Lord A, Macleod A, Mooney PJ, Moule S, Martin DM, Morgan GW, Mungall K, Norbertczak H, Ormond D, Pai G, Peacock CS, Peterson J, Quail MA, Rabbinowitsch E, Rajandream MA, Reitter C, Salzberg SL, Sanders M, Schobel S, Sharp S, Simmonds M, Simpson AJ, Tallon L, Turner CM, Tait A, Tivey AR, Van Aken S, Walker D, Wanless D, Wang S, White B, White O, Whitehead S, Woodward J, Wortman J, Adams MD, Embley TM, Gull K, Ullu E, Barry JD, Fairlamb AH, Opperdoes F, Barrell BG, Donelson JE, Hall N, Fraser CM, Melville SE, El-Sayed NM. Science. 2005;309:416–422. [Google Scholar]
- 14.Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, Kerhornou A, Ivens A, Fraser A, Rajandream MA, Carver T, Norbertczak H, Chillingworth T, Hance Z, Jagels K, Moule S, Ormond D, Rutter S, Squares R, Whitehead S, Rabbinowitsch E, Arrowsmith C, White B, Thurston S, Bringaud F, Baldauf SL, Faulconbridge A, Jeffares D, Depledge DP, Oyola SO, Hilley JD, Brito LO, Tosi LR, Barrell B, Cruz AK, Mottram JC, Smith DF, Berriman M. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payne SH, Loomis WF. Eukaryot Cell. 2006;5:272–276. doi: 10.1128/EC.5.2.272-276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gagnon D, Foucher A, Girard I, Ouellette M. Mol Biochem Parasitol. 2006;150:63–71. doi: 10.1016/j.molbiopara.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Walker JL, Oliver DJ. J Biol Chem. 1986;261:2214–2221. [PubMed] [Google Scholar]
- 18.Douce R, Bourguignon J, Neuburger M, Rebeille F. Trends Plant Sci. 2001;6:167–176. doi: 10.1016/s1360-1385(01)01892-1. [DOI] [PubMed] [Google Scholar]
- 19.Schirch L. Folates and Pterins. In: Blakley RL, Benkovic SJ, editors. Chemistry and Biochemistry of Folates. Vol. 1. John Wiley; New York: 1984. pp. 399–431. [Google Scholar]
- 20.Piper MD, Hong SP, Ball GE, Dawes IW. J Biol Chem. 2000;275:30987–30995. doi: 10.1074/jbc.M004248200. [DOI] [PubMed] [Google Scholar]
- 21.Engel N, van den Daele K, Kolukisaoglu U, Morgenthal K, Weckwerth W, Parnik T, Keerberg O, Bauwe H. Plant Physiol. 2007;144:1328–1335. doi: 10.1104/pp.107.099317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, Monack DM. Proc Natl Acad Sci U S A. 2007;104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong PC, Tsolis RM, Ficht TA. Infect Immun. 2000;68:4102–4107. doi: 10.1128/iai.68.7.4102-4107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sassetti CM, Rubin EJ. Proc Natl Acad Sci U S A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapler GM, Coburn CM, Beverley SM. Mol Cell Biol. 1990;10:1084–1094. doi: 10.1128/mcb.10.3.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knighton DR, Kan CC, Howland E, Janson CA, Hostomska Z, Welsh KM, Matthews DA. Nat Struct Biol. 1994;1:186–194. doi: 10.1038/nsb0394-186. [DOI] [PubMed] [Google Scholar]
- 27.Zufferey R, Allen S, Barron T, Sullivan DR, Denny PW, Almeida IC, Smith DF, Turco SJ, Ferguson MA, Beverley SM. J Biol Chem. 2003;278:44708–44718. doi: 10.1074/jbc.M308063200. [DOI] [PubMed] [Google Scholar]
- 28.Sacks DL, Perkins PV. Science. 1984;223:1417–1419. doi: 10.1126/science.6701528. [DOI] [PubMed] [Google Scholar]
- 29.Späth GF, Beverley SM. Exp Parasitol. 2001;99:97–103. doi: 10.1006/expr.2001.4656. [DOI] [PubMed] [Google Scholar]
- 30.Titus RG, Marchand M, Boon T, Louis JA. Parasite Immunol (Oxf) 1985;7:545–555. doi: 10.1111/j.1365-3024.1985.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 31.Robinson KA, Beverley SM. Mol Biochem Parasitol. 2003;128:217–228. doi: 10.1016/s0166-6851(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 32.Medina-Acosta E, Cross GA. Mol Biochem Parasitol. 1993;59:327–329. doi: 10.1016/0166-6851(93)90231-l. [DOI] [PubMed] [Google Scholar]
- 33.Ha DS, Schwarz JK, Turco SJ, Beverley SM. Mol Biochem Parasitol. 1996;77:57–64. doi: 10.1016/0166-6851(96)02580-7. [DOI] [PubMed] [Google Scholar]
