

Abstract
Pleiotropy refers to the phenomenon of a single mutation or gene affecting multiple distinct phenotypic traits and has broad implications in many areas of biology. Due to its central importance, pleiotropy has also been extensively modeled, albeit with virtually no empirical basis. Analyzing phenotypes of large numbers of yeast, nematode, and mouse mutants, we here describe the genomic patterns of pleiotropy. We show that the fraction of traits altered appreciably by the deletion of a gene is minute for most genes and the gene–trait relationship is highly modular. The standardized size of the phenotypic effect of a gene on a trait is approximately normally distributed with variable SDs for different genes, which gives rise to the surprising observation of a larger per-trait effect for genes affecting more traits. This scaling property counteracts the pleiotropy-associated reduction in adaptation rate (i.e., the “cost of complexity”) in a nonlinear fashion, resulting in the highest adaptation rate for organisms of intermediate complexity rather than low complexity. Intriguingly, the observed scaling exponent falls in a narrow range that maximizes the optimal complexity. Together, the genome-wide observations of overall low pleiotropy, high modularity, and larger per-trait effects from genes of higher pleiotropy necessitate major revisions of theoretical models of pleiotropy and suggest that pleiotropy has not only allowed but also promoted the evolution of complexity.
Keywords: genetics, adaptation, modularity, yeast, phenotype
Pleiotropy occurs when a single mutation or gene affects multiple distinct phenotypic traits (1). Pleiotropy has broad implications in genetics (1–3), development (4, 5), senescence (6), disease (7, 8), and many evolutionary processes such as adaptation (2, 9–13), maintenance of sex (14), preservation of redundancy (15), and stabilization of cooperation (16). For example, the antagonistic pleiotropy theory of senescence asserts that alleles beneficial to development and reproduction are deleterious after the reproductive age and cause senescence, which may explain why all species have a limited life span (6). Pleiotropy is the main theoretical reason behind the hypothesis that morphological evolution occurs more frequently through cis-regulatory changes than through protein sequence changes (9), as the former are thought to be less pleiotropic than the latter. Pleiotropy also has important implications in human disease, because many genetic defects each affect multiple phenotypic traits. For instance, mutations in the homeobox gene ARX cause ambiguous genitalia and lissencephaly (whole or parts of the surface of the brain appear smooth) (Online Mendelian Inheritance in Man no. 300215).
Due to pleiotropy's central importance in biology, several mathematical models of pleiotropy have been developed and important theoretical results have been derived from the analyses of these models (10, 11, 17, 18). For example, Fisher proposed that every mutation affects every trait and the effect size of a mutation on a trait is uniformly distributed (10). On the basis of this model and the assumption that the total effect size of a mutation is constant in different organisms, Orr derived that the rate of adaptation of a population to an environment quickly declines with the increase of the organismal complexity, which is defined by the total number of traits (12). This “cost of complexity” would likely prohibit the origins of complex organisms and hence is puzzling to evolutionary biologists (19, 20).
Although pleiotropy has been examined in detail in a few genes (21, 22), its genomic pattern is largely unknown, which seriously limits us from evaluating mathematical models of pleiotropy, verifying theoretical inferences from these models, and testing various pleiotropy-related hypotheses in many fields of biology. In this work, we compile from existing literature and databases phenotypes of large numbers of yeast, nematode, and mouse mutants. We describe the genomic patterns of pleiotropy in these organisms and show that these patterns drastically differ from any mathematical model of pleiotropy. We further demonstrate that the cost of complexity is substantially alleviated when the empirical patterns of pleiotropy are taken into consideration and that the observed value of a key parameter of pleiotropy falls in a narrow range that maximizes the optimal complexity.
Results and Discussion
Most Genes Affect only a Small Fraction of Traits.
To uncover the genomic patterns of pleiotropy, we compiled three large datasets of gene pleiotropy for the baker's yeast Saccharomyces cerevisiae, one for the nematode worm Caenorhabditis elegans, and one for the house mouse Mus musculus (SI Methods). The first dataset, yeast morphological pleiotropy, is based on the measures of 279 morphological traits in haploid wild-type cells and 4,718 haploid mutant strains that each lack a different nonessential gene (23). The second dataset, yeast environmental pleiotropy, is based on the growth rates of the same collection of yeast mutants relative to the wild type in 22 different environments (24). The third dataset, yeast physiological pleiotropy, is based on 120 literature-curated physiological functions of genes recorded in the Comprehensive Yeast Genome Database (CYGD). The fourth dataset, nematode pleiotropy, is based on the phenotypes of 44 early embryogenesis traits in C. elegans treated with genome-wide RNA-mediated interference (25). The fifth dataset, mouse pleiotropy, is based on the phenotypes of 308 morphological and physiological traits in gene-knockout mice recorded in Mouse Genome Informatics (MGI). These five datasets provide qualitative information about the traits that are affected appreciably by each gene. In addition, the first dataset also includes the quantitative information of the effect size of each gene on each trait. Even after the removal of genes that do not affect any trait and traits that are not affected by any gene, these five datasets each include hundreds to thousands of genes and tens to hundreds of traits (Fig. 1). They are thus suitable for examining genome-wide patterns of pleiotropy.
