

Abstract
Prenylated Rab acceptor (PRA1) is a protein that binds Rab GTPases and the v-SNARE VAMP2. The protein is localized to the Golgi complex and post-Golgi vesicles. To determine its functional role, we generated a number of point mutations and divided them into three classes based on cellular localization. Class A mutants were retained in the endoplasmic reticulum (ER) and exerted an inhibitory effect on transport of vesicular stomatitis virus envelope glycoprotein (VSVG) from the ER to Golgi as well as to the plasma membrane. Class B mutants exhibited a highly condensed Golgi complex and inhibited exit of anterograde cargo from this organelle. Class C mutants exhibited an intermediate phenotype with Golgi and ER localization along with extensive tubular structures emanating from the Golgi complex. There was a direct correlation between the cellular phenotype and binding to Rab and VAMP2. Class A and C mutants showed a significant decrease in Rab and VAMP2 binding, whereas an increase in binding was observed in the class B mutants. Thus, PRA1 is required for vesicle formation from the Golgi complex and might be involved in recruitment of Rab effectors and SNARE proteins during cargo sequestration.
Rab GTPases constitute the largest group within the Ras superfamily. They regulate vesicle trafficking by cycling through active membrane-bound GTP- and inactive cytosolic GDP-bound states. Membrane localization requires modification of the cysteine-containing motif at the carboxyl terminus by prenyl residues. Cycling between the membrane and cytosol is mediated by GDP dissociation inhibitor (GDI),1 which extracts GDP-bound Rab from the membrane. Activation through guanine nucleotide exchange at the membrane is catalyzed by a guanine nucleotide exchange factor, of which a number have been identified in mammals (1, 2).
Vesicular transport through the secretory pathway undergoes a number of discrete steps each involving budding, membrane remodeling, targeting, docking, and fusion. In ER to Golgi transport, anterograde cargo proteins such as VSVG are selectively transported to the Golgi along with resident ER proteins with the latter retrieved by a salvage process that recognizes distinct motifs within the protein (3). These transport vesicles contain an electron-dense coat assembled under the control of the small GTPase ARF (4). The fungal metabolite brefeldin A (BFA) inhibits ARF activation by stabilizing the inactive ARF-guanine nucleotide exchange factor (GEF) complex (5) resulting in retrograde transport of Golgi content to the ER. At the Golgi complex, cargo proteins destined for post-Golgi locations are sorted into distinct carriers upon exit from the trans face, whereas Golgi resident proteins such as mannosidase II (Man II) are selectively retrieved in COPI-coated vesicles and returned to the cis face (6).
Vesicle fusion is mediated by the core SNARE complex consisting of the vesicle protein VAMP, or synaptobrevin, and two other membrane proteins, syntaxin and SNAP-25 (7). Rab effectors play a regulatory role in this process either through direct interaction with t-SNAREs (8) or recruitment of other SNARE regulatory proteins (9–12). We have isolated previously (13) a Rab and VAMP2-interacting protein called prenylated Rab acceptor or PRA1. PRA1 inhibits the removal of Rab from the membrane by GDI (14) suggesting that recycling of Rab depends on the opposing action of PRA1 and GDI, with PRA1 favoring membrane retention and GDI favoring solubilization. PRA1 has also been shown to interact with a variety of proteins as follows: the presynaptic cytoskeletal matrix protein Piccolo (15); other Ras GTPases (16); the Epstein-Barr Bcl-2 homologue BHRF1 (17); and the SIV envelope protein gp41 (18). However, the functional significance of these interactions remains unclear. To determine the physiological function of PRA1, we generated a number of point mutations and examined their effect on cellular localization, organelle morphology, protein trafficking, and binding to Rab and VAMP2.
MATERIALS AND METHODS
PRA1 Mutagenesis
PRA1 mutations were generated by the PCR amplification using pQE11/HA-tagged PRA1 as template (13). For bacterial expression, the PCR products were inserted between the ApaI and SpeI sites of a modified pQE10* vector (Qiagen). For the mammalian expression of the PRA1 mutants, the constructs were subcloned into the pIRESpuro vector (CLONTECH) between the ClaI and EcoRI sites.
Cell Culture and Immunocytochemistry
Chinese hamster ovary (CHO) cells were maintained in minimum Eagle’s medium α (Invitrogen) supplemented with 5% fetal bovine serum and 100 units/ml penicillin, 100 μg/ml streptomycin. For transient expression, 6 × 104 CHO cells were seeded overnight on 12-mm coverslips. Cells were transfected with LipofectAMINE (Invitrogen) and fixed 36–48 h after transfection with 4% paraformaldehyde in phosphate-buffered saline for 1 h followed by incubation in blocking buffer (1% bovine serum albumin, 2% normal goat serum, and 0.4% saponin in phosphate-buffered saline) for 15 min. Mouse monoclonal anti-HA (Roche Molecular Biochemicals) and rabbit anti-Man II (a generous gift from Dr. M. Farquhar) antibodies were used as primary antibodies, and after washing washed with 100 mM glycine in phosphate-buffered saline, Alexa 488 or 595-labeled secondary antibodies (Molecular Probes) were used. Coverslips were mounted with SlowFade Light antifade (Molecular Probes). For sensitivity to BFA, cells were treated with 10 μg/ml BFA for 30 min before fixing. For stable transfection, CHO were transfected with pIRESpuro/HA-PRA1 constructs and clonal lines selected for puromycin resistance at 10 μg/ml. Several clonal lines were isolated, and positive clones were identified by immunocytochemistry.
