

Summary
An understanding of how animal size is controlled requires knowledge of how positive and negative growth regulatory signals are balanced and integrated within cells. Here we demonstrate that the activities of the conserved growth promoting transcription factor Myc and the tumor-suppressing Hippo pathway are co-dependent during growth of Drosophila imaginal discs. We find that Yorkie (Yki), the Drosophila homolog of the Hippo pathway transducer, Yap, regulates the transcription of Myc, and that Myc functions as a critical cellular growth effector of the pathway. We demonstrate that in turn, Myc regulates the expression of Yki as a function of its own cellular level, such that high levels of Myc repress Yki expression through both transcriptional and post-transcriptional mechanisms. We propose that the co-dependent regulatory relationship functionally coordinates the cellular activities of Yki and Myc and provides a mechanism of growth control that regulates organ size and has broad implications for cancer.
Introduction
Animal growth is controlled by regulatory pathways that promote tissue expansion and also by those that limit it. A major challenge to our understanding of size control is how the positive and negative forces are balanced to prevent aberrant growth and restrict organ and body size to a norm. Two key mediators of size control are the cellular growth promoter Myc and the Hippo (Hpo) tumor suppressor pathway. Their critical roles in regulating organ size are well conserved and both are frequently de-regulated in human cancer (de la Cova and Johnston, 2006; Pan, 2007; Zeng and Hong, 2008), but if and how these pathways intersect during growth regulation is unknown.
Myc is a functionally conserved transcriptional regulator that drives cellular growth by activating numerous target genes (de la Cova and Johnston, 2006; Vita and Henriksson, 2006). In mammals Myc functions as an oncogene in a vast array of tumors, and is also required for growth during normal development and in regeneration. Much of Myc’s power as a growth promoter stems from its regulation of genes that promote ribosome assembly (Grandori et al., 2005; Grewal et al., 2005). The Hpo pathway, in both flies and mammals, restricts growth by retaining the transcriptional co-activator, Yorkie (Yki)/Yap in the cytoplasm through a phosphorylation cascade regulated by the conserved kinases Hpo/Mst1 and Warts/Lats, in association with the scaffolding proteins Sav/WW45 and Mats (Pan, 2007). Reduced Hpo activity allows nuclear translocation of Yki/Yap and its association with DNA binding proteins, and transcriptional activation of target genes that regulate cell proliferation and tissue growth. Much of the Hpo pathway was first elucidated in Drosophila, where its targets include the anti-apoptotic factor dIAP1, the cell cycle regulator Cyclin E, and bantam, a Drosophila-specific microRNA with anti-apoptotic and growth-promoting functions (Huang et al., 2005; Nolo et al., 2006; Tapon et al., 2002; Thompson and Cohen, 2006; Wu et al., 2003). Additional targets include upstream regulators such as the FERM domain proteins Expanded (Ex)/FRMD6 and Merlin/NF2, and the Golgi kinase Four Jointed (FJ)/Fjx1 (Badouel et al., 2009b; Reddy and Irvine, 2008). A recent report indicates that the patterning system of the Drosophila wing imaginal disc modulates Hpo activity through regulation of the association of Fat with Ds (Rogulja et al., 2008).
The cell survival and cell division targets regulated by the Hpo pathway clearly contribute to its control of size, yet specific and conserved growth regulatory targets have remained elusive. The extensive functional conservation of components of the Hpo pathway suggests that a generally conserved growth factor is subject to its regulation. Here we provide evidence that Drosophila Myc is a transcriptional target of Yorkie and functions as a critical cellular growth effector of the pathway. Furthermore, we demonstrate that in a negative feedback loop, dMyc modulates Yki expression and activity as a function of its own levels, suggesting that dMyc can adjust overall growth rates by affecting Hpo pathway activity. Based on our results, we propose that a negative feedback loop between Hpo pathway signaling and dMyc provides a stabilizing, homeostatic mechanism that contributes to regulation of organ size.
