

Abstract
Transcription of interleukin (IL)-12 p40 in myeloid cells is attributed to the recruitment of multiple activated transcription factors such as nuclear factor κB (NFκB), CCAAT enhancer-binding protein β, ets-2, PU.1, and so forth. We now provide the first description of the human erythroid Kruppel-like factor (EKLF) in human primary macrophages and identify the role of EKLF in IL-12 p40 expression. EKLF-specific binding to the CACCC element (−224 to −220) on the human IL-12 p40 promoter was observed in resting human primary macrophages. Functional analysis of the CACCC element revealed a dependent role for EKLF binding in activating IL-12 p40 transcription in resting RAW264.7 cells, whereas EKLF overexpression in the presence or absence of this element repressed IL-12 p40 transcription in interferon γ/lipopolysaccharide-stimulated RAW264.7 cells. Murine endogenous IL-12 p40 mRNA was consistently induced by overexpressed EKLF in resting RAW264.7 cells, whereas EKLF suppressed IL-12 p40 expression in activated RAW264.7 cells. Modulation of nuclear binding activities at the IL-12 p40 NFκB half-site was induced by EKLF for down-regulation of IL-12 p40 transcription in activated RAW264.7 cells, but no effect of EKLF on NFκB activity was observed in resting RAW264.7 cells. Taken together, we identify EKLF as a transcription factor in macrophages able to regulate IL-12 p40 transcription depending on the cellular activation status. The bifunctional control of IL-12 p40 by EKLF and its modulation of NFκB support a potential function for this factor in orchestrating IL-12 p40 production in macrophages.
Interleukin (IL)-12 1 p40 expression is a critical component of heterodimer cytokines IL-12 and IL-23, both of which activate innate immunity and contribute to the development of type 1 T-helper responses (1–3). IL-12 p40 is a shared component with IL-12 p35 and IL-23 p19 to form biologically active IL-12 and IL-23, respectively. Similar to IL-12 p35, p19 is poorly secreted when expressed alone and requires IL-12 p40 for optimal expression. Although IL-12 and IL-23 share functions, IL-23 has preferential activity on memory T cells, suggesting that it may function to sustain a long-term cellular immune response in contrast to the activity of IL-12 on naïve cells at the onset of the response. The positive and negative transcriptional regulation of IL-12 p40 is an important component of the control of inflammatory responses by antigen-presenting cells (3). Analysis of how transcription factors control IL-12 p40 expression has revealed several trans-activators such as nuclear factor κB (NFκB), CCAAT enhancer-binding protein (C/EBP) β/LAP, ets-2, PU.1, AP-1, IRF-1 and ICSBP in human and/or murine macrophages (4–10). However, inhibitory mechanisms for IL-12 p40 transcription in human macrophages have not been elucidated, except for the identification of a cis-element named GA-12 in the human IL-12 p40 promoter that interacts with a nuclear factor induced by IL-4 or prostaglandin E2 in human monocytes and that mediates IL-4-mediated repression of IL-12 p40 promoter activity (11).
Analysis of β-globin and tryptophan oxygenase promoters demonstrates that the CACCC box in these promoters is essential for gene activation because mutation of this box can lead to dysfunctional transcription (12). Recognition and binding to CACCC sequences have been shown to be mediated predominantly by a family of proteins referred to as Kruppel-like factors (KLFs) (13–15). KLFs have been reported as both activators (16) and repressors (17, 18) of gene expression, depending on the type of KLF, cell type, and other transcription factors with which it may interact. For example, erythroid Kruppel-like factors (EKLFs) activate β-globin gene expression when binding to the CACCC element of its promoter (19) but can repress gene expression by recruiting co-repressors such as histone deacetylases (20). A CACCC box-binding protein named htβ was found to be expressed in the macrophage cell line U937 (21). KLFs have a broad biological function such as cellular proliferation/differentiation, apoptosis, angiogenesis, and tumorigenesis as reviewed recently (22). Recent evidence suggests that KLFs regulate the promoter activities of complement C4 (23) and iNOS (24), implicating a role of KLFs in immune system due to the gene-regulatory role via the CACCC cis-element. However, expression or function of KLFs has not been described in primary myeloid cells. We now identify the presence of EKLF in human macrophages as a factor able to activate the IL-12 p40 promoter in a CACCC element-dependent manner in resting macrophages and repress IL-12 p40 promoter in association with inhibition of NFκB nuclear localization in activated macrophages.
Experimental Procedures
Cell Culture
Human elutriated macrophages from healthy donors were obtained by standard leukopheresis and counterflow centrifugation and cultured at a density of 2 × 106 cells/ml RPMI 1640 (Mediatech, Herndon, VA) supplemented with 10% fetal calf serum (HyClone, Logan, UT), 100 μg/ml penicillin, 100 μg/ml streptomycin, and 200 mM glutamine at 37 °C and 5% CO2. RAW264.7 cells (American Type Culture Collection, Manassas, VA) were split every 2–3 days and also maintained in RPMI 1640 medium at a density between 0.2 and 0.5 × 106 cells/ml before transfection. To activate them, cells were stimulated by murine IFN-γ (10 ng/ml) overnight followed by lipopolysaccharide (LPS; 1 μg/ml) for the amount of time as indicated.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed as described previously (25). The wild type oligonucleotides 5′-ATTCCCCACCCAAAAGTCATTTCCTCTTAGT-3′ (−230 to −200 relative to the transcription start site on human IL-12 p40 promoter; Ref. 26) harboring the CACCC element (−224/−220) and its CACCC element mutant oligonucleotides with replacement of the CACCC element by nucleotides CTGCC (Fig. 1A) and the oligonucleotides harboring the NFκB half-site sequence ACTTCTTGAAATTCCCCCAGAAGG derived from IL-12 p40 promoter were synthesized by Sigma Genosys (The Woodlands, TX). The consensus NFκB and C/EBP oligonucleotides were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Cells were stimulated as indicated in figure legend before nuclear extractions. For supershift assay, 5-μg nuclear extracts were incubated with 1 μl or 2 μg of antibodies on ice for 30 min before incubation with p32-labeled oligonucleotides.