To examine VSVG-GFP trafficking time course, CHO and PRA1 stable cell lines were seeded overnight on 12-mm coverslips, transfected with pCDM8.1/VSVGts045-GFP (19), and 18 h after transfection shifted to 42 °C for 5 h. Cycloheximide (20 μg/ml) was added 10 min before shifting back to 37 °C, and cells were then fixed after 0, 15, 30, 60, and 90 min. Cells were analyzed by confocal microscopy and were scored by phenotype (ER, Golgi-trans-Golgi network, and plasma membrane) with a minimum of 100 cells scored per coverslip. The values presented were from three independent experiments and verified on at least two clonal lines. For surface labeling, cells were chased for 0, 60, and 120 min and fixed with ice-cold 4% paraformaldehyde for 30 min. The cells were processed for immunocytochemistry as described above except in the absence of saponin, and all steps were performed at 4 °C. The monoclonal antibody BW8G65 directed against the extracellular domain of VSVG (a generous gift from Dr. M. Farquhar) was used to label cell surface-exposed VSVG followed by Alexa 594-labeled secondary antibodies.
In Vitro Binding Assays
The PRA1 mutants were subcloned into the modified pGAD424X prey vector (13) at the EcoRI and XhoI sites. They were then co-transformed as described (20) with Rab3A or VAMP2 bait plasmids (13) and grown on Trp and Leu drop-out plates for 3–5 days. The cells were patched onto filter paper, lysed by brief liquid nitrogen treatment, and incubated with 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) (21). The intensity as well as the time of onset was used to assess the strength of the interactions.
To verify the yeast two-hybrid results, in vitro binding studies were performed with recombinant His6-tagged Rab3A as described previously (14). GST-tagged VAMP2 (from Dr. W. S. Trimble) and His6-HA-tagged PRA1 wild type and mutants were purified as described previously (13), except that the PRA1 used in the PRA1/Rab3A binding assay was eluted with 50 mM EDTA. All recombinant proteins were quantified by densitometric analysis of Coomassie Blue-stained gels using bovine serum albumin as a standard. His6-tagged, purified PRA1s were covalently cross-linked to CNBr-activated Sepharose 4B (Amersham Biosciences). A typical PRA1-Rab3A binding assay contained 10.8 pmol of His6-HA-PRA1 cross-linked to CNBr-activated Sepharose 4B and 320 nM His6-Rab3A in 25 mM Tris-HCl, pH 7.5, 150 mM KCl, 0.5 mM MgCl2, 0.25 mM GDPβS, 10% glycerol, and 0.005% Triton X-100 in a total volume of 250 μl, and incubated for 1 h at 4 °C. The beads were then washed with ice-cold binding buffer. Denaturing loading buffer was added to the beads, and proteins purified with the beads were subjected to Western immunoblot analysis using anti-Rab3A antibodies (Santa Cruz Biotechnology) with Alexa Fluor 488-labeled goat anti-rabbit IgG as secondary antibodies (Molecular Probes). The assays were done in triplicate, and the signals were quantified using the Typhoon 8600 imager (Amersham Biosciences) and averaged for comparative analysis. Glutathione-agarose beads were used to pulldown GST-VAMP2 as described previously (13). A typical PRA1-VAMP2 binding assay contained 40 nM GST-VAMP2 or GST control and 30 nM His6-HA-PRA1s in 25 mM Tris-HCl, pH 7.5, 150 mM KCl, 10% glycerol, and 0.005% Triton X-100 in a total volume of 250 μl and was incubated for 1 h at 4 °C. Glutathione-agarose was used to recover the GST-VAMP2, and PRA1 was detected using anti-HA antibodies (Roche Molecular Biochemicals). The signals obtained were quantified as described above.
RESULTS
Mutation of PRA1 Alters Its Cellular Localization
To determine the function of PRA1, we employed a mutagenic approach by introducing point mutations to the two most highly conserved regions: one proximal to the first hydrophobic domain (residues 40 –104) and a second spanning residues 134 –171. The resulting amino-terminal HA-tagged PRA1 mutants were transfected into CHO and classified based on cellular localization (Table I). The wild type PRA1 is primarily localized to the Golgi complex (Fig. 1A) but retained in the ER in the truncated PRA1-(1–164) (Fig. 1B) (14). The class A mutants displayed an ER phenotype similar to PRA1-(1–164), suggesting that the protein was either retained in or retrogradely transported to the ER (Fig. 1C). The class B mutants were characterized by a highly condensed Golgi complex, which is suggestive of a defect in transport through this compartment (Fig. 1, D and E). Finally, in class C, PRA1 showed both Golgi and ER localization but with extensive tubular structures emanating from the Golgi complex (Fig. 1F). Two other PRA1 mutations, N77A and W154A, showed little or no significant change in their Golgi localization compared with the wild type. Thus, mutations targeted to the two conserved regions of PRA1 can alter the cellular localization of the protein as well as Golgi morphology.
