

Abstract
The robustness of proteins against point mutations implies that only a small subset of residues determines functional properties. We test this prediction using photoactive yellow protein (PYP), a 125-residue prototype of the PER-ARNT-SIM (PAS) domain superfamily of signaling proteins. PAS domains are defined by a small number of conserved residues of unknown function. We report high-throughput biophysical measurements on a complete Ala scan set of purified PYP mutants. The dataset of 1,193 values on active site properties, functional kinetics, stability, and production level reveals that 124 mutants retain the characteristic photocycle of PYP, but that the majority of substitutions significantly alter functional properties. Only 35% of substitutions that strongly affect function are located at the active site. Unexpectedly, most PAS-conserved residues are required for maintaining protein production. PAS domain activation often involves conformational changes in α-helices linked to the PAS core. However, the mechanism of transmission and kinetic regulation of allosteric structural changes from the PAS domain to these helices is not clear. The Ala scan data reveal interactions governing allosteric switching in PYP. The photocycle kinetics is significantly altered by substitutions at 58 positions and spans a 3,000-fold range. Nine residues that dock the N-terminal α-helices of PYP to its PAS core regulate signaling kinetics. Ile39 and Asn43 are identified as part of a mechanism for regulating allosteric switching that is conserved among PAS domains. These results show that PYP combines robustness with a high degree of evolvability and imply production level as an important factor in protein evolution.
Keywords: allosteric signal transmission, molecular evolution, protein structure-function relationship
Two central challenges in understanding proteins are to predict (i) the three-dimensional structure adopted by a protein with a given amino acid sequence (the folding problem), and (ii) the functional properties of a protein with a given three-dimensional structure (the “function problem”). The vastness of amino acid sequence space presents a formidable challenge to addressing these problems. One approach to these questions is evolutionary analysis of sequence and structure databases. This strategy has led to the important insight that only a limited number of protein folds exists. In addition, it has revealed superfamilies of protein domains defined by a small number of conserved residues (1, 2). These residues could either constitute the active site or be required for folding. The role of conserved residues in folding is debated (3, 4). Large-scale database analysis has revealed that down to 30% sequence identity active site chemistry in protein superfamilies is maintained (5). However, the level of sequence conservation in most superfamilies is much lower than this. A related discovery is that enzyme superfamilies can be mechanistically diverse and catalyze different chemical reactions (6, 7). Therefore, the few residues that are conserved in protein superfamilies do not appear to be part of a conserved actives site, which leaves the functional role of the small number of conserved residues in protein superfamilies unexplained.
A second approach to exploring amino acid sequence space utilizes mutagenesis. Such studies typically evaluate the effects of mutations through in vivo processes that are easily detected, such as plaque formation (8) and antibiotics resistance (9). However, these processes can be affected by many different properties of the protein, including its folding and functional activity. Here we resolve this complication by dissecting the effects of mutations on specific in vitro biochemical properties of a library of purified mutant proteins. Extensive mutagenesis studies have revealed a high degree of robustness in proteins, where only ∼15% of point mutations cause loss of function (8–10). Influential ideas that guide thinking about protein structure–function relationships are that structurally critical residues are often buried and part of the hydrophobic core, whereas functionally critical residues are largely confined to the protein active site. The latter view is challenged by the results reported here.
Robustness against point mutations is a general feature of proteins and prevents frequent catastrophic loss of function during protein evolution. It is often assumed that this robustness implies that most substitutions do not significantly alter the functional properties of a protein. This consideration is in line with Kimura’s neutral theory of evolution, in which most mutations do not alter the properties of the organism (11). We test this proposal using high-throughput methods to examine functional properties of all mutants from a complete Ala mutagenesis scan of a PER-ARNT-SIM (PAS) domain protein.
The PAS domain is a ubiquitous protein module with a characteristic three-dimensional fold (12, 13). The term PAS is derived from the first letter of the name of the three founding members of the superfamily: the Drosophila period protein, the aryl hydrocarbon receptor nuclear translocator (ARNT), and the Drosophila single-minded protein. Over 20,000 different PAS domains have been identified in a wide variety of signaling proteins in organisms ranging from bacteria to humans. PAS domains consist of ∼100 residues, only nine of which are well conserved (12) (Fig. S1A). PAS domains can bind a variety of cofactors (12) and exhibit a wide range of active site chemistries. Cofactors at the active sites of PAS domains including p-coumaric acid (pCA) in photoactive yellow protein (PYP), heme in the oxygen sensor FixL, and flavin in LOV photoreceptors, but some function in the absence of cofactors. In addition, PAS domains exhibit a wide range of active site chemistries, exemplified by pCA isomerization and protonation in PYP (14) and transient formation of a Cys-flavin adduct in LOV (15). Thus, PAS domains do not share a conserved active site, and the molecular role played by the residues that are conserved in this superfamily is not clear. The diversity of the PAS domain superfamily provides an opportunity to examine how proteins evolve to perform a wide range of functions. What is the percentage of mutations that alter functional properties? How do these mutations relate to the three-dimensional structure of the protein? These questions are also relevant to the debate on the extent to which amino acid substitutions are selectively neutral or adaptive (11). Here we quantify these key aspects of protein evolvability in a photosensory PAS domain.