Fig. 1.

Frequency distributions of degree of gene pleiotropy in (A) yeast morphological, (B) yeast environmental, (C) yeast physiological, (D) nematode, and (E) mouse pleiotropy data. Mean and median degrees of pleiotropy and their SDs are indicated. The numbers in parentheses are the mean and median degrees of pleiotropy divided by the total number of traits. After the removal of genes that do not affect any trait and traits that are not affected by any gene, the total numbers of genes and traits in these datasets are (A) 2,449 genes and 253 traits, (B) 774 genes and 22 traits, (C) 1,256 genes and 120 traits, (D) 661 genes and 44 traits, and (E) 4,915 genes and 308 traits.
In all five datasets, we observed that most genes affect only a small fraction of traits and only a minority of genes affect many traits (Fig. 1). The median degree of pleiotropy varies from one to seven traits (or 1–9% of the traits considered) in these datasets. The degree of pleiotropy measured by the percentage of examined traits is expected to be more accurate for the three datasets in which the same set of traits was examined in all mutants (Fig. 1 A, B, and D). By bootstrapping traits, we found that the SDs of our estimated median and mean degrees of pleiotropy are generally small, indicating that these estimates are precise (Fig. 1). To examine the impact of the number of examined traits on the estimated pleiotropy, we randomly removed 50 and 90% of the traits from each dataset, respectively. We found that the mean and median degrees of pleiotropy, measured by the percentage of traits examined, remain largely unchanged (Table S1), suggesting that further additions of traits to our data would not substantially alter our results. We thus predict that the median number of traits affected by a gene is no greater than a few percent of the total number of traits in an organism. Furthermore, because gene pleiotropy is largely owing to the involvement of the same molecular function in multiple different biological processes rather than the presence of multiple molecular functions per gene (26), random mutations in a gene will likely affect the same traits as the deletion of the gene does, although the magnitude of the phenotypic effects should be much smaller. Consequently, the observable degree of pleiotropy is expected to be even lower for random mutations than for gene deletions. Our genome-wide results echo recent small-scale observations from fish and mouse quantitative trait locus (QTL) studies (27, 28) and an inference from protein sequence evolution (29) and reveal a general pattern of low pleiotropy in eukaryotes, which is in sharp contrast to some commonly used theoretically models (10, 18) that assume universal pleiotropy (i.e., every gene affects every trait) (Table S2).
Gene–Trait Relationships Are Highly Modular.
The genome-wide data also allow us to test the modular pleiotropy hypothesis, which is important for a number of theories of development and evolution (30). Gene–trait relationships can be represented by a bipartite network of genes and traits, in which a link between a gene node and a trait node indicates that the gene affects the trait (Fig. 2A). Modular pleiotropy refers to the phenomenon that links within modules are significantly more frequent than those across modules (Fig. 2B). Given that cellular functions are modularly and hierarchically organized (30), modular pleiotropy likely exists, although it is not considered in commonly used models of pleiotropy (10, 12, 18) (Table S2). Using a bipartite-network–specific algorithm (31), we identified modules and estimated the modularity of each gene–trait network. Because random networks of certain structures also have nonzero modularity (32), we compared the modularity of an observed network with that of its randomly rewired networks, which have randomized links but an unchanged number of links per node (32) (Fig. 2C). We then calculated the scaled modularity of a network, which is the difference between the observed modularity of a network and the mean modularity of its randomly rewired networks in terms of the number of SDs (32) (Methods). Our results show large scaled modularity (34–238) in each of the five gene–trait networks examined (Fig. 2 D–H), providing definitive evidence for the modular pleiotropy hypothesis. Our results remain qualitatively unchanged even when a random 50% of the traits in each dataset are removed (Table S3). The modularity would be overestimated if the genetic correlations among traits are biased upward in our datasets compared with the complete datasets that include all possible traits. Although we do not know whether this bias exists, to be conservative, we merged traits whose genetic correlation coefficients are >0.7 (Methods). We found that highly significant modularity is still present in each of the five gene–trait networks (Table S3).
Fig. 2.

High modularity of gene–trait bipartite networks. (A) A hypothetical gene–trait bipartite network. A link between a gene and a trait indicates that the gene affects the trait, and the thickness of the link indicates the effect size. (B) Two modules are identified in the hypothetical gene–trait network after the quantitative links are transformed to qualitative links (i.e., presence/absence) on the basis of whether an effect size is significantly different from 0. (C) A randomly rewired network that has the same degree distribution as the original hypothetical network shows no detectable modular structure. The modularity and scaled modularity of the hypothetical bipartite network are 0.41 and 3.9, respectively. D–H show the observed modularity (blue arrows) and distribution of modularity for 250 randomly rewired networks (red histograms) for the gene–trait networks of the (D) yeast morphological, (E) yeast environmental, (F) yeast physiological, (G) nematode, and (H) mouse pleiotropy datasets.