Table I. Classification of PRA1 mutants according to cellular localization.
The HA-tagged constructs were transfected into CHO cells and stained with anti-HA antibodies.
| PRA1 | Class | Cellular localization |
|---|---|---|
| WT | Golgi | |
| N77A | Golgi | |
| W154A | Golgi | |
| N70T | A | ER |
| Y78A | A | ER |
| S76A | B | Condensed Golgi |
| S76V | B | Condensed Golgi |
| V161A | B | Condensed Golgi |
| Y73A | C | Golgi, ER, and tubular structures |
| H166A | C | Golgi, ER, and tubular structures |
Fig. 1. Cellular localization of PRA1 mutants.
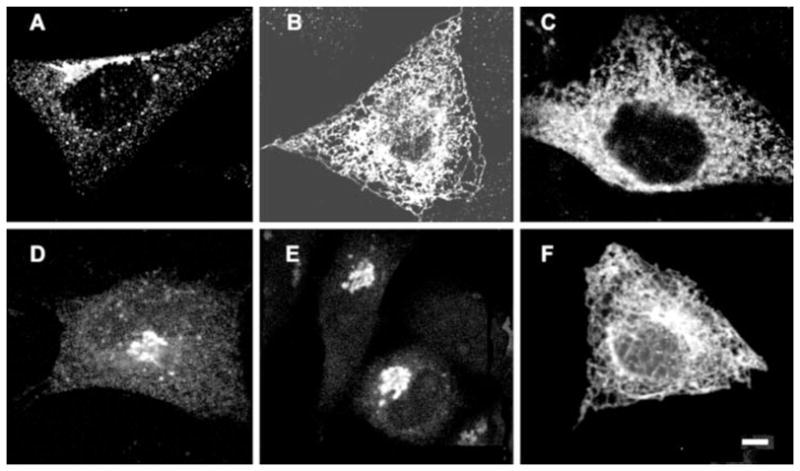
HA-tagged PRA1 was transfected into CHO cells and detected with anti-HA antibodies. A, wild type PRA1. B, truncated PRA1-(1–164). C, class A mutant N70T. D and E, class B mutants S76A and V161A, respectively. F, class C mutant H166A. Scale bar, 10 μm.
PRA1 Mutants Exhibit Altered Golgi Morphology
To determine whether mislocalization of PRA1 has any effect on Golgi morphology, we examined the localization of the prototypical Golgi resident enzyme Man II. As shown in Fig. 2, Man II appeared as a well defined perinuclear crescent structure in CHO transfected with the wild type PRA1. However, in both class A (N70T) and C (H166A) mutants, Man II labeling appeared more dispersed at the perinuclear region with extensive punctate vesicular staining throughout the cell body. Because Man II recycles via COPI-containing vesicles from the trans-Golgi (6), the dispersed Golgi appearance in class A and C mutants is suggestive of impairment in recycling of these Man II-containing vesicles. In the class B (S76A) mutants, Man II tightly co-localized with the mutant PRA1 in the highly condensed Golgi complex (Fig. 2). This highly condensed Golgi complex remained sensitive to disruption by BFA (Fig. 3), which was also observed in the wild type and in all mutants (data not shown).
Fig. 2. Morphology of the Golgi complex.

Cellular localization of PRA1 was identified with anti-HA and the Golgi complex with anti-Man II as indicated. CHO cells were transfected with the wild type PRA1, a representative of the class A mutant N70T, class B mutant S76A, or class C mutant H166A.
Fig. 3. Disruption of the Golgi complex by BFA.
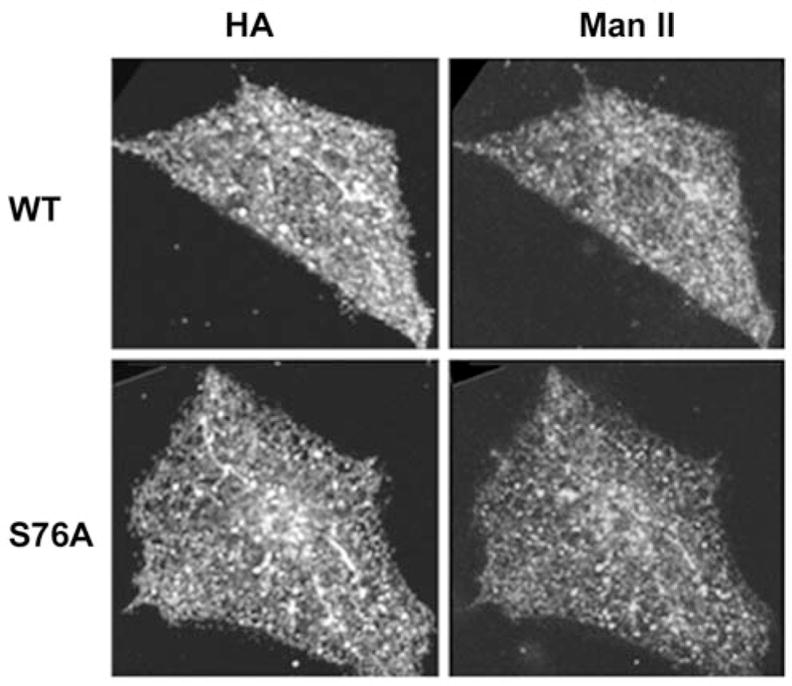
Cellular localization of PRA1 was identified with anti-HA and the Golgi complex with anti-Man II in cells transfected with the wild type and S76A mutant PRA1. The cells were treated with 10 μg/ml of BFA for 30 min before fixing.