PYP is a photoreceptor for negative phototaxis in the halophilic Proteobacterium Halorhodospira halophila (16–18). The protein is regarded as a prototype PAS domain (19) and was the first PAS domain to have its high-resolution structure determined (20). PYP exhibits a light-triggered photocycle (17) based on its pCA chromophore (21, 22). The pCA is enclosed by the protein (20) and is covalently attached to Cys69 (23). The function of PYP involves strong shifts in the pKa and absorbance maximum of the pCA compared to its properties in solution (24). PYP consists of 125 residues divided into two regions: a typical PAS domain fold (13) with a central antiparallel six-stranded β-sheet flanked by three α-helices (the PAS domain core), and two N-terminal α-helices (residues 1–27, the N-terminal region). The N-terminal helices pack against the central β-sheet, forming a second, small hydrophobic core. Structure–function relationships in PYP have been studied by site-directed mutagenesis, revealing a number of residues that are critical to its function (24). Photoexcitation causes the conversion of the initial pG dark state of PYP into the long-lived pB state. The pB state has a blue-shifted absorbance spectrum and is partially unfolded (25–29). Its formation involves dissociation of the N-terminal region from the core of PYP (30, 31). The pB state is believed to be the signaling state of PYP, and spontaneously returns to the pG state in a few hundred milliseconds.
Results
Robustness in PYP Determined by 96-Well Biophysics.
We have developed 96-well-based methods for the heterologous expression, purification, and spectroscopic characterization of PYP mutants (32). Here, we use this microscale high-throughput approach to study a complete Ala scan library of PYP mutants. The residue at each position was substituted with Ala; positions containing Ala in WT PYP were substituted with Gly. Each of the 125 resulting point mutants was quantitatively examined with respect to six properties: (i) visible absorbance maximum (λmax), (ii) pCA 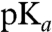 , (iii) pCA fluorescence quantum yield (Φfl), (iv) lifetime of the pB state (τpB), (v) thermodynamic stability against unfolding (ΔGU), and (iv) protein production level (see SI Materials and Methods). The total dataset reported here consists of 1,193 values (Table S1). These microscale high-throughout data are in very good agreement with published results (Table S2). In addition, we examined selected mutants by mass spectrometry. In all cases, the measured mass corresponded to that predicted for the mutant, and no cases of cross-contamination of different samples during growth in 96-well plates were identified.
, (iii) pCA fluorescence quantum yield (Φfl), (iv) lifetime of the pB state (τpB), (v) thermodynamic stability against unfolding (ΔGU), and (iv) protein production level (see SI Materials and Methods). The total dataset reported here consists of 1,193 values (Table S1). These microscale high-throughout data are in very good agreement with published results (Table S2). In addition, we examined selected mutants by mass spectrometry. In all cases, the measured mass corresponded to that predicted for the mutant, and no cases of cross-contamination of different samples during growth in 96-well plates were identified.
We examined the in vitro functional activity of the 125 PYP mutants (Fig. 1). All but one of the mutants retain a photocycle with a pB intermediate. The only mutation causing loss of function is C69A, in which the chromophore is no longer covalently attached to the protein (23). In addition, all mutants (see Materials and Methods) retain a significant ΔGU. This result demonstrates a very high degree of robustness of the structure and function of PYP against point mutations. The level of robustness of PYP against point mutations is approximately 20-fold higher for that reported for other proteins (8–10), likely because in previous studies functional proteins with greatly reduced kinetics or production level would be scored as nonfunctional based on in vivo essays.
Fig. 1.

Mapping changes in functional and structural properties caused by point mutations on the amino acid sequence of PYP. The effect of Ala substitutions on visible absorbance maximum λmax, pCA pKa, pCA fluorescence quantum yield Φfl, pB lifetime τpB, thermodynamic stability  , and protein production level are shown as the value of [(value of mutant – value of WT)/value of WT]. The secondary structure of PYP is also shown, with α-helices in red, β-strands in blue, and the π-helix in yellow. Positions at which substitutions cause large increases in values are indicated in red, small increases in yellow, large decreases in dark blue, and small decreases in light blue. The same color scheme is used in Figs. 2 and 3 to facilitate visual inspection of the dataset.