Fig. 1. Identification of EKLF as the CACCC-binding protein bound to IL-12 p40 promoter in elutriated human primary macrophages.
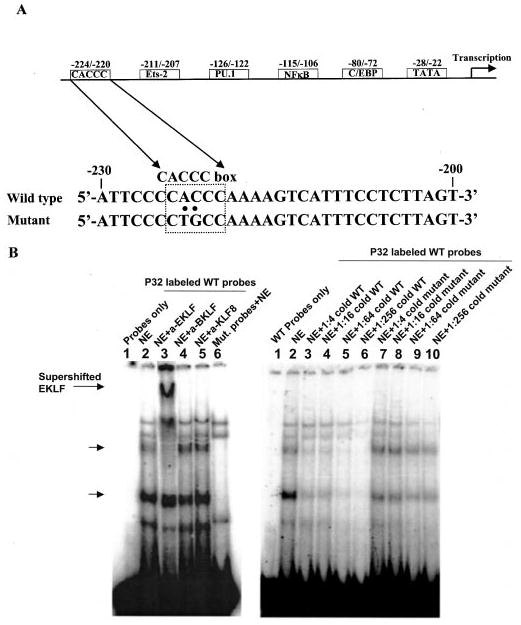
A, sequence of wild type IL-12 p40 promoter encompassing the CACCC element and its CACCC element mutant oligonucleotides used in EMSA, together with other identified functional cis-elements in human IL-12 p40 promoter. B, left panel shows a supershift EMSA in which lane 1 shows probe only control and lanes 2 (wild type probe) and lane 6 (mutant probe) show probe binding in absence of antisera (no antibodies). Nuclear extracts (NE) were incubated with antisera against human Kruppel-like factor family members including 1 μl of EKLF (lane 3), BKLF (lane 4), or KLF8 (lane 5). Note that only EKLF antisera were able to supershift the CACCC-binding protein. Right panel, analysis of binding site specificity through competition and mutation EMSAs. Lanes with competition EMSA reaction had cold wild type (WT) or mutant oligonucleotides in molar excess from 4- to 256-fold (in 4-fold increments) over the 32P-labeled wild type oligonucleotides before protein-DNA interaction reactions. The arrows indicate the specific CACCC element binding product. The EMSA is a representative of three similar experiments.
Site-directed Mutagenesis
To generate the IL-12 p40 promoter harboring a mutated CACCC element (−224/−220) linked with a luciferase gene as a reporter for transfection assay, the CACCC element mutant sense oligonucleotides (5′-CCTTCCTTATTCCCCTGCCAAAAGTCATTTCCT-3′, −238 to −206 relative to the transcription start site of human IL-12 p40 promoter (26)) and the corresponding antisense primers were synthesized by Invitrogen with mutation of CACCC (−224/−220) to CTGCC. The CACCC mutant plasmid of human IL-12 p40-luciferase was made by the Genomic Core Facility (University of Pennsylvania, Philadelphia, PA), following the instructions of QuikChange™ Site-Directed Mutagenesis Kit (Stratagene). Mutations were confirmed by DNA sequencing.
Transfection Assay
All plasmids used in transfection assay were prepared with the endo toxin-free plasmid Maxi-kit (Qiagen, Valencia, CA) and resuspended with endo-free 0.1× Tris/EDTA buffer to a concentration of 1 μg/μl. Transfections of RAW264.7 cells were performed as described previously (26). The truncated IL-12 p40 promoter constructs Hp40–122 and Hp40–222 harboring NFκB half-site element TGAAATTCCCC were generated by PCR as described previously (6). Luciferase activity was generated with luciferase substrate purchased from Promega according to the procedures of Luciferase Assay System (Promega) and read out in opaque 96-well plates using a plate reading luminometer (PerkinElmer Life Sciences). CMV-β-galactosidase was co-transfected to normalize transfection efficiency. β-Galactosidase activity was read with β-galactosidase enzyme assay system from Promega. Experiments were performed in triplicate.
Western Blot
Western blots were performed as described previously (25). Briefly, 5 μg of nuclear extracts were fractionated in 10% SDS-PAGE gels for each sample. The separated proteins were transferred to the polyvinylidene difluoride membrane (Millipore) and detected by 1:1000 diluted rabbit antiserum against EKLF, basic KLF, and KLF8 (27); 1 μg/ml anti-NFκB p50, p65, c-Rel, and histone H1 purchased from Santa Cruz Biotechnology, Inc.; and 1:500 diluted anti-actin (Sigma) followed by 1:4500 diluted secondary antibodies of donkey anti-rabbit Ig and horseradish peroxidase-linked whole antibody (Amersham Biosciences). The bound horseradish peroxidase was detected by ECL™ detection reagents (Amersham Biosciences). The produced chemiluminescent signals were captured by Kodak radiography film (Eastman Kodak Co.).