Anterograde Transport of VSVG through the Golgi Complex Is Impaired
To test whether the altered Golgi morphology is due to impairment in Golgi transport, we examined the transport of the anterograde-directed VSVGts045 tagged with GFP (19) through the secretory pathway. We took a representative from each class of mutants and established stable clonal lines. The cells were transfected with VSVGts045-GFP and shifted to the non-permissive temperature of 42 °C for 5 h to trap the GFP-tagged protein at the ER. This prolonged incubation also allowed complete clearance of preexisting VSVGts045-GFP from the plasma membrane and other intracellular compartments. Protein synthesis was then blocked with cycloheximide, and the cells were returned to the permissive temperature. The cells were fixed with paraformaldehyde at various times, after release from the non-permissive temperature, and scored for the presence of VSVGts045-GFP in the various intracellular compartments. In CHO cells, VSVGts045-GFP was evident in dispersed perinuclear structures 15 min after release (Fig. 4). At 30 min, VSVGts045-GFP has reached the Golgi complex with significant accumulation at the plasma membrane at 60 min release. Almost all of the cells exhibited VSVGts045-GFP at the plasma membrane after 90 min. Cells overexpressing the wild type PRA1 showed a slight delay in transport of VSVGts045-GFP to the Golgi complex such that a significant amount of the protein remained in the ER or in dispersed intermediate compartment at 30 min release. However, the bulk of the protein was transported to the Golgi complex at 60 min and eventually reached the plasma membrane after 90 min. In contrast there was a significant delay in transport of VSVGts045-GFP out of the ER in both the class A mutant N70T and C mutant H166A after 15 min release. The protein finally cleared the ER compartment after 30 min with some reaching the plasma membrane at 90 min in the class A mutant N70T. Finally, VS-VGts045-GFP was transported rapidly from the ER to Golgi in the class B mutant S76A after 15 min release. However, the protein remained at the Golgi complex with little or no transport to the plasma membrane after 90 min.
Fig. 4. Transport of VSVGts045-GFP in CHO and cells stably transfected with wild type or mutant PRA1.
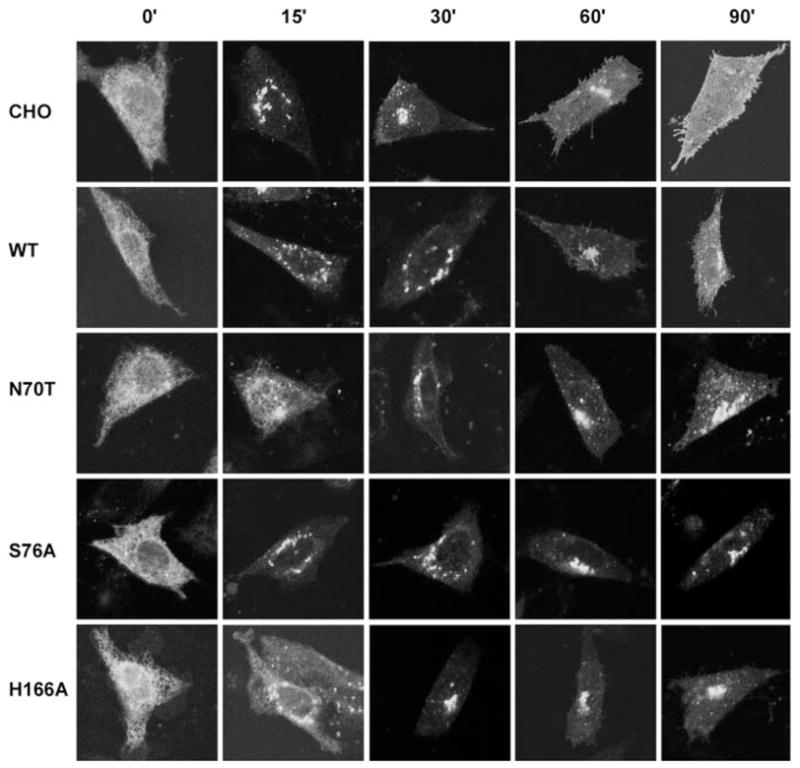
Representative confocal images of VSVGts045-GFP in CHO expressing wild type or mutant PRA1 (as indicated in left margin). Cells were incubated at the non-permissive temperature for 5 h and shifted to the permissive temperature for time intervals indicated at the top.