, and protein production level are shown as the value of [(value of mutant – value of WT)/value of WT]. The secondary structure of PYP is also shown, with α-helices in red, β-strands in blue, and the π-helix in yellow. Positions at which substitutions cause large increases in values are indicated in red, small increases in yellow, large decreases in dark blue, and small decreases in light blue. The same color scheme is used in Figs. 2 and 3 to facilitate visual inspection of the dataset.
Quantifying Evolvability in PYP.
The Ala scan dataset allows us to quantify the evolvability of PYP with respect to the number of residues that alter functional properties and the range of values that these properties adopt. Although substitutions at only 0.8% of all positions cause the loss of photochemical activity, ∼60% significantly alter one or more of the six properties studied here. Active site properties are affected by ∼15% of the residues in PYP. Mutations at 58 residues affect τpB (Fig. S2) by more than 50% compared to WT PYP. Substitutions at 53 positions alter the ΔGU of PYP by more than 1 kcal/mol. Finally, 51 substitutions change the production level of PYP by more than 50%.
The values of the functional properties in the PYP mutant set varies over a wide range (Fig. 2 and Table 1). Visible λmax values range from 439 to 468 nm, the pKa of the pCA ranges from 3 to 8, and pCA Φfl values vary 13-fold (from 0.07% to 0.94%). The value of τpB is altered more than 3,000-fold (from 0.5 s to 30 min). ΔGU values from 7.2 to 13.9 kcal/mol were observed, whereas protein production levels varied from a factor 8 lower to a factor 2 higher than WT PYP. Thus, although PYP displays strong robustness against mutations, it simultaneously exhibits a high degree of evolvability: A typical mutation likely does not abolish function, but will alter the functional properties of the protein. This result indicates that modification of functional properties of a protein during evolution can readily occur.
Fig. 2.

Range of functional properties observed in the Ala scan of PYP. The arrows indicate the values of WT PYP, whereas the horizontal bar indicates the error in the measurement. Positions for which substitutions cause large increases in values are indicated in red, small increases in yellow, large decreases in dark blue, and small decreases in light blue.
Table 1.
Quantifying the evolvability of PYP
| λmax | 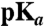 |
Φfl | τpB | ΔGU | Production | |
| % affected mutants* | 20% | 7% | 23% | 52% | 51% | 74% |
| % strongly affected mutants* | 12%† | 7%‡ | 9%§ | 20%¶ | 28%∥ | 41%§ |
| Range of values observed | 439–470 nm | 3–8 | 0.07–0.94% | 0.5 s–30 min | 7.2–13.9 kcal/mol | 13–200% |
| Value of WT PYP | 447 nm | 3.0 | 0.14 | 0.9 s | 12.5 kcal/mol | 100% |
*From a total of 125 substituted positions.
†Altered by at least 3 nm compared to WT PYP.
‡Altered by at least 1.5 pH units.
§Altered by at least ± 50%.
¶Altered by at least a factor 5.
∥Altered by at least 2 kcal/mol.
The properties measured here would be expected to be under selective pressure (see SI Text). Thus, the finding that substitutions at the majority of the residues in PYP (60%) cause readily detectable effects in these properties argues against the prevalent view (11) that the majority of substitutions are described by Kimura’s neutral theory of evolution. The percentage of selectively nearly neutral substitutions in Drosophila was recently reported to be 46% (33), very similar to the 40% of substitutions in PYP that do not significantly alter the properties measured here. The results reported here imply that the majority of point mutations in proteins cause changes in functional properties expected to be under selective pressure.
Protein Production Level as a Key Factor in Amino Acid Sequence Conservation.
Unexpectedly, substitutions at 39 positions decrease PYP production level by more than 60%. Protein production levels vary by 16-fold in the PYP mutants reported here. SDS-PAGE analysis (Fig. S3) of the soluble and insoluble fractions of the Escherichia coli cells expressing these PYP derivatives indicated the insolubility of some mutants and proteolytic degradation (little PYP detected) of others, similar to published results on bacteriophage λ Cro (34).
We examined the possibility that the observed reduced PYP production levels are due to mutations in the plasmid or due to the 96-well format of protein expression and purification. We selected the 10 mutants with very low PYP production levels (D34A, G59A, K64A, A67G, P68A, F75A, E93A, K104A, G115A, and F121A) and recloned the pyp genes encoding these mutants. After confirmation of the DNA sequence of the inserts, these 10 mutants and WT PYP were expressed in individual 15 mL cultures and purified by batch chromatography using Ni-NTA agarose. Expression levels were evaluated based on the pCA absorbance band of the purified proteins at neutral pH. The results confirmed that the 10 mutants indeed have strongly reduced protein production levels (Fig. S3E).