Results
dMyc is required for Yki to promote growth
Growth control in Drosophila imaginal discs, the epithelial primordia that give rise to the body of the adult fly, requires the activity of the Hpo pathway and of dMyc, encoded by the diminutive (dm) gene. Disc cells homozygous for the dm4 null mutation proliferate very poorly and form small, infrequently recovered clones (Fig. 1B, E), whereas dMyc over-expression increases cellular growth and leads to an increase in tissue mass due to larger cell size (Johnston et al., 1999; Wu and Johnston, 2010). Cell clones that over-express Yki or carry mutations that inactivate its inhibitor, Wts, grow large via increased cell proliferation and can occupy large territories of wing and eye discs (Fig. 1C, H, Fig. 2B–C) (Wu et al., 2003). We tested the possibility that the increase in Yki-driven growth required the function of dMyc. We used the dm4 null allele in MARCM (mosaic analysis with a repressible marker) experiments, which couples Gal4-UAS control of gene expression to the generation of cell clones by mitotic recombination (Lee and Luo, 1999), to assess whether Yki expression could promote growth of clones in which dMyc function was specifically deleted. Measurement of clone size after defined periods of time indicated that while UAS-yki stimulated the growth of wildtype cells (Fig. 1C, E), it was completely unable to promote growth in the absence of dMyc. dm4 mutant cell clones expressing UAS-yki were no larger than dm4 clones expressing UAS-GFP, even when a dominant negative form of the Dronc caspase (UAS-droncCD) (Meier et al., 2000) was expressed with Yki to prevent their death (Fig. 1D–E). Thus, dMyc function is necessary for Yki expression to stimulate tissue growth, suggesting that dMyc is an important effector of growth downstream of the Hpo pathway.
Figure 1. Yki requires dMyc for growth and local differences in Hpo activity induce cell competition.
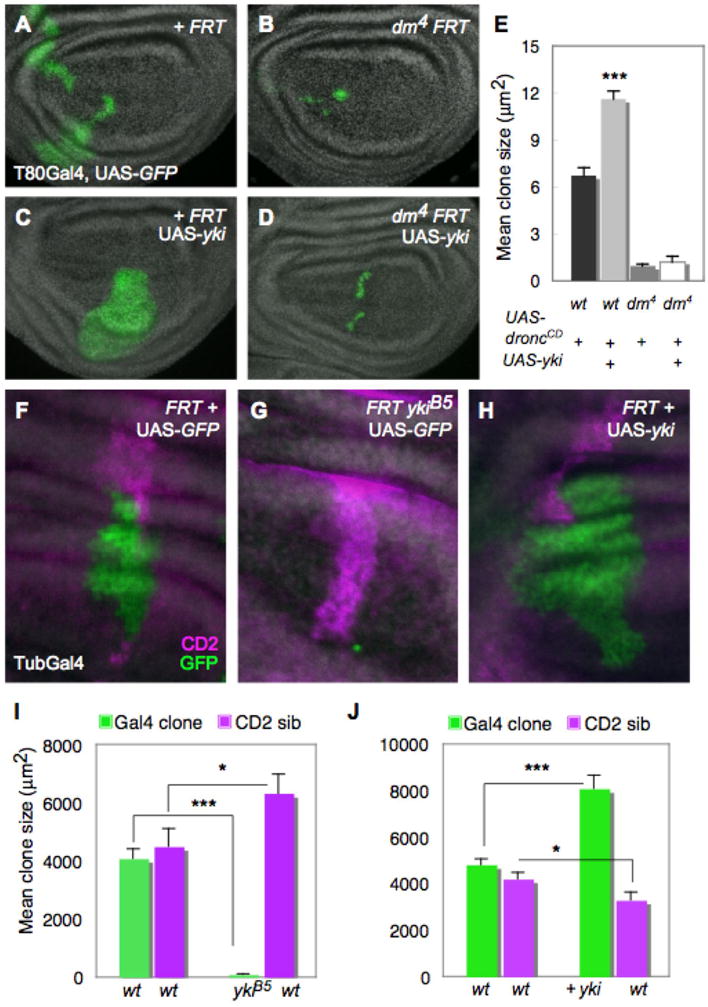
(A–E) Loss of dMyc prevents Yki-induced growth. MARCM clones generated in wing discs, marked by T80Gal4>UAS-GFP expression (green). (A) control clones; (B) dm4 mutant clones; (C) UAS-yki-expressing clones; (D) dm4 mutant clones that express UAS-yki. (E) Quantification of mean clone size. UAS-yki-expressing clones (grey bar) induce significant growth relative to control clones (black bar), but its expression fails to induce growth in dm4 