Multiplex Reverse Transcription-PCR (RT-PCR)
Total RNA was extracted using TRI-Reagent (Molecular Research Center, Cincinnati, OH). 1 μg of RNA was treated by DNA-free™ kit (Ambion, Austin, TX) to remove genomic DNA contamination followed by reverse transcription using the RETROscript kit (Ambion) according to the manufacturer's instructions. Human EKLF forward primer (5′-GTTCGGAGGATCACTCGGGT-3′) and reverse primer (5′-GGTGTAGCTCTTGCCGCAAC-3′) were synthesized by Sigma Genosys. Mouse IL-12 p40 primer pairs (amplicon size, 396 bp), Classic 18S primer™ pairs (amplicon size, 488 bp), Classic II 18S primer™ pairs (amplicon size, 324 bp), and the corresponding competimor primer pairs were purchased from Ambion. 0.5 μl of 10 μCi/μl [α-32P]dCTP was added in each reaction. The cDNA products were amplified using the PCR Core Kit (Roche Applied Science). The amplification program was performed as follows: samples were heated at 95 °C for 5 min, followed by 28 (human EKLF) or 30 (murine IL-12 p40) cycles of 94 °C for 20 s, 55 °C (human EKLF) or 57 °C (murine IL-12 p40) for 30 s, and 72 °C for 40 s.
Transfection/Cell Sorting
10 million RAW264.7 cells were transfected with 10 μg of pSG5-EKLF and 4 μg of pLEIN-GFP in each cuvette. A total amount of 80–100 million RAW264.7 cells were transfected each time. Cells transfected with pSG5-EKLF/pLEIN-GFP were sorted for green fluorescent protein (GFP) expression 1 day after transfection by flow cytometry. GFP-positive and GFP-negative cells were then treated with IFN-γ/LPS as indicated.
Statistical Analysis
Statistical software of JMP4 (SAS Institute Inc., Cary, NC) was used to describe data and test for differences by two-tailed paired Student's t test. p values of <0.05 were considered significant.
Results
EKLF: CACCC-binding Protein on Human IL-12 p40 Promoter
A study was carried out to investigate whether CACCC-binding proteins were present in human primary macrophages and could interact with the IL-12 p40 promoter at the CACCC element (−224/−220) (Fig. 1A) by EMSA. As shown in Fig. 1B, five nuclear protein bands (left panel, lane 2) were identified to bind to the probes representing constitutively expressed proteins in elutriated human primary macrophages. The two bands labeled by arrows stand for the proteins binding to the CACCC element because the signals were lost in the nuclear extracts incubated with the oligonucleotides mutated at the CACCC element (Fig. 1B, left panel, lane 6).
The identity of the CACCC-binding proteins binding to the CACCC element of IL-12 p40 promoter in elutriated human macrophages was investigated by supershift EMSA. Because previous work showed that the majority of identified CACCC element-binding proteins belong to Sp1/Kruppel-like factor (17, 19, 22, 28), we tested for the presence of EKLF, BKLF, and KLF8 in the CACCC-binding proteins. As shown in Fig. 1B, only sera against EKLF acted to retard migration of the complex c (left panel, lane 3), suggesting that this complex is likely to be EKLF. The specificity of this complex was further examined by competition with increasing amounts of cold wild type probes (Fig. 1B, right panel, lanes 3–6), leading to the loss of signal. Furthermore, competition with increasing amounts of cold mutant probes had minimal effect on this complex (Fig. 1B, right panel, lanes 7–10). Based on the fact that the anti-EKLF antisera was made against N-terminal EKLF, the faster-moving band that is specific to the CACCC region is interpreted as a breakdown product of the upper protein band such as the C-terminal EKLF containing the three zinc finger domains.
Expression of EKLF in elutriated human primary macrophages was confirmed by RT-PCR and Western blot analysis. EKLF expression by mRNA was clearly identified in primary macrophages, whereas it was weak in the macrophage cell lines THP1 and U937 (Fig. 2A). EKLF protein was also abundant in the nuclei of primary macrophages, consistent with its mRNA expression and observations with EMSA, but EKLF protein was not detectable in THP1 and U937 cells (Fig. 2B, top panel). The other two KLF family members, BKLF and KLF8, were also examined in macrophages. Interestingly, BKLF showed an opposite expression pattern to EKLF in macrophages. BKLF protein was weakly expressed in primary macrophages (Fig. 2B, middle panel), in contrast to the abundant level of BKLF protein in macrophage cell lines THP1 and U937. KLF8 was not detected in primary macrophages or THP1 or U937 cells (data not shown). Taken together, these data identify EKLF as the transcription factor binding to the CACCC cis-element on the IL-12 p40 promoter in human primary macrophages.
Fig. 2. EKLF mRNA and protein expression in elutriated human primary macrophages and human monocyte-like macrophage cell lines.