The data reported here on substitutions that alter the production level of PYP concern the artificial heterologous overexpression of the PYP from H. halophila in E. coli. However, a number of observations provide strong support that this system yields relevant information on structural features that affect protein production. First, the residues that significantly reduce production are structurally cohesive: They are present as a cluster in the core of PYP (see below). Secondly, 67% and 61% of the residues in PYP that are highly conserved in the PYP family and the PAS domain superfamily significantly reduce the protein overproduction level, respectively (see below). For comparison, less than 30% of all residues in PYP result in a similar level of reduced production. This link of production level to conserved residues supports the physiological relevance of the production levels reported here.
The existence of complex regulatory mechanisms in all organisms to control the expression level of proteins implies that this is a property under strong selective pressure. The strong effect of 31% of the residues in PYP on its production suggests that constraints imposed by maintaining an adequate production level are an important factor in protein evolution.
Functional Anatomy of PYP.
The high-resolution crystal structure of PYP (20) allows a detailed structural analysis of the Ala scan results (Fig. 3 and Fig. S4). Residues at the active site of a protein often exhibit unusual properties, such as strongly shifted pKa values, that are functionally important. How do proteins achieve such tuning of active site residues? Our data identify all residues that are central to functional tuning in the active site of PYP.
Fig. 3.

Mapping the degree to which point mutations alter functional and structural properties on the crystal structure of PYP. The orientation of PYP was chosen to illustrate the packing of the two N-terminal helices to the central β-sheet of PYP. The pCA chromophore and Cys69 are shown in stick representation. Substitutions that significantly alter the six indicated functional properties are color coded according to the effect of substitutions at these sites, with large increases in values in red, small increases in yellow, large decreases in dark blue, and small decreases in light blue.
Substitution of residues F62, V66, P68, T95, Y98, which are within 5 Å of the pCA, does not significantly alter the active site properties of PYP. The residues with the largest effects on the properties of the pCA are Gly29, Ile31, Tyr42, Asn43, Glu46, Ile49, Arg52, and Phe96. Of the 20 residues (Table S3) that significantly affect one or more property of the pCA, 15 are buried or partially buried and 5 are exposed. Of these side chains, 12 are within 5 Å of the pCA, 5 are between 5 and 10 Å, and 3 are more than 10 Å removed from the pCA. Thus, most residues that affect the pCA are in its immediate vicinity and are buried, matching the traditional view of a protein active site. The interactions between these residues and the pCA include hydrogen bonding, charge–charge interactions, and aromatic ring–ring interactions (Table S3). However, substitutions at four positions far removed from the pCA alter its properties by long-range effects: F28A, G77A, G82A, and Y94A. The F28A mutation may shift the pCA λmax through altered docking of the N-terminal α-helices against the β-sheet. Gly77 and Gly82 are located in α-helix 5; Ala substitution at these positions may decrease pCA Φfl and increase τpB by reducing the structural flexibility of this helix. The Y94A mutation removes the side-chain hydrogen bond from residue 94 to the backbone carbonyl oxygen of Cys69, to which the pCA is attached. In summary, the interactions that result in active site tuning are complex and heterogeneous.
The lifetime of the pB state is important for its signaling function (see SI Text). The 58 substitutions (45% of PYP) that significantly affect τpB are distributed throughout the protein (Fig. 3). This dispersed distribution is in line with the partially unfolded nature of the pB state (25–29): Its decay involves refolding of many regions of the protein, and can thus be affected by mutations at many different locations. The statistically significant correlation of 0.59 between ΔGU and τpB in the Ala scan dataset (Table S4) supports a link (25) between protein folding and pB decay. Analysis of the location of the residues that affect τpB reveals the importance of interactions between the N-terminal region of PYP and its central β-sheet (Table S3). First, the side-chain hydrogen bonds of Asp24 and Asn43 form functionally important connections between the N-terminal domain and the photoactive core of PYP. Second, mutations in 7 of the 10 residues that form the secondary hydrophobic core of PYP greatly increase τpB. Interestingly, τpB is also increased in deletion mutants of PYP lacking the N-terminal region (30, 35). The Ala scan results indicate that both hydrogen bonding and hydrophobic interactions between the N-terminal region and the β-sheet in PYP regulate pB lifetime.
The Ala scan data also yield insights into the structural basis of the ΔGU of PYP. Of the 23 mutations that reduce ΔGU by ≥2 kcal/mol, the majority is buried (68%) or partially buried (23%), in line with results on other proteins (8, 9). Of these residues, 57% are part of the two hydrophobic cores of PYP. A reduction in ΔGU is caused by substitution of each residue that is part of the minor hydrophobic core, but only 4 of the 20 residues at the major hydrophobic core. We propose that the thermodynamic fragility of the minor hydrophobic core is functionally important by priming the stimulus-induced release of the N-terminal region. NMR measurements of PYP have revealed that the two α-helices in the N-terminal region are structurally dynamic (36), suggesting that the N-terminal region is not strongly associated with the PAS domain core of PYP. This issue is relevant to signaling by PYP, because dissociation of the N-terminal region is a key event in pB formation (30, 31). Substitutions in the secondary hydrophobic core formed by the packing of the N-terminal region against the central β-sheet not only destabilize PYP by ∼4.0 kcal/mol, but also increase τpB. This result shows that although the N-terminal region is dynamic, it is thermodynamically coupled to the PAS core, and thus influences both τpB and ΔGU.