mutant cells (dark grey bar) even when death is prevented by UAS-droncCD expression (white bar). Error bars in this and all subsequent figures are standard error of the mean (SEM). (F–J) Measurement of cell competition using MARCM in wing discs to generate a Gal4/UAS-GFP-expressing clone (green) and its sibling clone (marked with 2 copies of CD2, magenta). (F) Control TubGal4>UAS-GFP clone and wildtype (wt) CD2 sibling showing that wt sibling clones grow equally well; (G) ykiB5 mutant TubGal4>UAS-GFP clone and wt CD2 sibling; (H) UAS-yki-expressing TubGal4>UAS-GFP clone and wt CD2 sibling. (I, J) Quantification of cell competition. (I) ykiB5 mutant TubGal4>UAS-GFP clones grow less than control wt TubGal4>UAS-GFP clones (green bars) and are competed against by their wt and heterozygous neighbors, while their wt CD2 sibling clones grow significantly more than control CD2 sibling clones (magenta bars), an indication of “winner” status. (J) UAS-yki-expressing TubGal4>UAS-GFP clones grow more than control TubGal4>UAS-GFP clones (green bars), while their wt sibling CD2 clones are smaller than control wt sibling CD2 clones (magenta bars), indicative of “loser” status. See also Figure S1. P-values, * p < 0.05; *** p < 0.001. In this and all subsequent figures, discs are oriented with dorsal up and posterior to the right.
Figure 2. Hpo signaling activity regulates dMyc expression.
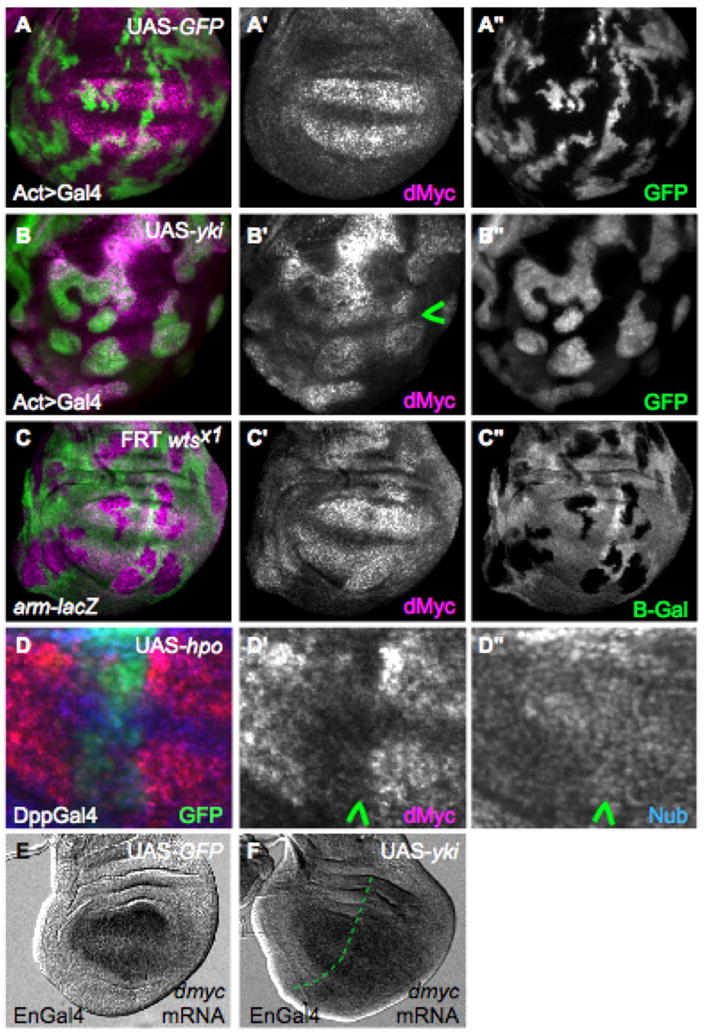
(A, A′) Control wing disc with ActGal4>GFP-expressing clones (green) showing wildtype dMyc expression (magenta) (A′). (B, B′) Clonal expression of UAS-yki (green) up-regulates dMyc expression (magenta) in endogenous regions and induces it ectopically. Note that clones at the dorso-ventral boundary (arrowhead in B′) do not express dMyc due to dominant patterning cues (Johnston et al 1999). (C, C′) Clones of wtsx1 mutant cells (absence of green) induce expression of dMyc. (D–D″) Wing disc expressing UAS-hpo and UAS-p35 under DppGal4 control (green). dMyc is specifically down-regulated by UAS-hpo expression (arrowhead, D′), whereas Nub expression (blue), which is expressed throughout the wing pouch, is unaffected (arrowhead, D″). See also Figure S2. (E–F) dmyc mRNA in control wing disc (E) or wing disc that expresses UAS-yki in posterior cells with EnGal4 (right side of dotted line in F). dmyc mRNA is increased in posterior cells in response to UAS-yki expression (F).