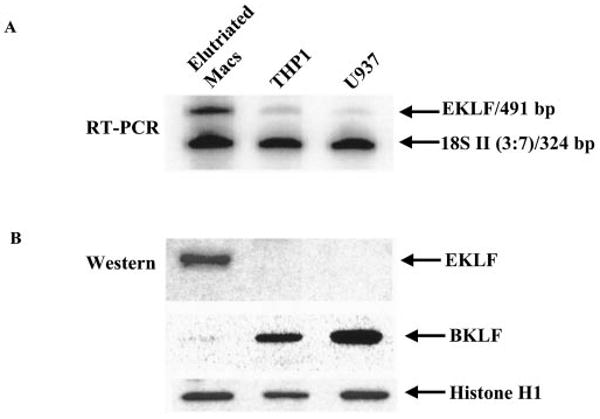
A, multiplex RT-PCRs were performed to detect EKLF mRNA. 18S competimer primer pairs that were modified at the 3′ ends to block product extension by DNA polymerase were used to compete with 18S primer pairs to capture two mRNA signals whose abundance varies considerably in the same experiment. The ratio of 18S primer pairs to 18S competimer primer pairs was 3:7. B, Western blots were used to detect the EKLF and BKLF protein levels in the nuclei of primary macrophages and cell lines THP1 and U937. The same nuclear extracts were immunoblotted with anti-histone H1 as a control. The RT-PCR and Western pictures are representatives of three similar experiments.
Activation of Human IL-12 p40 by EKLF in Resting RAW264.7 Cells
The functional role of human EKLF at the CACCC element on the human IL-12 p40 promoter was examined by co-transfection of human EKLF with human IL-12 p40 promoter linked with a luciferase reporter gene in the murine macrophage cell line RAW264.7 due to its similarity to IL-12-producing human cell types and reliable transfection efficiency. The suitability of this transfection system for the interaction between human EKLF and the CACCC element on the human IL-12 p40 promoter was evaluated upon overexpression of EKLF. As shown in Fig. 3A, EKLF was not detected in RAW264.7 cells with rabbit EKLF anti-serum reactive with both human and murine EKLF (29) (lanes 1 and 2). IFN-γ/LPS treatment did not have a noticeable effect on the endogenous or overexpressed EKLF (Fig. 3A, lane 1 versus lane 2 and lane 3 versus lane 4). Transfection of human EKLF in the absence of stimulation (with or without polymyxin B as an added control against exogenous LPS) induced human IL-12 p40 transcription activity in resting RAW264.7 cells (Fig. 3B, columns 1 and 3), suggesting that EKLF functions as an activator of IL-12 p40 expression. The activation of IL-12 p40 by EKLF is shown to be dependent on its binding to the CACCC element (−224/−220) because EKLF could not activate the mutant p40 promoter with a CTGCC sequence in replacement of the CACCC element (Fig.3B, columns 5 and 7). Note that the absence of EKLF in RAW264.7 cells as shown in Fig. 3A is interpreted to correlate with the lack of a change in IL-12 p40 transcription activity between columns 1 and 5 and columns 2 and 6 (Fig. 3B), irrespective of the presence or absence of the ability to bind EKLF.
Fig. 3. Effect of EKLF on transcription activity of IL-12 p40 promoter in RAW264.7 cells.
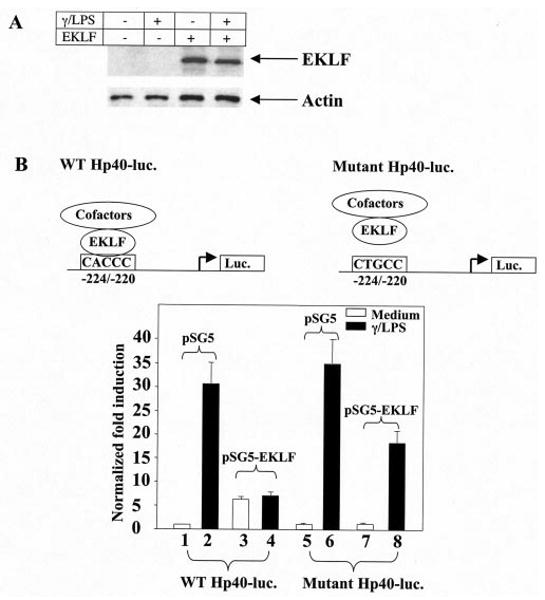
A, detection of endogenous and overexpressed EKLF in RAW264.7 cells by Western blots. Total proteins from each culture were immunoblotted with anti-actin as a control. B, the normalized fold induction obtained from RAW264.7 cells transfected with either wild type human IL-12 p40 (WT Hp40-luc.) or its CACCC element mutant form (Mutant Hp40-luc.) is shown. Transfection efficiency was normalized against co-transfected CMV-β-galactosidase plasmids. The mean value of the basal unstimulated wild type IL-12 p40 promoter activity was set at 1. The transcriptional activity was expressed as fold induction from the wild type IL-12 p40 promoter activity. Error bars represent the S.D. for n = 6.
Repression of Human IL-12 p40 by EKLF in Activated RAW264.7 Cells
In contrast to EKLF activating IL-12 p40 expression in resting macrophages, EKLF repressed IL-12 p40 transcription activity in IFN-γ/LPS-stimulated macrophages (Fig. 3B, columns 2 and 4). The EKLF inhibition of transcription activity of wild type IL-12 p40 promoter was partially reversed on the mutant IL-12 p40 promoter (Fig 3B, columns 4 and 8) in IFN-γ/LPS-stimulated macrophages, supporting a partial role for the CACCC element (−224/−220) on the IL-12 p40 promoter in mediating inhibition. However, the lack of complete reversal of inhibition by EKLF when unable to bind to the CACCC element identified the presence of an activity outside of this motif.