How do proteins achieve their remarkable robustness of structure and function against point mutations? Regarding protein stability, the prevalent view is that the strongest effects of single substitutions usually are at most a modest loss of ΔGU, whereas an influential notion on protein function is that only substitutions of the few residues involved in active site chemistry strongly alter functional properties. The PYP Ala scan dataset confirms the above view on protein stability, with substitutions at 28% of the positions causing a significant reduction in ΔGU and losses in stability of up to 5.3 kcal/mol. However, 40% of the residues that alter PYP active site functional properties are not located at the active site (Table S3), whereas only 12% of substitutions that cause strong changes in the kinetics of pB decay are at the active site (Table S3). These results show that most residues (65%) with the largest effects on PYP function are not located at the active site.
The results reported here reveal that many point mutations significantly reduce protein production level. The structural factors that affect protein production are of considerable biotechnological interest. The side chains of 26 residues whose substitution significantly reduce expression level are fully or partially buried and form a cluster in the center of PYP, suggesting their contribution to a folding nucleus. Mutations in any of the residues that form the π-helix in PYP (residue 61–67) strongly diminish protein production level. No correlation was observed between production level and ΔGU (Table S4). We propose that protein aggregation of mutants in the highly crowded cytoplasm may impose significant restrictions on changes in amino acid sequence.
Discussion
The data reported here vividly illustrate that important challenges remain in understanding protein structure–function relationships. First, none of the six PAS-conserved residues in PYP were selected in structure-based mutagenesis studies of PYP (24). However, all of these residues significantly alter the functional properties of PYP (Table 2). Secondly, there are only six positions in PYP at which a substitution simultaneously affects λmax, pKa, and τpB: Gly29, Ile31, Tyr42, Asn43, Glu46, and Ile49 (Fig. 1 and Tables S1 and S3). Of these functionally most critical residues only Tyr42 and Glu46 were identified in site-directed mutagenesis studies based on the crystal structure of PYP (24). The efficient microscale high-throughput measurements used here to quantitatively characterize the properties of libraries of purified mutants offers an effective approach to obtain rich datasets that provide insights into the functional anatomy of proteins.
Table 2.
Summary of effects of substitution of residues in PYP that are highly conserved in the PAS domain superfamily
| Residue | Dist. to pCA* | λmax | Φfl | pKa | τpB/%bl† | ΔGU‡ | Production level |
| WT | NA | 447 | 0.14 | 3 | 0.9/24 | 12.5 | 1.0 |
| Asp34 | III | 445 | 0.18 | ND | 0.9/15 | 10.9 | 0.27§ |
| Gly37 | III | 445 | 0.17 | 3 | 2/30 | 10.3 | 0.24 |
| Ile39 | II | 446 | ND | 3 | 7/75 | ND | 0.18 |
| Asn43 | II | 441 | 0.94 | 5.5 | 59/91 | 9.3 | 2.0 |
| Gly51 | II | 447 | 0.22 | 3 | 2/32 | 10.8 | 1.04 |
| Gly59 | III | 446 | 0.16 | 3 | 1/21 | 11.6 | 0.28 |
NA, not applicable; ND, not determined.
*II indicates 5 and 10 Å; III indicates more than 10 Å.
†τpB indicated in seconds.
‡ΔGU is indicated in kcal/mol.
§Underlined values deviate significantly from WT PYP.
Future elaboration of the high-throughput approach described here can provide additional functional information. One important example is the use of the curved temperature dependence of the kinetics of pB decay (27) or the use of fluorescent dyes (37, 38) to achieve high-throughput detection of the extent of light-induced conformational changes. More generally, the approach reported here can be applied to any protein for which (i) sufficient amount of protein can be overproduced and purified by Ni-NTA affinity chromatography, and (ii) the properties of the protein can be detected spectroscopically either by an intrinsic chromophore (aromatic side chains or cofactors) or by attached fluorescent probes or chromogenic substrate derivatives.