- 34.Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakai T, Nakagawa N, Maoka N, Masui R, Kuramitsu S, Kamiya N. EMBO J. 2005;24:1523–1536. doi: 10.1038/sj.emboj.7600632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 37.Coombs GH, Tetley L, Moss VA, Vickerman K. Parasitology. 1986;92:13–23. doi: 10.1017/s0031182000063411. [DOI] [PubMed] [Google Scholar]
- 38.Clayton CE. EMBO J. 2002;21:1881–1888. doi: 10.1093/emboj/21.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christensen KE, MacKenzie RE. BioEssays. 2006;28:595–605. doi: 10.1002/bies.20420. [DOI] [PubMed] [Google Scholar]
- 40.Ferraro P, Nicolosi L, Bernardi P, Reichard P, Bianchi V. Proc Natl Acad Sci U S A. 2006;103:18586–18591. doi: 10.1073/pnas.0609020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNeil JB, Bognar AL, Pearlman RE. Genetics. 1996;142:371–381. doi: 10.1093/genetics/142.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu E, Callender MA, Farrell MP, Schmitz JC. Cancer Chemother Pharmacol. 2003;52(Suppl 1):80–89. doi: 10.1007/s00280-003-0625-9. [DOI] [PubMed] [Google Scholar]
- 43.Bogdan C, Donhauser N, Doring R, Rollinghoff M, Diefenbach A, Rittig MG. J Exp Med. 2000;191:2121–2130. doi: 10.1084/jem.191.12.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 45.Späth GF, Epstein L, Leader B, Singer SM, Avila HA, Turco SJ, Beverley SM. Proc Natl Acad Sci U S A. 2000;97:9258–9263. doi: 10.1073/pnas.160257897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang K, Hsu FF, Scott DA, Docampo R, Turk J, Beverley SM. Mol Microbiol. 2005;55:1566–1578. doi: 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Capul AA, Hickerson S, Barron T, Turco SJ, Beverley SM. Infect Immun. 2007;75:4629–4637. doi: 10.1128/IAI.00735-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagarajan L, Storms RK. J Biol Chem. 1997;272:4444–4450. doi: 10.1074/jbc.272.7.4444. [DOI] [PubMed] [Google Scholar]
- 49.Horne DW, Patterson D, Cook RJ. Arch Biochem Biophys. 1989;270:729–733. doi: 10.1016/0003-9861(89)90556-0. [DOI] [PubMed] [Google Scholar]
- 50.Horne DW, Holloway RS, Said HM. J Nutr. 1992;122:2204–2209. doi: 10.1093/jn/122.11.2204. [DOI] [PubMed] [Google Scholar]
- 51.Chandok MR, Ytterberg AJ, van Wijk KJ, Klessig DF. Cell. 2003;113:469–482. doi: 10.1016/s0092-8674(03)00350-7. [DOI] [PubMed] [Google Scholar]
- 52.Klessig DF, Ytterberg AJ, van Wijk KJ. Cell. 2004;119:445. doi: 10.1016/j.cell.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Bauwe H, Kolukisaoglu U. J Exp Bot. 2003;54:1523–1535. doi: 10.1093/jxb/erg171. [DOI] [PubMed] [Google Scholar]
- 54.Stover P, Schirch V. J Biol Chem. 1991;266:1543–1550. [PubMed] [Google Scholar]
- 55.de Koning TJ, Snell K, Duran M, Berger R, Poll-The BT, Surtees R. Biochem J. 2003;371:653–661. doi: 10.1042/BJ20021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Opperdoes FR, Coombs GH. Trends Parasitol. 2007;23:149–158. doi: 10.1016/j.pt.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 57.Chaudhary K, Roos DS. Nat Biotechnol. 2005;23:1089–1091. doi: 10.1038/nbt0905-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kastanos EK, Woldman YY, Appling DR. Biochemistry. 1997;36:14956–14964. doi: 10.1021/bi971610n. [DOI] [PubMed] [Google Scholar]
- 59.Belkaid Y, Mendez S, Lira R, Kadambi N, Milon G, Sacks D. J Immunol. 2000;165:969–977. doi: 10.4049/jimmunol.165.2.969. [DOI] [PubMed] [Google Scholar]
- 60.Remick DG. Am J Pathol. 2007;170:1435–1444. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melchior D, Seve B, Le Floc’h N. J Anim Sci. 2004;82:1091–1099. doi: 10.2527/2004.8241091x. [DOI] [PubMed] [Google Scholar]
- 62.McConville MJ, de Souza D, Saunders E, Likic VA, Naderer T. Trends Parasitol. 2007;23:368–375. doi: 10.1016/j.pt.2007.06.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.