In addition to its cell autonomous growth-regulatory functions dMyc influences tissue size non-cell autonomously by driving cell competition, a homeostatic process that arises from local cellular differences in expression of dMyc or genes encoding ribosomal proteins (Rp) (Johnston, 2009). During competition in Drosophila, “loser” cells are instructed by “winner” cells to undergo apoptosis and are eliminated from the epithelium. Interestingly, mutations that inactivate the Hpo pathway can suppress the loss of rpl36/+ loser cells in competitive mosaics (Tyler et al., 2007). In competition induced by Rp differences, winner cells replace dying loser cells by proliferating at a normal rate during an extended growth period (Martin et al., 2009). In contrast, winner cells in dMyc-induced competition are stimulated to proliferate faster to replace loss of the loser cells (Senoo-Matsuda and Johnston, 2007; Wu and Johnston, 2010). To test whether inactivation of the Hpo pathway led to cell competition, we generated marked wildtype and ykiB5 null mutant sibling cell clones by MARCM and measured their growth (de la Cova et al., 2004). During a given growth period ykiB5 mutant clones grew extremely poorly, but their wildtype sibling clones grew significantly more than control sibling clones (Fig. 1F–G, I). This faster rate of growth is a hallmark of winner behavior in dMyc-induced competition (Johnston, 2009). In addition, wildtype cell clones grew poorly when next to cell clones expressing UAS-yki (Fig. 1H, J). The loser behavior of the wildtype cells was most likely due to a significant increase in cell death (Supp. Fig. 1). The apoptosis of losers and enhanced growth of winners when cells differ in Hpo activity specifically phenocopies competition induced by differences in dMyc expression (de la Cova et al., 2004; Senoo-Matsuda and Johnston, 2007; Wu and Johnston, 2010). In light of the requirement for dMyc in clonal growth promoted by Yki expression, these data suggest that local perturbations of the Hpo pathway can create differences in dMyc expression between neighboring cells, and provoke cell competition. This possibility led us to examine whether Hpo activity regulates expression of dMyc.
Hpo signaling activity regulates dMyc expression
To determine whether changes in Hpo signaling activity affect dmyc expression, we repressed Hpo pathway activity by generating clones of wts mutant cells or bypassed it with flp-out Gal4 clones of cells expressing UAS-yki. In the majority of these clones dMyc protein was substantially upregulated in multiple tissues, including the wing, leg, antenna and eye imaginal discs. dMyc was also induced in cells that normally express little or no dMyc (Fig. 2A–C and Supp. Fig. 2A–B). Furthermore, dmyc mRNA was induced in cells over-expressing UAS-yki (Fig. 2E, F), suggesting that loss of Hpo activity leads to transcriptional activation of the dm gene. Conversely, expression of UAS-hpo, along with the caspase inhibitor P35 to prevent cell death, sharply decreased endogenous dmyc expression (Fig. 2D–D″). Thus low Hpo activity is sufficient to activate dmyc expression and high Hpo activity to repress it. These observations confirm that endogenous expression of dmyc is controlled by the activity of the Hpo signaling pathway.