The results reported here allow an examination of the role of the residues that are highly conserved in the PYP family (24) and in the PAS domain superfamily (12), and thus of possible reasons for their evolutionary conservation. The PYP family of receptors shows considerable sequence divergence, with pairwise sequence identities down to 19% (24). Of the 23 residues that are highly conserved in PYP (Fig. S1B and Table S5) only eight are located at the photoactive site of PYP (24). Of these residues, only Tyr42, Glu46, and Cys69 play a classical role as active site residues in that they serve as hydrogen bonding partners (20), proton donor (39), and cofactor attachment site (23). Fifteen highly conserved side chains are far removed from the active site. Substitutions at many of these sites either significantly reduce the ΔGU of PYP (six mutants, average ΔΔG of -3.8 kcal/mol) and/or strongly reduced its protein production (14 mutants, average reduction in production by 76%). This finding indicates that protein stability and production level are major factors in causing high conservation of residues (Table 3).
Table 3.
Role of conserved residues in PYP
| Conserved in PYPs |
Conserved in PAS domains |
|
| Total | 23 | 6 |
| Active site | 8 | 1 |
| τpB | 14 | 2 |
| ΔGU | 6 | 2 |
| Production | 14 | 4 |
The number of residues affecting the indicated properties is shown.
The PAS domain superfamily is defined by their characteristic but very weak amino acid sequence similarity. However, the functional role of these conserved residues is not known. The Ala scan results yield insights into the role that these residues play. Of the nine most highly PAS-conserved residues (Fig. S1A), six are present in the PYP from H. halophila: Asp34, Gly37, Ile39, Asn43, Gly51, and Gly59. These residues are structurally inconspicuous. None are at the pCA binding pocket; three are buried and three are solvent exposed. In two mutants (I39A and N43A) τpB is strongly increased, whereas Asn43 also affects the properties of the pCA (Table S3). Substitutions at four PAS-conserved residues cause a ∼75% reduction in protein production level, while leaving the ΔGU of PYP essentially unchanged. These data yield insights into the role of five highly PAS-conserved residues (Table 2), and provide evidence that the majority of these residues affect protein production (Table 3).
The Ala scan data suggest a conserved role of Ile39 and Asn43 in signaling in PAS domains. The side chain of Ile39 is part of the main hydrophobic core of PYP, whereas the side-chain hydrogen bonds of Asn43 form a helical cap for α-helix 3 and restrain β-strand 1 at the N terminus of the PAS domain fold. Our in-depth studies of PYP mutants with substitutions at Asn43 indicate that its side-chain hydrogen bonds are important for regulating the kinetics of PAS domain signaling. We propose that these two residues form a conserved mechanism to control the kinetics of the allosteric structural changes upon PAS domain deactivation.
The kinetics of pB decay in PYP are also regulated by the N-terminal region (30, 35). The Ala scan data indicate that a significant fraction of the side chains in PYP is dedicated to tuning the interaction between the N-terminal region of PYP and its PAS domain core, and thus regulate τpB (Table S3). The C-terminal helical extensions of a PAS domain from the plant photoreceptor phototropin (40) and the Drosophila clock protein Period (41) and the N-terminal extension of a PAS domain in the fungal photoreceptor Vivid (42) all pack against the PAS core of the protein in a position similar to that of the N-terminal region of PYP. Functionally important conformational changes and partial unfolding occur in all of these extensions (30, 31, 40–42). This trend indicates a common theme in PAS domain signaling: partial unfolding of helical extensions that are attached to either the N or C termini of PAS domains. In the case of the heterodimeric transcription factor hypoxia-inducible factor (HIF), functionally important interactions between the PAS domains in HIFα and ARNT occur at the exposed face of the central β-sheet (43). This observation indicates the possibility that the light-induced release of helical extensions in many PAS domains expose the central β-sheet for subsequent interactions with downstream signaling partners.
Materials and Methods
Construction of PYP Mutants.
The PYP mutants were generated with 125 specific mutagenic primers using Stratagene’s QuikChange site-directed mutagenesis kit with the pyp gene from H. halophila in a pQE-80L plasmid (QIAGEN) as the template, and were confirmed by DNA sequencing.
Protein Purification.
The growth of the 125 E. coli strains overproducing the PYP mutants, their purification, and most of their spectroscopic analysis were performed in 96-well format. The proteins were purified using Ni-NTA agarose as described in (32), with the modifications described in the SI Text. Selected purified samples were examined by MALDI-TOF mass spectrometry.
Spectroscopic Characterization of PYP Mutants.
Absorbance spectra were measured in UV-transparent 96-well plates using a Synergy HT plate reader (BioTek). To probe the pKa of the pCA chromophore, the alanine scan library was purified at nine different pH values. The pCA Φfl was measured for each sample individually with excitation at 460 nm and detection at 520 nm in a SPEX Fluoromax-3 fluorometer (Jobin-Yvon). The protein production level of each substitution mutant was determined based on its absorbance at 280 nm after purification. To determine ΔGU values samples were titrated with Gdm-HCl and analyzed using a two-state model. The kinetics of the decay of the pB photocycle intermediate were measured in an HP 8453 diode array spectrophotometer upon illumination with a Cuda I-150 light source with a shutter.