We quantified the percentage of cells exhibiting the VS-VGts045-GFP signal at the different intracellular compartments after release from the non-permissive temperature. In untransfected CHO, only 10% of the cells retained a detectable VS-VGts045-GFP signal in the ER 15 min after release (Fig. 5A). VSVGts045-GFP rapidly exited the Golgi complex (Fig. 5B) and reached the plasma membrane at 90 min release (Fig. 5C). In cells transfected with the wild type PRA1, a significant percentage of the cells showed VSVGts045-GFP retention in the ER at 15 min release (Fig. 5A) and in the Golgi at 60 –90 min (Fig. 5B). This resulted in a decrease in the number of cells with detectable VSVGts045-GFP at the plasma membrane at 90 min (Fig. 5C). An even larger fraction of the cells showed ER accumulation of VSVGts045-GFP at 15 min release in the class A mutant N70T. There was also decreased localization of VS-VGts045-GFP in the plasma membrane at 90 min. Retention of VSVGts045-GFP in the ER at 15 min release was less severe in the class B mutant S76A (Fig. 5A), but the protein failed to exit the Golgi complex (Fig. 5B) resulting in very low plasma membrane localization at 90 min release (Fig. 5C). This was also seen in the class C mutant H166A except that this mutant also exhibited extensive accumulation in the ER at 15 min release (Fig. 5A). Thus, overexpression of wild type PRA1 has a mild inhibitory effect on transport of the anterograde cargo VS-VGts045-GFP to its final destination at the plasma membrane. Transport out of the ER was significantly affected in the class A mutant N70T, although final localization to the plasma membrane was also affected. This also appeared to be the case for the class C mutant H166A. Finally, exit from the Golgi complex was the major defect in the class B mutant S76A.
Fig. 5. Percentage of cells with VSVGts045-GFP in intracellular compartments and on the cell surface.

A, fraction of cells with VSVGts045-GFP in the ER at 15 min release. B, fraction of cells with VSVGts045-GFP in the Golgi complex at various times in the permissive temperature. C, fraction of cells with VSVGts045-GFP in the plasma membrane at various times in the permissive temperature. B and C, untransfected CHO (●), wild type PRA1 (■), class A mutant N70T (▲), class B mutant S76A (○), and class C mutant H166A (□).
Delayed Transport of VSVG to the Cell Surface
To verify that transport of VSVGts045-GFP to the plasma membrane was indeed delayed, we labeled cell surface-exposed VSVG with BW8G65, a monoclonal antibody against the extracellular domain, under non-permeabilized conditions. In all cases, VSVG was undetectable at the cell surface after 5 h of incubation at the non-permissive temperature, thus verifying that the luminal domain was indeed inaccessible to the antibody under our non-permeabilizing conditions (Fig. 6). VSVG was detectable in CHO as discrete patches on the cell surface at 60 min release, and the entire surface was labeled at 120 min release (Fig. 6). Patches of VSVG were detected on the plasma membrane in cells overexpressing wild type PRA1 at 60 and 90 min release. However, the overall intensities were significantly lower than the untransfected CHO at the corresponding time points. Even less VSVG reached the cell surface in cells transfected with mutant PRA1s at both 60 and 120 min release with S76A being the worst (Fig. 6). The green fluorescence signal was quite strong in all mutants indicating extensive intracellular accumulation of VSVGts045-GFP. Taken together, the data indicate that overexpression of wild type PRA1 delayed the transport of VSVGts045-GFP to the cell surface. This effect was exacerbated by the mutant PRA1s with S76A showing the highest level of inhibition.
Fig. 6. Delayed transport of VSVG to the cell surface.
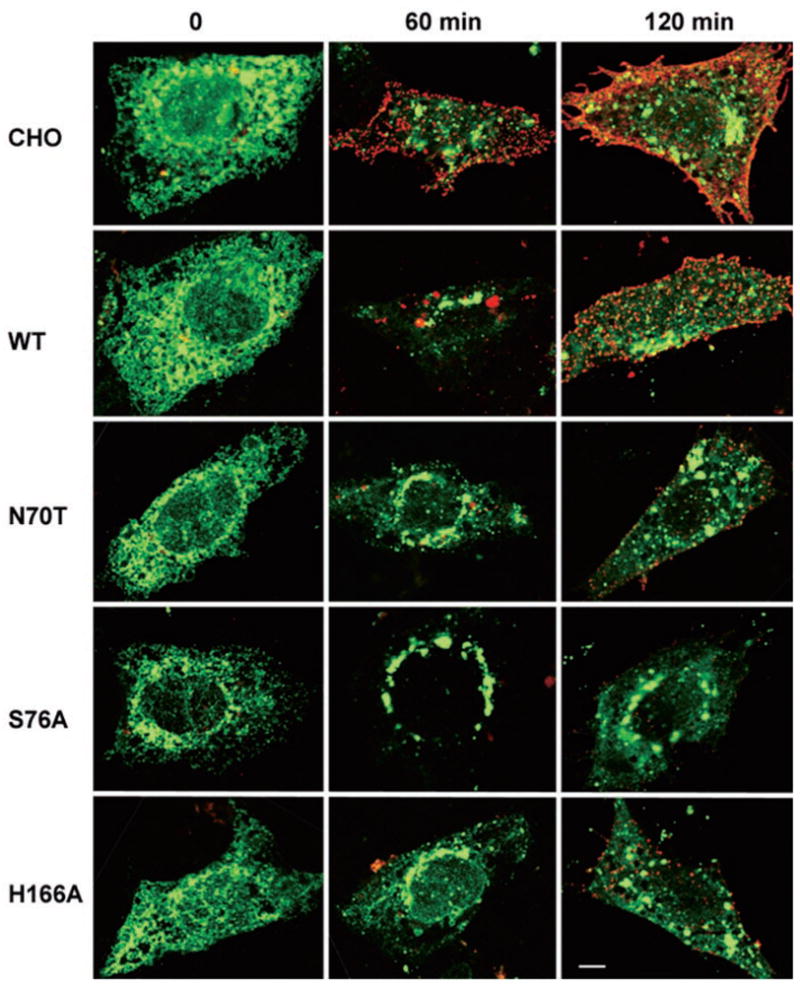
Representative confocal images of VSVGts045-GFP (green) at 0, 60, and 120 min release. Surface exposed VSVG was stained with BW8G65 (red), a monoclonal antibody directed against the luminal domain of VSVG, under non-permeabilizing conditions. Scale bar, 10 μm. WT, wild type.