Effects of EKLF on Murine IL-12 p40 Expression in RAW264.7 Cells
The effect of human EKLF on murine endogenous IL-12 p40 expression was investigated as described for the human promoter because a conserved CACCC cis-element is present (30). RAW264.7 cells were enriched for transfected EKLF/GFP via cell sorting for the presence (+EKLF) or absence (−EKLF) of GFP fluorescence and subsequently stimulated. Results are consistent with human IL-12 p40 promoter data described. EKLF enhanced the endogenous IL-12 p40 mRNA level (Fig. 4, lanes 1 and 3) in unstimulated cells while repressing expression upon IFN-γ/LPS stimulation (Fig. 4, lanes 2 and 4). Taken together, these data show a shared activity by human EKLF in modulating murine IL-12 p40 gene expression.
Fig. 4. Effect of EKLF on endogenous IL-12 p40 mRNA level in RAW264.7 cells.
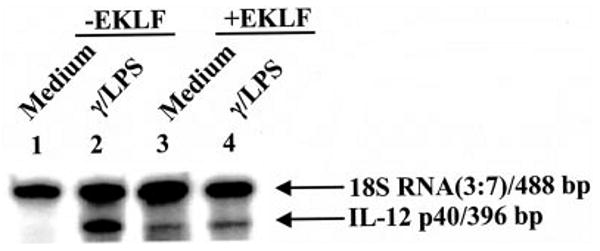
Multiplex RT-PCRs were performed to detect IL-12 p40 mRNA in EKLF-transfected cells enriched through cell sorting. The experiment shown is a representative of three similar experiments.
Induction of NFκB Modulation by EKLF in Activated RAW264.7 Cells
To dissect the additional factor involved in EKLF repression of IL-12 p40 transcription in activated RAW264.7 cells envisaged from the transfection experiments, NFκB and C/EBPβ were investigated based on the essential role of these factors in regulating IL-12 p40 transcription (4, 5, 10, 31). EKLF abrogated IFN-γ/LPS-stimulated transcription activity of two constructs derived from the human IL-12 p40 promoter containing either (a) upstream sequences up to −122 of the promoter (Hp40–122) inclusive of the NFκB half-site TGAAATTCCCC (−116/−106) and C/EBP motifs (6, 25) or (b) upstream sequences up to −222 of the promoter (Hp40–222) short of an intact CACCC cis-element (−224/−220) (Fig. 5A). Based on the potential for EKLF modulation of NFκB or C/EBPβ in IL-12 p40 transcription due to its retained inhibition potential on truncated promoter Hp40–122, EMSAs were used to identify the effect of EKLF on the binding activity of either NFκB or C/EBP with the IL-12 p40 promoter.
Fig. 5. EKLF recruits the activity of NFκB modulation in activated but not resting macrophages.
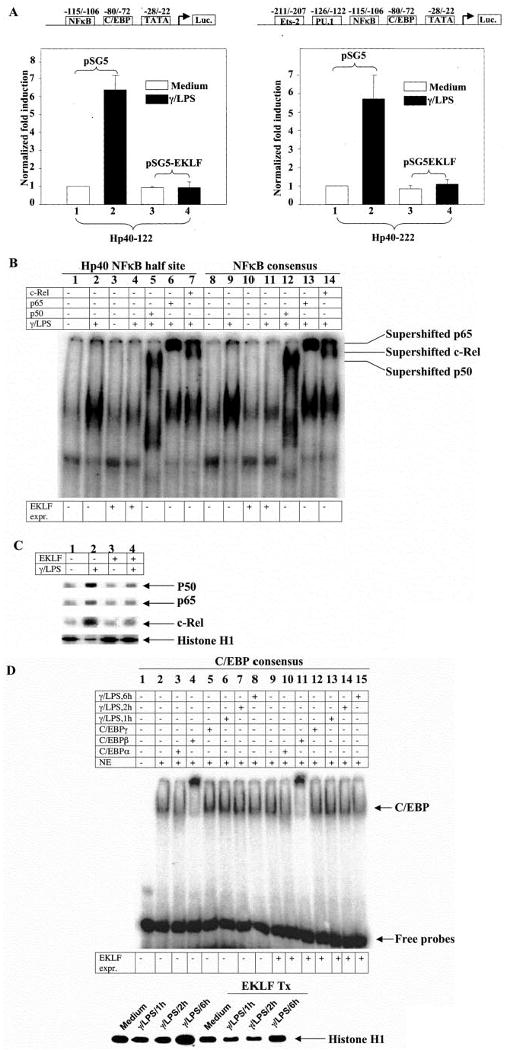
A, the effect of EKLF overexpression is shown on the luciferase reporter construct driven by Hp40–122 and Hp40–222 that harbor a putative NFκB half-site binding site TGAAATTCCCC (−116 to −106). Hp40–122 and Hp40–222 were derived from wild type Hp40 promoter with the sequences downstream of nucleotides −122 to −222 (relative to transcription start site), respectively. Transfection efficiency was normalized against co-transfected CMV-β-galactosidase plasmids. The mean value of the basal unstimulated promoter activity was set at 1. The transcriptional activity was expressed as fold induction from the unstimulated promoter activity. n = 4. B, supershift EMSA using the Hp40 NFκB half-site oligonucleotides and the NFκB consensus oligonucleotides is shown with the nuclear extracts from EKLF-enriched RAW264.7 cells. Cells were stimulated by murine IFN-γ (10 ng/ml) overnight followed by LPS (1 μg/ml) for 1 h. The experiment shown is representative of three similar experiments. C, the same nuclear extracts were immunoblotted with anti-p50, stripped, and re-probed with anti-p65 and anti-c-Rel, respectively. Re-probing with anti-histone H1 was performed as a control. The experiment shown is representative of three similar experiments. D, supershift EMSA using the C/EBP consensus oligonucleotides is shown with the nuclear extracts from EKLF-enriched RAW264.7 cells. Cells were stimulated by murine IFN-γ (10 ng/ml) overnight followed by LPS (1μg/ml) for 1, 2, or 6 h. Nuclear extracts were immunoblotted with anti-histone H1 as a control. The experiment shown is representative of three similar experiments.