Supplementary Material
Acknowledgments.
W.D.H. acknowledges support from National Institutes of Health Grant GM063805 and Oklahoma Center for the Advancement of Science and Technology Grant HR07-135S.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1004823107/-/DCSupplemental.
References
- 1.Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirny L, Shakhnovich E. Evolutionary conservation of the folding nucleus. J Mol Biol. 2001;308:123–129. doi: 10.1006/jmbi.2001.4602. [DOI] [PubMed] [Google Scholar]
- 4.Larson SM, Ruczinski I, Davidson AR, Baker D, Plaxco KW. Residues participating in the protein folding nucleus do not exhibit preferential evolutionary conservation. J Mol Biol. 2002;316:225–233. doi: 10.1006/jmbi.2001.5344. [DOI] [PubMed] [Google Scholar]
- 5.Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- 6.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: Mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 7.Glasner ME, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr Opin Chem Biol. 2006;10:492–497. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–87. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 9.Huang W, Petrosino J, Hirsch M, Shenkin PS, Palzkill T. Amino acid sequence determinants of β-lactamase structure and activity. J Mol Biol. 1996;258:688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 10.Wagner A. Robustness and Evolvability in Living Systems. Princeton, NY: Princeton Univ Press; 2005. pp. 63–65. [Google Scholar]
- 11.Nei M. Selectionism and neutralism in molecular evolution. Mol Biol Evol. 2005;22:2318–2342. doi: 10.1093/molbev/msi242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor BL, Zhulin IB. PAS domains: Internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63:479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hefti MH, Françoijs KJ, de Vries SC, Dixon R, Vervoort J. The PAS fold. A redefinition of the PAS domain based upon structural prediction. Eur J Biochem. 2004;271:1198–1208. doi: 10.1111/j.1432-1033.2004.04023.x. [DOI] [PubMed] [Google Scholar]
- 14.Hellingwerf KJ, Hendriks J, Gensch T. Photoactive yellow protein, a new type of photoreceptor protein: Will this “yellow lab” bring us where we want to go? J Phys Chem A. 2003;107:1082–1094. [Google Scholar]
- 15.Christie JM. Phototropin blue-light receptors. Annu Rev Plant Biol. 2007;58:21–45. doi: 10.1146/annurev.arplant.58.032806.103951. [DOI] [PubMed] [Google Scholar]
- 16.Meyer TE. Isolation and characterization of soluble cytochromes, ferredoxins and other chromophoric proteins from the halophilic phototrophic bacterium Ectothiorhodospira halophila. Biochim Biophys Acta. 1985;806:175–183. doi: 10.1016/0005-2728(85)90094-5. [DOI] [PubMed] [Google Scholar]
- 17.Meyer TE, Yakali E, Cusanovich MA, Tollin G. Properties of a water-soluble, yellow protein isolated from a halophilic phototrophic bacterium that has photochemical activity analogous to sensory rhodopsin. Biochemistry. 1987;26:418–423. doi: 10.1021/bi00376a012. [DOI] [PubMed] [Google Scholar]
- 18.Sprenger WW, Hoff WD, Armitage JP, Hellingwerf KJ. The Eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J Bacteriol. 1993;175:3096–3104. doi: 10.1128/jb.175.10.3096-3104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellequer JL, Wager-Smith KA, Kay SA, Getzoff ED. Photoactive yellow protein: A structural prototype for the three-dimensional fold of the PAS domain superfamily. Proc Natl Acad Sci USA. 1998;95:5884–5890. doi: 10.1073/pnas.95.11.5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borgstahl GEO, Williams DR, Getzoff ED. 1.4 Å structure of photoactive yellow protein, a cytosolic photoreceptor—Unusual fold, active site, and chromophore. Biochemistry. 1995;34:6278–6287. doi: 10.1021/bi00019a004. [DOI] [PubMed] [Google Scholar]
- 21.Hoff WD, et al. Thiol ester-linked p-coumaric acid as a new photoactive prosthetic group in a protein with rhodopsin-like photochemistry. Biochemistry. 1994;33:13959–13962. doi: 10.1021/bi00251a001. [DOI] [PubMed] [Google Scholar]
- 22.Baca M, et al. Complete chemical structure of photoactive yellow protein: Novel thioester-linked 4-hydroxycinnamyl chromophore and photocycle chemistry. Biochemistry. 1994;33:14369–14377. doi: 10.1021/bi00252a001. [DOI] [PubMed] [Google Scholar]