Mutation of PRA1 Affects Binding to Rab3A and VAMP2
We next examined the binding properties of the mutant PRA1s to determine whether this might be an underlying cause of the altered Golgi morphology and inhibition in transport of VS-VGts045-GFP. We and others (13, 16, 22) have shown previously that PRA1 binds to Rab GTPases and VAMP2. We first screened the binding properties of the mutant PRA1 in the yeast two-hybrid system by subcloning representatives from each into the prey vector. The resulting vectors were co-transformed into the Y190 tester strain with either Rab3A as a representative Rab GTPase or VAMP2 bait, and the transformants were scored for β-galactosidase activity. All class A mutants showed extremely weak or no interaction with either Rab3A or VAMP2, whereas the class B mutants showed increased interaction with both Rab3A and VAMP2 (Table II). The class C mutant H166A showed a weak interaction with VAMP2 but lost its interaction with Rab3A. Thus, the data suggest that interaction of the mutant PRA1 with Rab and VAMP2 may underlie mislocalization of the protein, the altered Golgi morphology, and defect in VSVGts045-GFP transport.
Table II. Yeast two-hybrid screen of PRA1 mutants against Rab3A or VAMP2.
The bait and prey plasmids were co-transformed into Y190 tester strain and selected on Trp, Leu drop-out plates. The transformants were tested for β-galactosidase activity on X-gal filter paper and intensity as well as the time of onset used to assess the strength of the interactions.
| PRA1 prey | Rab3A bait | VAMP2 bait |
|---|---|---|
| WT | ++ | ++ |
| PRA1-(1–164) | − | − |
| N70T | − | − |
| Y78A | − | − |
| S76A | +++ | +++ |
| V161A | +++ | +++ |
| H166A | − | + |
We verified the binding properties of the mutant PRA1s by in vitro pulldown assays using purified recombinant His6-HA-tagged PRA1s, GST-VAMP2, and His6-tagged Rab3A, with Rab3A expressed in yeast to ensure prenyl modification that is essential for PRA1 binding. Because both PRA1 and Rab3A were His6-tagged, purified PRA1s were covalently attached to CNBr-Sepharose beads. We first determined the saturating amount of wild type PRA1 needed to pulldown GDP-bound Rab3A. We then used the EC50 value to determine the amount of Rab3A recovered with the mutant PRA1s by Western immunoblot (Fig. 7A), and we normalized this to the wild type PRA1 (Fig. 7B). In all cases, we detected two immunoreactive Rab3A (Fig. 7A), which probably represents mono- and di-geranylgeranylated species. A similar approach was used to determine VAMP2 binding using glutathione-agarose beads. The class A mutant N70T showed only residual binding to Rab3A and VAMP2 when compared with wild type PRA1 (Fig. 7B). In contrast, there was a 3-fold increase in Rab3A and a 6-fold increase in VAMP2 binding in the class B mutant S76A. A significant reduction in Rab3A and VAMP2 binding was observed in the class C mutant H166A. Thus, there was a direct correlation between cellular phenotype and ability of PRA1 to bind Rab and VAMP2. Loss of Rab and VAMP2 binding in the class A mutants correlated with retention of the mutant PRA1 in the ER, whereas enhanced binding to both in the class B mutants correlated with a highly condensed Golgi. A decrease in both Rab and VAMP2 binding in the class C mutants correlated with an intermediate phenotype.
Fig. 7. In vitro binding of Rab3A and VAMP2 to PRA1.

A, representative Western immunoblot using anti-Rab3A (upper panel) and anti-HA (lower panel). Immobilized PRA1s was used for Rab3A pulldown, and glutathione-agarose was used to recover GST-VAMP2. Control beads (C), wild type PRA1 (W), N70T (N), S76A (S), or H166A (H). B, binding of Rab3A and VAMP2 to the mutant PRA1s normalized to that of the wild type PRA1. Values represent mean and S.E. (n = 3 with each performed in triplicate).