Genes Affecting More Traits Have Larger Per-Trait Effects.
The yeast morphological pleiotropy data contain quantitative information about the phenotypic effect size of mutations, which is another important parameter in genetics that has never been available at the genomic scale. Using a standardized measure of effect size for all traits (Z-score, defined by the phenotypic difference between a mutant and the mean of the wild type for a trait in terms of the number of SDs) (Methods), we obtained, for each yeast gene, the frequency distribution of the effect sizes on the 279 morphological traits. As exemplified in Fig. 3A, this distribution is approximately normal for most genes; the actual distribution is not significantly different from a normal distribution for 85% of the genes examined (5% false discovery rate in the goodness-of-fit test). This observation is consistent with a commonly used model (18), but is in contrast to another where the distribution is assumed to be uniform (10, 12) (Table S2). In fact, the uniform distribution can be rejected for every gene at the significance level of P = 5 × 10−7 (goodness-of-fit test). It is notable that the SD of the effect size distribution varies greatly among genes (Fig. 3B), in contrast to models that assume a constant SD among genes (10, 12, 18) (Table S2). It is also notable that the typical effect size distribution has a nearly zero mean, although a minority of genes exhibit positive or negative means (Fig. S1).
Fig. 3.

Scaling relationships between the total phenotypic effect size of a gene and the degree of pleiotropy in the yeast morphological pleiotropy data. (A) Examples showing the normal distribution of effect size over 279 traits. Two genes are chosen to show variable SDs of the normal distributions. (B) Distribution of the SD of the effect size for all 4,718 genes. Observed scaling relationships between the degree of pleiotropy n and the total phenotypic effect of a gene are measured by (C) Euclidean distance or (D) Manhattan distance. The orange curve is the best fit to the power function whose estimated parameters are shown in the upper left. The numbers after ± show the 95% confidence interval for the estimated scaling exponent. R2 indicates the square of the correlation coefficient. E and F are similar to C and D except that the effect sizes of each gene are randomly generated from a normal distribution with zero mean and observed SD. G and H are similar to C and D except that the effect sizes of each gene are randomly generated from a normal distribution with zero mean and a constant SD, which is the average of all SDs of all genes.
If one considers only those traits that are significantly affected by a gene, the total size of the phenotypic effects of the gene can be calculated by the Euclidean distance 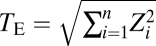 , where n is the gene's degree of pleiotropy defined by the number of significantly affected traits and Zi is the gene's effect on trait i measured by the Z-score (27). We estimated from the yeast morphological pleiotropy data that the exponent b in the scaling relationship of
, where n is the gene's degree of pleiotropy defined by the number of significantly affected traits and Zi is the gene's effect on trait i measured by the Z-score (27). We estimated from the yeast morphological pleiotropy data that the exponent b in the scaling relationship of 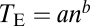 equals 0.601, with its 95% confidence interval of (0.590, 0.612) (Fig. 3C). This exponent is significantly greater than that assumed in any theoretical model (Table S2). For example, the invariant total effect model (12) assumes a constant total effect size (b = 0), whereas the Euclidian superposition model (11, 17, 18) assumes a constant effect size per affected trait (b = 0.5). Our results thus indicate that the per-trait effect of a gene, estimated by
equals 0.601, with its 95% confidence interval of (0.590, 0.612) (Fig. 3C). This exponent is significantly greater than that assumed in any theoretical model (Table S2). For example, the invariant total effect model (12) assumes a constant total effect size (b = 0), whereas the Euclidian superposition model (11, 17, 18) assumes a constant effect size per affected trait (b = 0.5). Our results thus indicate that the per-trait effect of a gene, estimated by 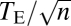 on the basis of the definition of the Euclidian distance, is larger when the gene affects more traits. One can also measure the total effect size by the Manhattan distance (33)
on the basis of the definition of the Euclidian distance, is larger when the gene affects more traits. One can also measure the total effect size by the Manhattan distance (33) 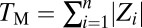 . We found the exponent d in the scaling relationship of
. We found the exponent d in the scaling relationship of 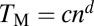 to be 1.095, with its 95% confidence interval of (1.083, 1.107) (Fig. 3D). Again, the observed d significantly exceeds that assumed in all current models (0.5 in the invariant total effect model and 1 in the Euclidian superposition model; Table S2) and indicates larger per-trait effects estimated by TM/n for genes affecting more traits. To examine the robustness of the above results, we randomly removed 50 and 90% of the traits from the data, respectively. Our results that b and d are slightly smaller when the number of traits used is smaller (Fig. S2 A–D) suggest that, when more traits are examined in the future, b and d would become slightly greater than the current estimates. Our results are also robust to merging traits with genetic correlations (Fig. S2 E and F). Because 279 morphological traits were measured in each yeast mutant, in the above analyses, a 5% false discovery rate was used as a cutoff to control for multiple testing in determining whether a trait is affected by a gene. Our results remain qualitatively unchanged when the more conservative P = 5% after Bonferroni correction is used to correct for multiple testing (Fig. S3).