IFN-γ/LPS stimulation of RAW264.7 cells showed NFκB binding activity with the oligonucleotides harboring the NFκB half-site sequence derived from IL-12 p40 promoter, as expected, with a clear repression of binding upon EKLF expression (Fig. 5B, lanes 2 and 4). Interestingly, EKLF expression did not affect NFκB binding in the absence of stimulation (Fig 5B, lanes 1 versus 3 and 8 versus 10), in agreement with an activation-dependent effect. To address a mechanism for decreased binding to NFκB in the presence of activation and EKLF, nuclear levels of p65, p50, and c-Rel were measured. EKLF expression was found to decrease the amount of p65, p50, and c-Rel in nuclear extracts (Fig. 5C). It is interpreted that nuclear export is enhanced by EKLF because all three factors are confirmed to bind this region in activated cells (Fig 5B, lanes 5–7). Similar observations were made with NFκB consensus oligonucleotides (Fig. 5B, lanes 8–14). A specificity for modulating NFκB activity was found when evaluating EKLF effects on C/EBP. Minimal effects were observed in the binding of candidate factors to the consensus C/EBP motif in the presence of activation and EKLF. Among the factors investigated, C/EBPβ was found to be the major form of family member that constitutively exists in RAW264.7 cells in the presence or absence of EKLF (Fig. 5D, lanes 2, 4, 9, and 11). Interestingly, nuclear binding activity of C/EBPβ increased to some extent after a 1-h incubation with LPS (Fig. 5D, lanes 2, 6, 8, and 13), as expected, yet the effects on this increase were minimal in the presence of EKLF (Fig. 5D, lanes 6–8 versus 13–15; Fig 5B, lanes 1–4). Thus, repression of IL-12 p40 expression in activated RAW264.7 cells by EKLF occurs at the NFκB binding site in association with an inhibition of NFκB nuclear localization.
Discussion
We identify a novel cis-element on the IL-12 p40 promoter, the CACCC box, and the nuclear protein that binds to this site as EKLF, which was previously unidentified as a transcription factor in primary human macrophages. Our data are consistent with a bifunctional role for EKLF in modulating IL-12 p40 expression. The effects of EKLF activity on IL-12 p40 expression and its modulation of NFκB binding suggest a role of EKLF in innate immunity.
The identification of EKLF as the binding factor to the CACCC element (−224/−220) was unexpected based on prior data showing a lack of expression for this transcription factor in monocyte-derived cell lines (13). Indeed, we confirmed low to no mRNA or protein expression levels in U937 and THP1 cell lines, whereas levels of both mRNA and protein were high in primary human macrophages (Fig. 2). Although our data show that differentiated macrophages express EKLF, it remains to be determined how additional factors may affect total expression levels for this protein (macrophage differentiation, cytokine environments, and so forth) and thus act to regulate IL-12 p40 expression. As with the transcription factor PU-1, which was originally described in erythroid cells and later identified as an important factor in myeloid cell transcription of immunoregulatory genes (32–34), EKLF was initially described as an erythroid cell-specific factor (13, 19) essential in erythroid development and responsible for regulating β-globin expression (35).
As a TLR4 ligand, LPS is a powerful stimulus derived from Gram-negative bacteria for NFκB activation (36) and IL-12 p40 production by macrophages. Our data showed that EKLF over-expression inhibited IFN-γ/LPS-stimulated IL-12 p40 expression (Figs. 3–5) in EKLF-deficient RAW264.7 cells in association with modulation of binding to the NFκB binding site. EKLF promoted a large decrease in the total amount of NFκB p50, p65, and c-Rel present in the nucleus, which is interpreted to have limited NFκB binding and activation (Fig. 5). Based on the activity of histone deacetylase 3 to promote NFκB nuclear export (37), the recruitment of histone deacetylases by EKLF in activated macrophages may present a potential selective step to its mechanisms of action. Our findings on EKLF modulation of IL-12 p40 promoter by activity on the CACCC and NFκB motifs raised the hypothesis for a role for this factor in contributing to inflammation and innate immunity, given the role NFκB plays in the expression of cytokine and chemokine genes such as IL-12 p40 (5, 6, 38), IL-1α (39), IL-1β (40, 41), IL-6 (42, 43), IL-8 (44), tumor necrosis factor α (45, 46), MCP-1 (47), and MIP-1α (48). As a potentially important mechanism to dampen inflammatory responses, the suppressive role of EKLF may provide the host with a constitutive mechanism to control against the expression of a powerful immune activator in the presence of chronic stimulation.