Yki and Scalloped physically associate with the dm locus
Yki is a potent transcriptional co-activator but requires a DNA binding partner to activate target gene transcription. Two DNA binding factors, the TEAD family protein, Scalloped (Sd) and the Hox co-factor, Homothorax (Hth) are known to form complexes with Yki to promote target gene activation in Drosophila (Peng et al., 2009; Wu et al., 2008). Sd is required for the growth and survival of cells in the central portion of the disc (the wing pouch) where dMyc is most highly expressed, and clones of sd null mutant cells are rarely recovered there (data not shown)(Liu et al., 2000). We therefore examined the role of Sd in regulation of dMyc using a short-hairpin (sh) RNA to reduce sd expression (Fig. 3A–C). UAS-dicer2 was included to potentiate shRNA silencing. When UAS-sd shRNA was expressed medially in the disc with DppGal4, dMyc expression was substantially reduced (Fig. 3A, A′). This effect was specific to dMyc because expression of Nubbin, another nuclear factor expressed in the wing pouch, was not substantially altered (Fig. 3A″). In addition, expression of UAS-sd shRNA and UAS-yki together in flp-out clones prevented the ability of UAS-yki to increase dMyc expression in its endogenous expression domain, and also prevented its ectopic induction (Fig. 3B–C). In contrast, expression of a UAS-shRNA against hth did not alter dMyc expression in any region of the disc (data not shown). Thus both Yki and Sd are required for control of dmyc expression in the wing disc, raising the possibility that the two factors cooperate in its transcriptional activation.
Figure 3. dMyc is a transcriptional target of the Hpo pathway.
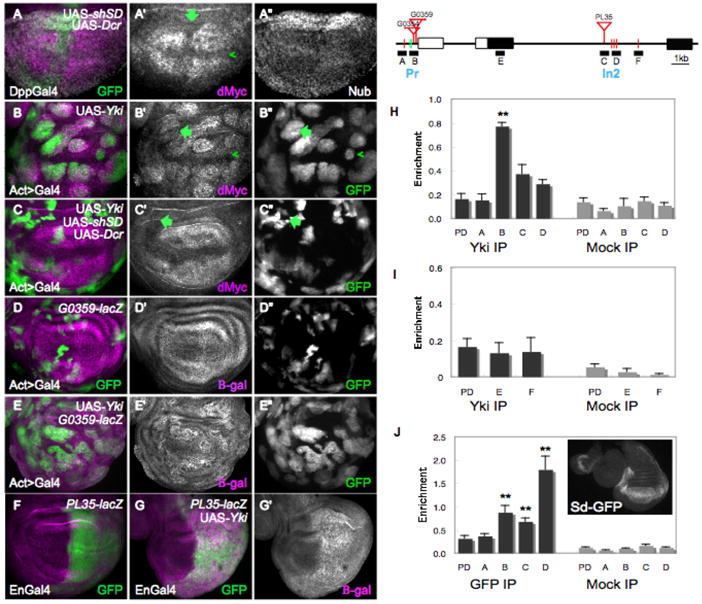
(A–A″) Wing disc expressing UAS-GFP, UAS-dcr-2 and a UAS- short-hairpin (sh) sd under DppGal4 control (green). Reduction of sd expression reduces dMyc expression in the wing pouch (arrow, A′), but Nub expression is generally unaffected (A″). (B–B″) Clones expressing UAS-GFP (B″) and UAS-yki upregulate dMyc (magenta) in the wing pouch and ectopically activate dMyc in proximal cells (arrow in B′, B″). Arrowhead points to a clone at the D–V boundary that does not alter dMyc expression. (C–C″) GFP-marked clones expressing UAS-yki, UAS-shsd, and UAS-dcr-2. Reduced Sd expression prevents dMyc upregulation (magenta) in its endogenous domain and ectopic activation in proximal cells (arrow in C′, C″). (D–E) Beta-galactosidase (β-gal) expression (magenta) from a P-element insertion (G0359) into the proximal promoter region of the dm locus. See also Figure S3. (D–D″) Control wing disc. (E–E″) Wing disc expressing UAS-yki in clones. G0359-lacZ responds strongly to UAS-yki expression. GFP marked clones are shown in E″. (F) β-gal expression from a P-element insertion (PL35) into intron 2 of the dm locus. (G, G′) Expression of Yki under Engal4 control leads to strong upregulation of β-gal (magenta). (H–J) Schematic representation of the dm locus. Black rectangles are coding regions, white rectangles are non-coding regions; lines denote introns. Red bars indicate consensus Sd binding sites and green lines are degenerate Sd binding sites in the locus; red triangles represent Yki-responsive P-element insertions. Blocks labeled A–F represent regions amplified in the chIP experiments; Pr, Promoter; In2, Intron2. (H) Yki chIP on the dm locus. Anti-Yki (Yki IP) or IgG (mock IP) antibodies were used to precipitate chromatin from wt discs; quantitative RT-PCR was done on regions A–D and on a negative control locus, pyruvate dehydrogenase (PD). Graph shows the enrichment of signal over input for A, B, C, D and PD. Amplicons B, C and D were all consistently enriched over PD in each of 4 independent experiments, although statistical significance was obtained only for amplicon B, due to noise between experiments. (I) ChIP data covering amplicon E, in the first coding exon (a negative control) and amplicon F, covering a single Sd site. Neither amplicon was enriched relative to PD in the experiments. (J) Sd-GFP chIP on the dm locus. Amplicons B, C, and D showed significant enrichment after precipitation of wt disc chromatin with antibodies against GFP. The strongest enrichment occurred of amplicon D, where a cluster of three highly conserved Sd sites is located (not shown; www.modencode.org). Inset shows expression from Sd-GFP line. P-values, ** p<0.01.
To determine whether the dm locus is directly controlled by the Hpo pathway we first searched for Sd binding sites in the dm gene and found potential matches at various positions along the locus (Fig. 3 and data not shown). Three sites mapped to the proximal promoter region of dm, and sites were identified within intron 2 (Fig. 3). The promoter-proximal sites and a cluster of three sites within intron 2 mapped near three independent P-element insertions that report lacZ expression from the dm locus (Bourbon et al., 2002; Peter et al., 2002). Each of these P-lacZ insertions responded robustly to UAS-yki by increasing lacZ expression, supporting the idea that regulatory regions in their vicinity are under transcriptional control by Hpo signaling (Fig. 3D–G and Supp. Fig. 3A–B).
Next, we carried out chromatin immunoprecipitation (chIP) experiments on the dm locus with chromatin isolated from wildtype imaginal discs. We designed primers to amplify regions of the dm locus that covered all of the potential Sd binding sites as well as the insertion regions of each P-element reporter. DNA amplified from chromatin precipitated with antibodies against Yki was specifically and significantly enriched in the region that harbors the two promoter-proximal P-element insertions (Fig. 3H, amplicon B). This region includes two putative Sd sites that are fairly divergent from the consensus CATTCC sequence, while an upstream perfect match to the consensus Sd site was not enriched (Fig. 3H, amplicon A). In addition, two amplicons within intron 2 showed enrichment above background in four independent experiments, although the combined average was not significant (p=0.07; Fig. 3H, amplicons C, D). These amplicons include the intronic cluster of three Sd consensus sites. An amplicon covering an additional single consensus Sd site just downstream was never enriched in the experiments, nor was an internal negative control region in the first coding exon (Fig. 3I). None of the regions was enriched when precipitated with control IgG antiserum, and a region in the pyruvate dehydrogenase (PD) gene, which served as a negative control, was not enriched in the Yki-precipitated chromatin, confirming the specificity of the reactions (Fig. 3H–I). Our results demonstrate that specific binding of endogenous Yki protein occurs at the dm promoter (Pr) region and in intron 2 (In2) in growing wildtype discs (Fig. 3H).