to be 1.095, with its 95% confidence interval of (1.083, 1.107) (Fig. 3D). Again, the observed d significantly exceeds that assumed in all current models (0.5 in the invariant total effect model and 1 in the Euclidian superposition model; Table S2) and indicates larger per-trait effects estimated by TM/n for genes affecting more traits. To examine the robustness of the above results, we randomly removed 50 and 90% of the traits from the data, respectively. Our results that b and d are slightly smaller when the number of traits used is smaller (Fig. S2 A–D) suggest that, when more traits are examined in the future, b and d would become slightly greater than the current estimates. Our results are also robust to merging traits with genetic correlations (Fig. S2 E and F). Because 279 morphological traits were measured in each yeast mutant, in the above analyses, a 5% false discovery rate was used as a cutoff to control for multiple testing in determining whether a trait is affected by a gene. Our results remain qualitatively unchanged when the more conservative P = 5% after Bonferroni correction is used to correct for multiple testing (Fig. S3).
We observed that the phenomenon of larger per-trait effects for genes affecting more traits disappeared when the effect sizes of all genes on all traits are randomly shuffled (Fig. S4). Thus, the phenomenon is a property of the actual data rather than an artifact of our analysis. It turns out that this phenomenon results from two genome-wide features of pleiotropy described above: (i) a normal distribution of effect sizes of a gene on different traits and (ii) variable SDs of the normal distributions among different genes. Comparing two genes both having normal distributions of effect sizes but with different SDs, we proved mathematically that the gene with the larger SD affects more traits (when a fixed effect-size cutoff is applied) and has on average a larger per-trait effect (SI Methods). In fact, the scaling relationships with the observed b and d values can be largely recapitulated by using randomly generated effect size data, provided that a normal distribution with the actual SD is used in generating such data for each gene (Fig. 3 E and F; SI Methods). By contrast, when the same SD is used in generating the random effect size data for all of the genes (SI Methods), we no longer observe larger per-trait effects for genes affecting more traits (b < 0.5 in Fig. 3G and d < 1 in Fig. 3H).
It is interesting to note that a recent mouse QTL study (27) also reported b > 0.5, but a subsequent analysis (33) showed that, owing to the likelihood of the inclusion of multiple genes per QTL, the data can establish only b > 0, but not b > 0.5. Because our yeast morphological pleiotropy data were collected from strains that each lack only one gene, they are immune from the above multiple-gene problem. Additionally, our observation that the estimated b tends to be smaller when fewer traits are used (Fig. S2) may also explain the unreliability (33) of the mouse QTL study (27), which was based on a much smaller dataset. Furthermore, because our data were generated by examining all yeast nonessential genes and a large number of traits, they are more likely to reveal the general patterns of pleiotropy. It is important to recognize that our results are based on pleiotropic effects of genes (i.e., null mutations) rather than random mutations. However, our results likely apply to random mutations because the effect sizes of random mutations in a gene are expected to be proportional to the effect sizes of the gene (Methods).
Cost of Complexity Is Diminished with the Actual Patterns of Pleiotropy.
One of the most puzzling results from theoretical analysis of pleiotropy is the cost of complexity conundrum (12). Using Fisher's geometric model (10), Orr (12) showed that the rate of adaptation of an organism is 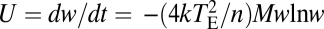 , where k is the product of the effective population size and the mutation rate per generation per genome (in the functional part), TE is the total effect size of a mutation defined earlier, n is the degree of pleiotropy of a mutation, w is the current mean fitness of the population relative to the optimal, and M is a function of TE and n (Methods). Although Orr assumed that each mutation affects all traits of an organism and therefore n is also a measure of organismal complexity, we have shown that deleting a gene affects only a small fraction of traits (Fig. 1). In other words, mutational pleiotropy is much smaller than organismal complexity. Nonetheless, if the fraction of traits affected by an average mutation is similar among different organisms (Fig. 1), mutational pleiotropy is still higher in complex organisms than in simple organisms, because complex organisms have more traits than simple organisms do. Hence, n in the above formula may be interpreted as the effective organismal complexity (i.e., the number of traits affected by an average mutation), which is much lower than the actual organismal complexity (i.e., the total number of traits). Empirical evidence suggests that k may increase slightly with the level of organismal complexity, but the exact relationship between them is unclear (19). To be conservative, we here assume k to be independent of n. Note that although we are using the original formula from Orr (12) for U, which was based on a fixed mutation size TE for a given n, this formula is known to be robust to variable mutation sizes (20). By comparing U among organisms of different levels of complexity, Orr implicitly assumed that the rate of adaptation is directly comparable among different organisms, which requires further theoretical and empirical support. Here, we follow Orr to compare U among different organisms.