The activity by EKLF on the CACCC element (−224/−220) of the IL-12 p40 promoter in absence of activation (Fig. 3) points to a mechanism of maintaining or bypassing the need for continuous stimulation to express IL-12 p40. We found this activity to be restricted to the proximal CACCC domain because mutation of an upstream CACCC element (−1662 to −1658) on the IL-12 p40 promoter in the presence or absence of the mutated downstream CACCC element (−224/−220) did not affect the mode of action of EKLF in regulating IL-12 p40 transcription accordingly (data not shown). Endogenous IL-12 p40 production by macrophages, albeit low, has been shown to be critical for Th1 development by studying the outcome of genetic mutation of IL-12 p40 (49) and IL-12 p40 receptor β1 (50, 51) in human and in IL-12 p40 knockout murine models (52, 53). Precedents exist for EKLF to function as activator by interacting directly with other proteins such as cAMP-response element-binding protein-binding protein (CBP) and p300 (54) and as repressor by interacting with mSin3A and histone deacetylases in association with its activity on CACCC elements (20). In support of EKLF regulating transcription by interaction with other proteins, interaction between EKLF and SWI/SNF has been identified in gene regulation (55). Based on our data and the data of others (4, 20), we can propose the following hypothetical model by which to interpret our data: (a) EKLF activates IL-12 p40 transcription by recruiting co-activators CBP and p300 in resting macrophages; and (b) EKLF represses IL-12 p40 transcription by recruiting co-repressors mSin3A and histone deacetylases in activated macrophages. For example, histone deacetylases may deacetylate acetylated NFκB family member c-Rel, leading to nuclear export of c-Rel dimerized with p50.
Acknowledgments
We acknowledge Drs. G. Trinchieri and A. Henderson for useful discussions.
Footnotes
This work was supported by The Philadelphia Foundation (Robert I. Jacobs Fund), M. Stengel-Miller, H. S. Miller, Jr., the Commonwealth of Pennsylvania, and National Institutes of Health Grants AI47760, AI51225, AI54891, and AI34412.
The abbreviations used are: IL, interleukin; NFκB, nuclear factor κB; C/EBP, CCAAT enhancer binding protein; KLF, Kruppel-like factor; EKLF, erythroid Kruppel-like factor; BKLF, basic Kruppel-like factor; LPS, lipopolysaccharide; IFN, interferon; RT-PCR, reverse transcription-PCR; EMSA, electrophoretic mobility shift assay; GFP, green fluorescent protein.
References
- 1.Chehimi J, Starr SE, Frank I, D'Andrea A, Ma X, MacGregor RR, Sennelier J, Trinchieri G. J Exp Med. 1994;179:1361–1366. doi: 10.1084/jem.179.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lankford CS, Frucht DM. J Leukocyte Biol. 2003;73:49–56. doi: 10.1189/jlb.0602326. [DOI] [PubMed] [Google Scholar]
- 3.Trinchieri G. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 4.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Proc Natl Acad Sci U S A. 2000;97:12705–12710. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Mol Cell Biol. 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gri G, Savio D, Trinchieri G, Ma X. J Biol Chem. 1998;273:6431–6438. doi: 10.1074/jbc.273.11.6431. [DOI] [PubMed] [Google Scholar]
- 7.Zhu C, Gagnidze K, Gemberling JH, Plevy SE. J Biol Chem. 2001;276:18519–18528. doi: 10.1074/jbc.M100440200. [DOI] [PubMed] [Google Scholar]
- 8.Maruyama S, Sumita K, Shen H, Kanoh M, Xu X, Sato M, Matsumoto M, Shinomiya H, Asano Y. J Immunol. 2003;170:997–1001. doi: 10.4049/jimmunol.170.2.997. [DOI] [PubMed] [Google Scholar]
- 9.Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K. J Immunol. 2000;165:271–279. doi: 10.4049/jimmunol.165.1.271. [DOI] [PubMed] [Google Scholar]
- 10.Plevy SE, Gemberling JH, Hsu S, Dorner AJ, Smale ST. Mol Cell Biol. 1997;17:4572–4588. doi: 10.1128/mcb.17.8.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker C, Wirtz S, Ma X, Blessing M, Galle PR, Neurath MF. J Immunol. 2001;167:2608–2618. doi: 10.4049/jimmunol.167.5.2608. [DOI] [PubMed] [Google Scholar]
- 12.Myers RM, Tilly K, Maniatis T. Science. 1986;232:613–618. doi: 10.1126/science.3457470. [DOI] [PubMed] [Google Scholar]
- 13.Bieker JJ. DNA Cell Biol. 1996;15:347–352. doi: 10.1089/dna.1996.15.347. [DOI] [PubMed] [Google Scholar]
- 14.Crossley M, Tsang AP, Bieker JJ, Orkin SH. J Biol Chem. 1994;269:15440–15444. [PubMed] [Google Scholar]
- 15.Lee JS, Ngo H, Kim D, Chung JH. Proc Natl Acad Sci U S A. 2000;97:2468–2473. doi: 10.1073/pnas.040476297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JS, Lee CH, Chung JH. Proc Natl Acad Sci U S A. 1999;96:10051–10055. doi: 10.1073/pnas.96.18.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Vliet J, Turner J, Crossley M. Nucleic Acids Res. 2000;28:1955–1962. doi: 10.1093/nar/28.9.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner J, Crossley M. EMBO J. 1998;17:5129–5140. doi: 10.1093/emboj/17.17.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller IJ, Bieker JJ. Mol Cell Biol. 1993;13:2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Bieker JJ. Mol Cell Biol. 2001;21:3118–3125. doi: 10.1128/MCB.21.9.3118-3125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Kobori JA, Hood L. Mol Cell Biol. 1993;13:5691–5701. doi: 10.1128/mcb.13.9.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Black AR, Black JD, Azizkhan-Clifford J. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 23.Ulgiati D, Subrata LS, Abraham LJ. J Immunol. 2000;164:300–307. doi: 10.4049/jimmunol.164.1.300. [DOI] [PubMed] [Google Scholar]