DISCUSSION
PRA1 is a protein that is localized primarily to the Golgi complex (14) and post-Golgi compartments (15). We have shown here that point mutations of PRA1 can alter its cellular localization as well as vesicle trafficking. The class A mutants displayed an ER phenotype along with a significant decrease in binding to both Rab3A and VAMP2. A number of factors are known to affect protein transport from the ER to Golgi complex. Efficient exit of PRA1 from the ER has been shown to be dependent on the DXEE motif at the carboxyl terminus (23). Because this motif remained intact in the class A mutants, it is unlikely that a defect in the DXEE-mediated mechanism is the underlying cause of ER retention. Protein folding or oligomerization is another factor. Although it is possible that all class A point mutations might bring about complete denaturation of the protein, we believe that it is more plausible that functional interaction of PRA1 with Rab is required for anterograde transport. A number of studies have shown involvement of Rab GTPases in vesicle formation at various stages of the secretory and endocytic pathways (24 –27). Such loss of Rab binding would affect either membrane recruitment of Rab or Rab effectors leading to inhibition of transport. This would be consistent with both accumulation of recycling Man II-containing vesicles and delay in ER to Golgi transport of the anterograde cargo VSVG in these cells. Thus, the significant decrease in binding to Rab and VAMP2 implies that functional interaction of PRA1 with these proteins is tightly linked to vesicular transport. It remains to be seen whether the effect on ER to Golgi transport is solely due to PRA1 or interference of PRA2, which is the isoform localized exclusively to the ER compartment (23).
The condensed Golgi morphology in the class B mutant S76A indicates that PRA1 is also involved in trafficking through this compartment. The mutation significantly inhibited transit of the anterograde cargo VSVGts045-GFP through the Golgi and its subsequent incorporation into the plasma membrane. Because PRA1 is known to interact with multiple Rab GTPases (13, 16, 22, 23, 28), it is likely that Golgi-localized Rab GT-Pases, which in yeast are required for formation of transport vesicles out of the Golgi apparatus (29), are affected. We surmise that the increased binding of Rab and VAMP2 by the class B mutant S76A might affect either recycling or functional interaction with Rab effector molecules that ultimately lead to decreased transport out of the Golgi apparatus.
Our data suggest a direct involvement of PRA1 in antero-grade transport to and out of the Golgi complex. Aside from its interaction with Rab GTPases (13, 16, 22, 23, 28), PRA1 also binds GDI (14) and SNAREs (13, 30), which is consistent with its assigned transport function based on interaction of the yeast homologue Yip3p with proteins in the secretory pathway (31, 32). The ability of PRA1 to form at least a dimer (13, 32) suggests that it might recruit Rab GTPases and their effectors to membrane domains along with functional SNAREs to ensure proper sequestration in the budding vesicle. ER to Golgi transport was affected in the class A mutants possibly due to incorporation of the mutant protein to the ER, which interferes with proper functioning through oligomerization with endogenous protein. Defect at later transport steps might arise from reduced exit of endogenous PRA1 and other components of the secretory machinery from the ER. This is likely to be the same underlying mechanism in the class C mutants although at lower severity. In the class B mutants, the increased binding to Rab GTPases may either interfere with functional interaction with their effectors or their retrieval from membranes. Exit of proteins such as VAMP2 might also be inhibited thereby affecting the fusion of secretory vesicles with the plasma membrane. The localization of the mutant PRA1 to the Golgi complex is consistent with its effect on exit of anterograde cargo from this compartment. The condensed Golgi appearance is similar to that observed in cells expressing dominant negative dynamin 2, which blocks vesicle formation from the trans-Golgi network (33). Thus, our data suggest that PRA1 is required for vesicle formation from the Golgi complex and that PRA1 might influence the recruitment of Rab effectors during cargo sequestration as well as proteins required for subsequent vesicle docking and fusion. This may serve to ensure that a budding vesicle has all the necessary proteins for subsequent docking and fusion.
Acknowledgments
We thank Dr. M. G. Farquhar for the generous gift of Man II and BW8G65 antibodies.
Footnotes
This work was supported by a grant from the Canadian Institute of Health Research.
The abbreviations used are: GDI, GDP dissociation inhibitor; PRA1, prenylated Rab acceptor; BFA, brefeldin A; Man II, mannosidase II; VAMP2, vesicle-associated membrane protein-2 or synaptobrevin-2; HA, hemagglutinin; CHO, Chinese hamster ovary; ER, endoplasmic reticulum; X-gal, 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside; GFP, green fluorescent protein; GDPβS, guanyl-5′-yl thiophosphate; GST, glutathione S-transferase; SNARE, soluble NSF attachment protein receptors, where NSF is N-ethylmaleimide-sensitive factor; HA, hemagglutinin; WT, wild type; ARF, ADP-ribosylation factor.