, where k is the product of the effective population size and the mutation rate per generation per genome (in the functional part), TE is the total effect size of a mutation defined earlier, n is the degree of pleiotropy of a mutation, w is the current mean fitness of the population relative to the optimal, and M is a function of TE and n (Methods). Although Orr assumed that each mutation affects all traits of an organism and therefore n is also a measure of organismal complexity, we have shown that deleting a gene affects only a small fraction of traits (Fig. 1). In other words, mutational pleiotropy is much smaller than organismal complexity. Nonetheless, if the fraction of traits affected by an average mutation is similar among different organisms (Fig. 1), mutational pleiotropy is still higher in complex organisms than in simple organisms, because complex organisms have more traits than simple organisms do. Hence, n in the above formula may be interpreted as the effective organismal complexity (i.e., the number of traits affected by an average mutation), which is much lower than the actual organismal complexity (i.e., the total number of traits). Empirical evidence suggests that k may increase slightly with the level of organismal complexity, but the exact relationship between them is unclear (19). To be conservative, we here assume k to be independent of n. Note that although we are using the original formula from Orr (12) for U, which was based on a fixed mutation size TE for a given n, this formula is known to be robust to variable mutation sizes (20). By comparing U among organisms of different levels of complexity, Orr implicitly assumed that the rate of adaptation is directly comparable among different organisms, which requires further theoretical and empirical support. Here, we follow Orr to compare U among different organisms.
If TE is independent of n, as assumed in the invariant total effect model (12), or if TE is proportional to n0.5, as assumed in the Euclidian superposition model (11, 17, 18) (Table S2), adaptation rate U decreases with the degree of pleiotropy (or level of effective organismal complexity) n (Fig. 4A), creating the cost of complexity. Interestingly, the relationship between U and n changes when the scaling exponent b > 0.5. It can be shown mathematically that, when b > 0.5, an intermediate level of complexity yields the highest adaptation rate (SI Methods) (Fig. 4A). The n value that corresponds to the highest adaptation rate (noptimal) depends on several parameters, including a (referred to as the mutation size, which is the expected TE for mutations that affect only one trait), b, and w. Smaller a and w values lead to larger noptimal (Fig. 4B). The null mutations in the yeast morphological pleiotropy data yield a = 2.9, but we expect natural random mutations to have a much smaller a, because most natural mutations affect the function of a gene only slightly and thus have on average much smaller phenotypic effects than gene deletions do (Methods). For example, if a = 0.01 for natural random mutations, b = 0.6 as we have shown, and w = 0.9, noptimal becomes 9 (Fig. 4A).
Fig. 4.

The “cost of complexity” is alleviated when the scaling exponent b > 0.5. (A) The relative adaptation rate as a function of the degree of pleiotropy (n) changes with the scaling exponent b. The relative adaptation rate is calculated using Orr's formula. The initial fitness w is set at 0.9 and the mutation size a is set at 0.01. (B) The optimal degree of pleiotropy noptimal, defined as the degree of pleiotropy that corresponds to the highest adaptation rate, changes with the mutation size a. Different curves are generated using different initial fitness (w) values but the same b = 0.6. (C) The optimal degree of pleiotropy noptimal changes with different b. Different curves are generated using different a but the same w = 0.9. (D) A heat map showing the b value that provides the maximal noptimal, given a and w.
Numerically, we found that, when a and w are given, noptimal reaches its maximum at an intermediate b value (Fig. 4C). By examining a large parameter space (10−8 ≤ a ≤ 10−2; 0.3 ≤ w ≤ 0.99), we observed that the b value that offers the maximal noptimal occurs in a narrow range between 0.56 and 0.79 (Fig. 4D), although b potentially can vary from negative infinity to positive infinity.
It is important to confirm two key assumptions that Orr made in deriving the formula for U (12). The first assumption is that phenotypic traits are independent from one another, as was originally described by Fisher in his geometric model (10). As questioned by Haldane and other authors, this assumption is often unmet (20, 34, 35). Nonetheless, it was later demonstrated that the geometric model can still be applied when nonindependent traits are linearly transformed to compound traits that are independent from one another (35). We therefore performed a principal component analysis of the wild-type phenotypic matrix (SI Methods) to make the resultant principal component traits orthogonal to one another. Applying the same linear transformation to the mutant phenotypic matrix to obtain the compound phenotypic effects for each gene (SI Methods), we found that our results of the Euclidian scaling coefficients b > 0.5 and the Manhattan scaling coefficient d > 1.0 remain unchanged (Fig. S5). This finding also demonstrates that the phenomenon that genes affecting more traits have larger per-trait effects holds even when the traits are independent from one another.