We then examined whether Sd bound to these regions of the locus. We used a protein trap line in which an in-frame insertion of GFP is spliced into Sd coding exons, producing a GFP-Sd fusion protein as the only source of Sd (Buszczak et al., 2007). This protein trap line is homozygous viable and allowed us to use anti-GFP antibodies to pull down Sd in chIP experiments, since antibodies against Sd were unavailable. We found that the enriched sequences of the dm locus from disc chromatin immunoprecipitated by anti-GFP were identical to those that bind Yki (Fig. 3J), suggesting that these two factors might co-associate on the DNA in vivo. Interestingly, the In2 region was most highly enriched in the experiments using anti-GFP antibodies, correlating well with fact that the cluster of Sd sites there is conserved among several Drosophila species (Fig. 3J). Our results thus demonstrate that endogenous Yki and Sd protein bind to the same DNA sequences at the dm locus. Along with the Yki-induced transcriptional activation of Plac-Z reporter insertions in these regions, the specific binding of Yki and Sd to these sites suggests a mechanism by which dMyc expression could be physiologically regulated by the Hpo pathway.
dMyc and Yki cooperate in growth regulation
Yki is required for cell proliferation (Fig. 1G, Fig. 4D) in part because its loss in cells prevents expression of diap1 and bantam, targets of Yki that promote cell survival (Nolo et al., 2006; Thompson and Cohen, 2006; Wu et al., 2003). Expression of UAS-diap1 or UAS-bantam in ykiB5 mutant clones can increase clone size to a limited extent (Fig. 4G–J) (Nolo et al., 2006; Thompson and Cohen, 2006). Since our results indicated that dMyc is a transcriptional target of Yki and is required for UAS-yki to promote growth, we asked whether dMyc expression would promote the growth of yki mutant cell clones. We expressed UAS-dmyc in ykiB5 mutant cell clones by itself, or with UAS-bantam or an activated form of diap1 (UAS-diap1*) to increase cell survival, and analyzed clone size after a defined growth period. Expression of UAS-dmyc either alone (Fig. 4F), with UAS-diap1* (Fig. 4H), or with UAS-bantam (Fig. 4I), was unable to rescue the growth defect of yki mutant clones. In fact, ykiB5 clones that co-expressed UAS-dmyc and UAS-diap1* or UAS-bantam grew to a similar size as those expressing either survival factor alone (Fig. 4J). The size of ykiB5 clones was also not rescued when UAS-dmyc was expressed in anterior cells with CiGal4, indicating that the lack of clonal growth was not an effect of Gal80 perdurance in the MARCM experiments (data not shown). These results suggested the possibility that dMyc’s ability to drive growth in yki mutant cells could be compromised.
Figure 4. Co-dependence of dMyc and Yki in growth regulation.
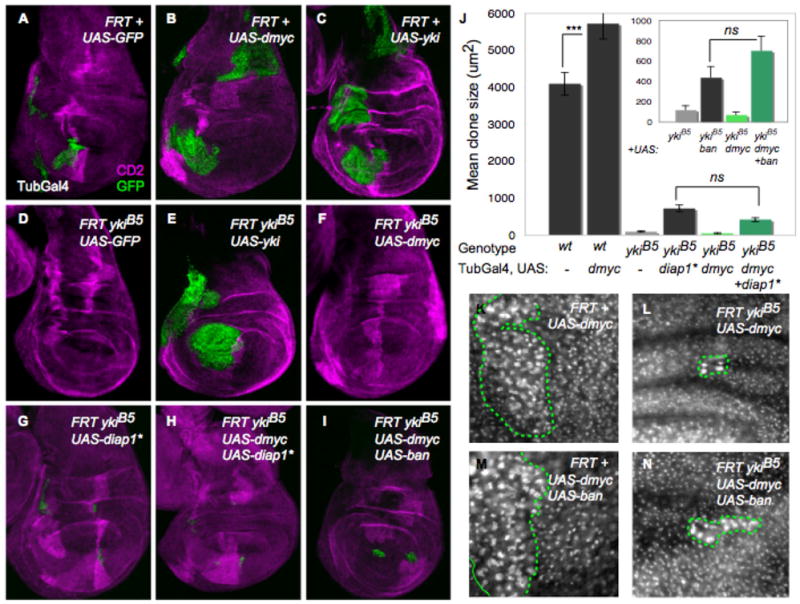
(A–I) MARCM TubGal4>UAS-GFP clones (green) that express UAS-dmyc (alone or together with other transgenes) in yki mutant clones; their wt CD2 sibling clones are magenta. (A) Discs with control clones; (B) clones expressing UAS-dmyc; (C) clones expressing UAS-yki; (D) ykiB5 mutant clones; (E) ykiB5 mutant clones rescued by yki expression; (F) ykiB5 mutant clones that express UAS-dmyc; (G) ykiB5 mutant clones that express UAS-diap1*; (H) ykiB5 mutant clones that co-express UAS-dmyc and UAS-diap1*; (I) ykiB5 mutant clones that co-express UAS-dmyc and UAS-bantam. (J) Clone size quantification. UAS-diap1* or UAS-bantam expression slightly rescues growth of ykiB5 clones. UAS-dmyc significantly increases size of wt clones but fails to increase ykiB5 mutant clone size, even with co-expression of UAS-diap1* or UAS-bantam. (K–N) Fibrillarin staining in wing imaginal discs with clones expressing dmyc (K); with ykiB5 mutant clones expressing dmyc (L); with clones expressing dmyc + bantam (M); and with ykiB5 mutant clones expressing UAS-dmyc + UAS-bantam. UAS-dmyc upregulates Fibrillarin and increases nucleolar size in wt cells (K) and in ykiB5 mutant cells (L, N), indicating that some growth-promoting targets of dMyc are independent of Yki. Green lines in K–N indicate clone boundaries. See also Figure S4. P-value, *** p<0.001.