- 24.Warke VG, Nambiar MP, Krishnan S, Tenbrock K, Geller DA, Koritschoner NP, Atkins JL, Farber DL, Tsokos GC. J Biol Chem. 2003;278:14812–14819. doi: 10.1074/jbc.M300787200. [DOI] [PubMed] [Google Scholar]
- 25.Ma X, Sun J, Papasavvas E, Riemann H, Robertson S, Marshall J, Bailer RT, Moore A, Donnelly RP, Trinchieri G, Montaner LJ. J Immunol. 2000;164:1722–1729. doi: 10.4049/jimmunol.164.4.1722. [DOI] [PubMed] [Google Scholar]
- 26.Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, Dzialo R, Trinchieri G. J Exp Med. 1996;183:147–157. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin SH. Mol Cell Biol. 1996;16:1695–1705. doi: 10.1128/mcb.16.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkins AC, Sharpe AH, Orkin SH. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 29.Southwood CM, Downs KM, Bieker JJ. Dev Dyn. 1996;206:248–259. doi: 10.1002/(SICI)1097-0177(199607)206:3<248::AID-AJA3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 30.Schoenhaut DS, Chua AO, Wolitzky AG, Quinn PM, Dwyer CM, McComas W, Familletti PC, Gately MK, Gubler U. J Immunol. 1992;148:3433–3440. [PubMed] [Google Scholar]
- 31.Bradley MN, Zhou L, Smale ST. Mol Cell Biol. 2003;23:4841–4858. doi: 10.1128/MCB.23.14.4841-4858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA. Cell. 1990;61:113–124. doi: 10.1016/0092-8674(90)90219-5. [DOI] [PubMed] [Google Scholar]
- 33.Celada A, Borras FE, Soler C, Lloberas J, Klemsz M, van Beveren C, McKercher S, Maki RA. J Exp Med. 1996;184:61–69. doi: 10.1084/jem.184.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott EW, Simon MC, Anastasi J, Singh H. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 35.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 36.Beutler B, Rietschel ET. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 37.Chen L, Fischle W, Verdin E, Greene WC. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 38.D'Ambrosio D, Cippitelli M, Cocciolo MG, Mazzeo D, Di Lucia P, Lang R, Sinigaglia F, Panina-Bordignon P. J Clin Investig. 1998;101:252–262. doi: 10.1172/JCI1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhat-Nakshatri P, Newton TR, Goulet R, Jr, Nakshatri H. Proc Natl Acad Sci U S A. 1998;95:6971–6976. doi: 10.1073/pnas.95.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, Fenton MJ. J Leukocyte Biol. 2001;69:1036–1044. [PubMed] [Google Scholar]
- 41.Goto M, Katayama KI, Shirakawa F, Tanaka I. Cytokine. 1999;11:16–28. doi: 10.1006/cyto.1998.0390. [DOI] [PubMed] [Google Scholar]
- 42.Paludan SR. J Virol. 2001;75:8008–8015. doi: 10.1128/JVI.75.17.8008-8015.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chu W, Gong X, Li Z, Takabayashi K, Ouyang H, Chen Y, Lois A, Chen DJ, Li GC, Karin M, Raz E. Cell. 2000;103:909–918. doi: 10.1016/s0092-8674(00)00194-x. [DOI] [PubMed] [Google Scholar]
- 44.Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K. J Biol Chem. 1992;267:22506–22511. [PubMed] [Google Scholar]
- 45.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z, Dziarski R, Wang Q, Swartz K, Sakamoto KM, Gupta D. J Immunol. 2001;167:6975–6982. doi: 10.4049/jimmunol.167.12.6975. [DOI] [PubMed] [Google Scholar]
- 47.Kelly RW, Carr GG, Riley SC. Biochem Biophys Res Commun. 1997;239:557–561. doi: 10.1006/bbrc.1997.7502. [DOI] [PubMed] [Google Scholar]
- 48.Grove M, Plumb M. Mol Cell Biol. 1993;13:5276–5289. doi: 10.1128/mcb.13.9.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altare F, Lammas D, Revy P, Jouanguy E, Doffinger R, Lamhamedi S, Drysdale P, Scheel-Toellner D, Girdlestone J, Darbyshire P, Wadhwa M, Dockrell H, Salmon M, Fischer A, Durandy A, Casanova JL, Kumararatne DS. J Clin Investig. 1998;102:2035–2040. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Doffinger R, Bernaudin F, Jeppsson O, Gollob JA, Meinl E, Segal AW, Fischer A, Kumararatne D, Casanova JL. Science. 1998;280:1432–1435. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- 51.Verhagen CE, de Boer T, Smits HH, Verreck FA, Wierenga EA, Kurimoto M, Lammas DA, Kumararatne DS, Sanal O, Kroon FP, van Dissel JT, Sinigaglia F, Ottenhoff TH. J Exp Med. 2000;192:517–528. doi: 10.1084/jem.192.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooper AM, Magram J, Ferrante J, Orme IM. J Exp Med. 1997;186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su Z, Stevenson MM. J Immunol. 2002;168:1348–1355. doi: 10.4049/jimmunol.168.3.1348. [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Bieker JJ. Proc Natl Acad Sci U S A. 1998;95:9855–9860. doi: 10.1073/pnas.95.17.9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kadam S, McAlpine GS, Phelan ML, Kingston RE, Jones KA, Emerson BM. Genes Dev. 2000;14:2441–2451. doi: 10.1101/gad.828000. [DOI] [PMC free article] [PubMed] [Google Scholar]