- 23.van Beeumen JJ, et al. Primary structure of a photoactive yellow protein from the phototrophic bacterium Ectothiorhodospira halophila, with evidence for the mass and the binding site of the chromophore. Protein Sci. 1993;2:1114–1125. doi: 10.1002/pro.5560020706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumauchi M, Hara M, Stalcup P, Xie A, Hoff WD. Identification of six new photoactive yellow proteins: Diversity and structure-function relationships in a bacterial blue light photoreceptor. Photochem Photobiol. 2008;84:956–969. doi: 10.1111/j.1751-1097.2008.00335.x. [DOI] [PubMed] [Google Scholar]
- 25.Lee B-C, Pandit A, Croonquist PA, Hoff WD. Folding and signaling share the same pathway in a photoreceptor. Proc Natl Acad Sci USA. 2001;98:9062–9067. doi: 10.1073/pnas.111153598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernard C, et al. The solution structure of a transient photoreceptor intermediate: Delta 25 photoactive yellow protein. Structure. 2005;13:953–962. doi: 10.1016/j.str.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 27.van Brederode ME, Hoff WD, van Stokkum IHM, Groot ML, Hellingwerf KJ. Protein folding thermodynamics applied to the photocycle of the photoactive yellow protein. Biophys J. 1996;71:365–380. doi: 10.1016/S0006-3495(96)79234-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoff WD, et al. Global conformational changes upon receptor stimulation in photoactive yellow protein. Biochemistry. 1999;38:1009–1017. doi: 10.1021/bi980504y. [DOI] [PubMed] [Google Scholar]
- 29.Zhao JM, et al. Single-molecule detection of structural changes during Per-Arnt-Sim (PAS) domain activation. Proc Natl Acad Sci USA. 2006;103:11561–11666. doi: 10.1073/pnas.0601567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Horst MA, van Stokkum IH, Crielaard W, Hellingwerf KJ. The role of the N-terminal domain of photoactive yellow protein in the transient partial unfolding during signalling state formation. FEBS Lett. 2001;497:26–30. doi: 10.1016/s0014-5793(01)02427-9. [DOI] [PubMed] [Google Scholar]
- 31.Imamoto Y, Kamikubo H, Harigai M, Shimizu N, Kataoka M. Light-induced global conformational change of photoactive yellow protein in solution. Biochemistry. 2002;41:13595–13601. doi: 10.1021/bi0264768. [DOI] [PubMed] [Google Scholar]
- 32.Philip AF, Eisenman KT, Papadantonakis GA, Hoff WD. Functional tuning of photoactive yellow protein by active site residue 46. Biochemistry. 2008;47:13800–13810. doi: 10.1021/bi801730y. [DOI] [PubMed] [Google Scholar]
- 33.Sawyer SA, Parsch J, Zhang Z, Hartl DL. Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila. Proc Natl Acad Sci USA. 2007;104:6504–6510. doi: 10.1073/pnas.0701572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pakula AA, Young VB, Sauer RT. Bacteriophage λ cro mutations: Effects on activity and intracellular degradation. Proc Natl Acad Sci USA. 1986;83:8829–8833. doi: 10.1073/pnas.83.23.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harigai M, et al. Amino acids in the N-terminal region regulate the photocycle of photoactive yellow protein. J Biochem. 2001;130:51–56. doi: 10.1093/oxfordjournals.jbchem.a002961. [DOI] [PubMed] [Google Scholar]
- 36.Düx P, et al. Solution structure and backbone dynamics of the photoactive yellow protein. Biochemistry. 1998;37:12689–12699. doi: 10.1021/bi9806652. [DOI] [PubMed] [Google Scholar]
- 37.Lee B-C, Croonquist PA, Sosnick TR, Hoff WD. PAS domain receptor photoactive yellow protein is converted to a molten globule state upon activation. J Biol Chem. 2001;276:20821–20823. doi: 10.1074/jbc.C100106200. [DOI] [PubMed] [Google Scholar]
- 38.Hendriks J, et al. Transient exposure of hydrophobic surface in the photoactive yellow protein monitored with Nile red. Biophys J. 2002;82:1632–1643. doi: 10.1016/S0006-3495(02)75514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie A, et al. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 2001;40:1510–1517. doi: 10.1021/bi002449a. [DOI] [PubMed] [Google Scholar]
- 40.Harper SM, Neil LC, Garner KH. Structural basis of a phototropin light switch. Science. 2003;301:1541–1544. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 41.Yildiz O, et al. Crystal structure and interactions of the PAS repeat region of the Drosophila clock protein PERIOD. Mol Cell. 2005;17:69–82. doi: 10.1016/j.molcel.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 42.Zoltowski BD, et al. Conformational switching in the fungal light sensor vivid. Science. 2007;316:1054–1057. doi: 10.1126/science.1137128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erbel PJA, Card PB, Karakuzu O, Bruick RK, Gardner KH. Structural basis for PAS domain heterodimerization in the basic helix-loop-helix-PAS transcription factor hypoxia-inducible factor. Proc Natl Acad Sci USA. 2003;100:15504–15509. doi: 10.1073/pnas.2533374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.