The second assumption of Orr is that the fitness effect of a mutation should increase with its phenotypic effect size (TE). We confirmed the validity of this assumption by showing that the fitness effect of deleting a gene is significantly positively correlated with its TE (Spearman's ρ = 0.377, P < 10−10). This positive correlation, coupled with the positive correlation between TE and the degree of pleiotropy n (Fig. 3C), may explain the phenomenon that deleting a more pleiotropic gene tends to cause a larger fitness reduction (36).
Concluding Remarks.
In summary, our genome-wide analysis of pleiotropy in yeast, nematode, and mouse revealed a generally low level of pleiotropy for most genes in a eukaryotic genome and a highly modular structure in the gene–trait relationship. Furthermore, the quantitative morphological data from yeast showed that genes affecting more traits tend to have larger per-trait phenotypic effects. Although an organism potentially contains many more traits than our data currently include, several analyses indicated that our results are robust and therefore our conclusions are expected to be largely unchanged even when most or all traits of an organism are considered. These findings necessitate a major revision of the current theoretical models that lack the above three empirical features of pleiotropy (Table S2) and require reevaluation of biological inferences derived from these models. For example, these three features substantially alleviate the cost of complexity in adaptive evolution. First, the generally low pleiotropy means that even mutations in organisms as complex as mammals do not normally affect many traits simultaneously. Second, high modularity reduces the probability that a random mutation is deleterious, because the mutation is likely to affect a set of related traits in the same direction rather than a set of unrelated traits in random directions (20, 37). These two properties substantially lower the effective complexity of an organism. Third, the greater per-trait effect size for more pleiotropic mutations (i.e., b > 0.5) causes a greater probability of fixation and a larger amount of fitness gain when a beneficial mutation occurs in a more complex organism than in a less complex organism. These effects, counteracting lower frequencies of beneficial mutations in more complex organisms (10), result in intermediate levels of effective complexity having the highest rate of adaptation. Together, they explain why complex organisms could have evolved despite the cost of complexity. Because organisms of intermediate levels of effective complexity have greater adaptation rates than organisms of low levels of effective complexity due to the scaling property of pleiotropy, pleiotropy may have promoted the evolution of complexity. Whether the intriguing finding that the empirically observed scaling exponent b falls in a narrow range that offers the maximal optimal complexity is the result of natural selection for evolvability or a by-product of other evolutionary processes (38) requires further exploration.
Methods
Modularity of Gene–Trait Bipartite Networks.
Gene–trait relationships can be represented by a bipartite network where the genes form one type of nodes and traits form the second type of nodes. A link between a gene node and a trait node indicates that the gene affects the trait. To separate modules, we used BRIM (31), which is modified from the widely used Newman definition of modularity (39) for bipartite networks. For a given module partition, this algorithm calculates the difference between the density of within-module links and its random expectation. It then attempts to find the module partition that yields the highest difference, which is called the modularity of the network. The raw modularity score obtained from the algorithm has a theoretical range between 0 and 1, with 0 meaning no modularity and 1 meaning very high modularity. However, because even a random network may have a nonzero modularity depending on the network size and degree distribution (32, 40), we measure the level of network modularity by scaled modularity (32), which is the difference between the observed modularity and the mean modularity of its randomly rewired networks divided by the SD in modularity among the randomly rewired networks. The randomly rewired networks were generated by randomly linking nodes while conserving the number of links of each node and the total number of nodes of the original network. We also calculated scaled modularity after merging traits whose genetic correlation coefficient is >0.7. We chose this cutoff because, after the merge, no trait can genetically explain more than one-half of the variance of another trait (0.72 = 0.49).
Scaling Relationships Between the Degree of Pleiotropy and the Total Effect Size.
Using the yeast morphological pleiotropy data, we calculated the number (n) of traits that are significantly affected by each gene. We then measured a gene's total phenotypic effect on these n traits, using either the Euclidian distance (TE) or the Manhattan distance (TM). We expect the scaling relationships of  and
and  . We estimated a, b, c, and d using the curve-fitting toolbox in MATLAB, which employs a nonlinear least-squares method to fit the observations with the power function. The confidence intervals of the estimated parameters were also calculated by MATLAB, which uses the decomposition of the Jacobian, the degree of freedom, and the root mean squared error.
. We estimated a, b, c, and d using the curve-fitting toolbox in MATLAB, which employs a nonlinear least-squares method to fit the observations with the power function. The confidence intervals of the estimated parameters were also calculated by MATLAB, which uses the decomposition of the Jacobian, the degree of freedom, and the root mean squared error.