References
- 1.Horiuchi H, Lippe R, McBride HM, Rubino M, Woodman P, Stenmark H, Rybin V, Wilm M, Ashman K, Mann M, Zerial M. Cell. 1997;90:1149–1159. doi: 10.1016/s0092-8674(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 2.Wada M, Nakanishi H, Satoh A, Hirano H, Obaishi H, Matsuura Y, Takai Y. J Biol Chem. 1997;272:3875–3878. doi: 10.1074/jbc.272.7.3875. [DOI] [PubMed] [Google Scholar]
- 3.Jackson MR, Nilsson T, Peterson PA. J Cell Biol. 1993;121:317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothman JE, Wieland FT. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- 5.Peyroche A, Antonny B, Robineau S, Acker J, Cherfils J, Jackson CL. Mol Cell. 1999;3:275–285. doi: 10.1016/s1097-2765(00)80455-4. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Menarguez JA, Prekeris R, Oorschot VM, Scheller R, Slot JW, Geuze HJ, Klumperman J. J Cell Biol. 2001;155:1213–1224. doi: 10.1083/jcb.200108029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YA, Scheller RH. Nat Rev Mol Cell Biol. 2001;2:98–106. doi: 10.1038/35052017. [DOI] [PubMed] [Google Scholar]
- 8.McBride HM, Rybin V, Murphy C, Giner A, Teasdale R, Zerial M. Cell. 1999;98:377–386. doi: 10.1016/s0092-8674(00)81966-2. [DOI] [PubMed] [Google Scholar]
- 9.Peterson MR, Burd CG, Emr SD. Curr Biol. 1999;9:159–162. doi: 10.1016/s0960-9822(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 10.Tall GG, Hama H, DeWald DB, Horazdovsky BF. Mol Biol Cell. 1999;10:1873–1889. doi: 10.1091/mbc.10.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. Proc Natl Acad Sci U S A. 2000;97:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen E, Christoforidis S, Uttenweiler-Joseph S, Miaczynska M, Dewitte F, Wilm M, Hoflack B, Zerial M. J Cell Biol. 2000;151:601–612. doi: 10.1083/jcb.151.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martincic I, Peralta ME, Ngsee JK. J Biol Chem. 1997;272:26991–26998. doi: 10.1074/jbc.272.43.26991. [DOI] [PubMed] [Google Scholar]
- 14.Hutt DM, Da-Silva LF, Chang LH, Prosser DC, Ngsee JK. J Biol Chem. 2000;275:18511–18519. doi: 10.1074/jbc.M909309199. [DOI] [PubMed] [Google Scholar]
- 15.Fenster SD, Chung WJ, Zhai R, Cases-Langhoff C, Voss B, Garner AM, Kaempf U, Kindler S, Gundelfinger ED, Garner CC. Neuron. 2000;25:203–214. doi: 10.1016/s0896-6273(00)80883-1. [DOI] [PubMed] [Google Scholar]
- 16.Figueroa C, Taylor J, Vojtek AB. J Biol Chem. 2001;276:28219–28225. doi: 10.1074/jbc.M101763200. [DOI] [PubMed] [Google Scholar]
- 17.Li LY, Shih HM, Liu MY, Chen JY. J Biol Chem. 2001;276:27354–27362. doi: 10.1074/jbc.M103821200. [DOI] [PubMed] [Google Scholar]
- 18.Evans DT, Tillman KC, Desrosiers RC. J Virol. 2002;76:327–337. doi: 10.1128/JVI.76.1.327-337.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 20.Gietz D, St Jean A, Woods RA, Schiestl RH. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel P, Chien CT, Sternglanz R, Fields S. BioTechniques. 1993;14:920–924. [PubMed] [Google Scholar]
- 22.Bucci C, Chiariello M, Lattero D, Maiorano M, Bruni CB. Biochem Biophys Res Commun. 1999;258:657–662. doi: 10.1006/bbrc.1999.0651. [DOI] [PubMed] [Google Scholar]
- 23.Abdul-Ghani M, Gougeon PY, Prosser DC, Da-Silva LF, Ngsee JK. J Biol Chem. 2001;276:6225–6233. doi: 10.1074/jbc.M009073200. [DOI] [PubMed] [Google Scholar]
- 24.Riederer MA, Soldati T, Shapiro AD, Lin J, Pfeffer SR. J Cell Biol. 1994;125:573–582. doi: 10.1083/jcb.125.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson BS, Nuoffer C, Meinkoth JL, McCaffery M, Feramisco JR, Balch WE, Farquhar MG. J Cell Biol. 1994;125:557–571. doi: 10.1083/jcb.125.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLauchlan H, Newell J, Morrice N, Osborne A, West M, Smythe E. Curr Biol. 1998;8:34–45. doi: 10.1016/s0960-9822(98)70018-1. [DOI] [PubMed] [Google Scholar]
- 27.Mammoto A, Sasaki T, Kim Y, Takai Y. J Biol Chem. 2000;275:13167–13170. doi: 10.1074/jbc.c000096200. [DOI] [PubMed] [Google Scholar]
- 28.Janoueix-Lerosey I, Jollivet F, Camonis J, Marche PN, Goud B. J Biol Chem. 1995;270:14801–14808. doi: 10.1074/jbc.270.24.14801. [DOI] [PubMed] [Google Scholar]
- 29.Benli M, Doring F, Robinson DG, Yang X, Gallwitz D. EMBO J. 1996;15:6460–6475. [PMC free article] [PubMed] [Google Scholar]
- 30.Ito T, Tashiro K, Muta S, Ozawa R, Chiba T, Nishizawa M, Yamamoto K, Kuhara S, Sakaki Y. Proc Natl Acad Sci U S A. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwikowski B, Uetz P, Fields S. Nat Biotechnol. 2000;18:1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- 32.Bader GD, Donaldson I, Wolting C, Ouellette BF, Pawson T, Hogue CW. Nucleic Acids Res. 2001;29:242–245. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao H, Thompson HM, Krueger EW, McNiven MA. J Cell Sci. 2000;113:1993–2002. doi: 10.1242/jcs.113.11.1993. [DOI] [PubMed] [Google Scholar]