Because gene pleiotropy is largely owing to the involvement of the same molecular function in multiple different biological processes rather than the presence of multiple molecular functions per gene (26), random mutations in a gene will likely affect the same traits as the deletion of the gene does, although the magnitude of the phenotypic effects should be much smaller. For simplicity, let us assume that the effect on a trait from a random mutation in a gene is on average h times the effect on the same trait from a null mutation in the gene, where the effect is again measured by Z-score and 0 < h << 1. Let 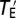 be the total effect size of the random mutation in Euclidean distance. It can be shown that
be the total effect size of the random mutation in Euclidean distance. It can be shown that 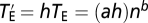 . Thus, the scaling relationship between the total effect size of a random mutation and pleiotropy is the same as that between the total effect size of a null mutation and pleiotropy, except that the mutation size parameter for random mutations is h times that for null mutations.
. Thus, the scaling relationship between the total effect size of a random mutation and pleiotropy is the same as that between the total effect size of a null mutation and pleiotropy, except that the mutation size parameter for random mutations is h times that for null mutations.
Calculating the Rate of Adaptation.
Assuming Fisher's geometric model, Orr (12) derived the formula for the rate of fitness increase during an adaptive walk to the optimal to be  where
where 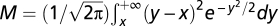 ,
, 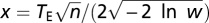 , and the other variables are defined in the main text. In Orr's calculation (12), TE was assumed to be independent of n. In our model, TE scales with the degree of pleiotropy by
, and the other variables are defined in the main text. In Orr's calculation (12), TE was assumed to be independent of n. In our model, TE scales with the degree of pleiotropy by 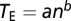 , where a is the mutation size parameter that corresponds to the mutation size when the degree of pleiotropy is 1 and b is the scaling exponent. We implemented numerical calculations of the above formulas in MATLAB.
, where a is the mutation size parameter that corresponds to the mutation size when the degree of pleiotropy is 1 and b is the scaling exponent. We implemented numerical calculations of the above formulas in MATLAB.
Supplementary Material
Acknowledgments
We thank Qi He for mathematical consultation and Meg Bakewell, Günter Wagner, Xionglei He, Wenfeng Qian, and Jian-Rong Yang for valuable comments. This work was supported by National Institutes of Health research grants (to J.Z.) and Taiwan National Health Research Institutes intramural funding (to B.-Y.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. G.P.W. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1004666107/-/DCSupplemental.
References
- 1.Tyler AL, Asselbergs FW, Williams SM, Moore JH. Shadows of complexity: What biological networks reveal about epistasis and pleiotropy. BioEssays. 2009;31:220–227. doi: 10.1002/bies.200800022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright S. Evolution and the Genetics of Populations. Chicago: Univ of Chicago Press; 1968. [Google Scholar]
- 3.Barton NH. Pleiotropic models of quantitative variation. Genetics. 1990;124:773–782. doi: 10.1093/genetics/124.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodgkin J. Seven types of pleiotropy. Int J Dev Biol. 1998;42:501–505. [PubMed] [Google Scholar]
- 5.Carroll SB. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 6.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 7.Albin RL. Antagonistic pleiotropy, mutation accumulation, and human genetic disease. Genetica. 1993;91:279–286. doi: 10.1007/BF01436004. [DOI] [PubMed] [Google Scholar]
- 8.Brunner HG, van Driel MA. From syndrome families to functional genomics. Nat Rev Genet. 2004;5:545–551. doi: 10.1038/nrg1383. [DOI] [PubMed] [Google Scholar]
- 9.Carroll SB. Evolution at two levels: On genes and form. PLoS Biol. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher RA. The Genetic Theory of Natural Selection. 2nd Ed. Oxford: Clarendon; 1930. [Google Scholar]
- 11.Waxman D, Peck JR. Pleiotropy and the preservation of perfection. Science. 1998;279:1210–1213. [PubMed] [Google Scholar]
- 12.Orr HA. Adaptation and the cost of complexity. Evolution. 2000;54:13–20. doi: 10.1111/j.0014-3820.2000.tb00002.x. [DOI] [PubMed] [Google Scholar]
- 13.Otto SP. Two steps forward, one step back: The pleiotropic effects of favoured alleles. Proc Biol Sci. 2004;271:705–714. doi: 10.1098/rspb.2003.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill JA, Otto SP. The role of pleiotropy in the maintenance of sex in yeast. Genetics. 2007;175:1419–1427. doi: 10.1534/genetics.106.059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Zhang J. Abundant indispensable redundancies in cellular metabolic networks. Genome Biol Evol. 2009;1:23–33. doi: 10.1093/gbe/evp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster KR, Shaulsky G, Strassmann JE, Queller DC, Thompson CR. Pleiotropy as a mechanism to stabilize cooperation. Nature. 2004;431:693–696. doi: 10.1038/nature02894. [DOI] [PubMed] [Google Scholar]
- 17.Wagner GP. The influence of variation and of developmental constraints on the rate of multivariate phenotypic evolution. J Evol Biol. 1988;1:45–66. [Google Scholar]
- 18.Turelli M. Effects of pleiotropy on predictions concerning mutation-selection balance for polygenic traits. Genetics. 1985;111:165–195. doi: 10.1093/genetics/111.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haygood R, SMBE Tri-National Young Investigators Proceedings of the SMBE Tri-National Young Investigators’ Workshop 2005. Mutation rate and the cost of complexity. Mol Biol Evol. 2006;23:957–963. doi: 10.1093/molbev/msj104. [DOI] [PubMed] [Google Scholar]
- 20.Welch JJ, Waxman D. Modularity and the cost of complexity. Evolution. 2003;57:1723–1734. doi: 10.1111/j.0014-3820.2003.tb00581.x. [DOI] [PubMed] [Google Scholar]
- 21.Brown KD, Barlow C, Wynshaw-Boris A. Multiple ATM-dependent pathways: An explanation for pleiotropy. Am J Hum Genet. 1999;64:46–50. doi: 10.1086/302223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hekerman P, et al. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J. 2005;272:109–119. doi: 10.1111/j.1742-4658.2004.04391.x. [DOI] [PubMed] [Google Scholar]
- 23.Ohya Y, et al. High-dimensional and large-scale phenotyping of yeast mutants. Proc Natl Acad Sci USA. 2005;102:19015–19020. doi: 10.1073/pnas.0509436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudley AM, Janse DM, Tanay A, Shamir R, Church GM. A global view of pleiotropy and phenotypically derived gene function in yeast. Mol Syst Biol. 2005;1 doi: 10.1038/msb4100004. 2005.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sönnichsen B, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434:462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 26.He X, Zhang J. Toward a molecular understanding of pleiotropy. Genetics. 2006;173:1885–1891. doi: 10.1534/genetics.106.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner GP, et al. Pleiotropic scaling of gene effects and the ‘cost of complexity’. Nature. 2008;452:470–472. doi: 10.1038/nature06756. [DOI] [PubMed] [Google Scholar]
- 28.Albert AY, et al. The genetics of adaptive shape shift in stickleback: Pleiotropy and effect size. Evolution. 2008;62:76–85. doi: 10.1111/j.1558-5646.2007.00259.x. [DOI] [PubMed] [Google Scholar]
- 29.Su Z, Zeng Y, Gu X. A preliminary analysis of gene pleiotropy estimated from protein sequences. J Exp Zool B Mol Dev Evol. 2010;314:115–122. doi: 10.1002/jez.b.21315. [DOI] [PubMed] [Google Scholar]
- 30.Wagner GP, Pavlicev M, Cheverud JM. The road to modularity. Nat Rev Genet. 2007;8:921–931. doi: 10.1038/nrg2267. [DOI] [PubMed] [Google Scholar]
- 31.Barber MJ. Modularity and community detection in bipartite networks. Phys Rev E Stat Nonlin Soft Matter Phys. 2007;76:066102. doi: 10.1103/PhysRevE.76.066102. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Zhang J. In search of the biological significance of modular structures in protein networks. PLOS Comput Biol. 2007;3:e107. doi: 10.1371/journal.pcbi.0030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hermisson J, McGregor AP. Pleiotropic scaling and QTL data. Nature. 2008;456:E3. doi: 10.1038/nature07452. [DOI] [PubMed] [Google Scholar]
- 34.Haldane J. The Causes of Evolution. London: Longmans Greens; 1932. [Google Scholar]
- 35.Waxman D, Welch JJ. Fisher's microscope and Haldane's ellipse. Am Nat. 2005;166:447–457. doi: 10.1086/444404. [DOI] [PubMed] [Google Scholar]
- 36.Cooper TF, Ostrowski EA, Travisano M. A negative relationship between mutation pleiotropy and fitness effect in yeast. Evolution. 2007;61:1495–1499. doi: 10.1111/j.1558-5646.2007.00109.x. [DOI] [PubMed] [Google Scholar]
- 37.Martin G, Lenormand T. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution. 2006;60:893–907. [PubMed] [Google Scholar]
- 38.Pigliucci M. Is evolvability evolvable? Nat Rev Genet. 2008;9:75–82. doi: 10.1038/nrg2278. [DOI] [PubMed] [Google Scholar]
- 39.Newman ME. Fast algorithm for detecting community structure in networks. Phys Rev E Stat Nonlin Soft Matter Phys. 2004;69:066133. doi: 10.1103/PhysRevE.69.066133. [DOI] [PubMed] [Google Scholar]
- 40.Guimerà R, Sales-Pardo M, Amaral LA. Modularity from fluctuations in random graphs and complex networks. Phys Rev E Stat Nonlin Soft Matter Phys. 2004;70:025101. doi: 10.1103/PhysRevE.70.025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.