Control of ribosome biogenesis is central to Myc’s growth-promoting function (Grandori et al., 2005; Grewal et al., 2005), and misexpression of dMyc activates expression of the nucleolar protein Fibrillarin and increases nucleolar and cell size (Figure S4 B) (Grandori et al., 2005; Grewal et al., 2005). These features are unique to dMyc, as expression of the Yki target bantam did not lead to these changes (Figure S4 C). To determine whether UAS-dmyc expression still enhanced ribosome biogenesis and cellular growth in cells lacking yki, we generated ykiB5 clones and co-expressed bantam to promote survival of the cells. Despite the small size of the clones (Fig. 4J–N), cell size, nucleolar size, and Fibrillarin expression were increased when UAS-dmyc, with or without UAS-bantam, was expressed in ykiB5 mutant cells (Fig. 4L, N and data not shown). This clearly shows that dMyc’s ability to promote ribosome biogenesis is unimpaired in yki mutant cells, but that cells lacking yki fail to respond growth-promoting inputs from dMyc in a manner that specifically allows tissue expansion. Collectively these results suggest that dMyc and Yki induce non-redundant targets, and that for appropriate tissue growth the coordinated function of both factors is required.
Yki expression is regulated by dMyc through multiple mechanisms
To investigate how the activities of Yki and dMyc are integrated during tissue growth we examined whether Hpo activity itself was subject to control by dMyc. We tested this idea by examining the expression, subcellular localization and activity of Yki in cells in which dmyc was either reduced or augmented. dMyc expression was eliminated by generating dm4 null mutant cell clones, or reduced by expression of UAS-dmyc shRNA in flp-out Gal4 clones. Strikingly, complete or partial loss of dmyc visibly increased the overall level of Yki protein within clones located in the wing pouch (Fig. 5B, D). Interestingly, the increase in Yki levels did not alter its subcellular localization (Fig. 6C″, arrow). Conversely, Yki expression was markedly reduced in cells of flp-out Gal4 clones in which UAS-dmyc was expressed (Fig. 5G). Yki expression was unchanged in control clones (Fig. 5A, C). Control of Yki expression in cells with high or low dMyc was cell autonomous, because it also occurred when dMyc expression was manipulated in whole compartments (Fig. 5E–F), The inhibitory effect of dMyc on Yki expression was specific, as expression of the well characterized growth regulators Cyclin D + Cdk4, its dedicated cyclin dependent kinase, or the catalytic subunit of the PI3K, Dp110, did not alter Yki expression (Figure S5 A–C) (Datar et al., 2000; de la Cova et al., 2004; Leevers et al., 1996). The increase in Yki upon loss of dMyc was restricted to the wing pouch, where dMyc is normally present at high levels (Fig. 5F, arrows; Fig. 2A′). Since Yki expression in wildtype discs is fairly uniform (Fig. 5C′), the specificity of this increase suggests that the endogenous pattern of dMyc expression has a role in keeping Yki expression uniform across the disc.
Figure 5. dMyc regulates Yki expression and activity.
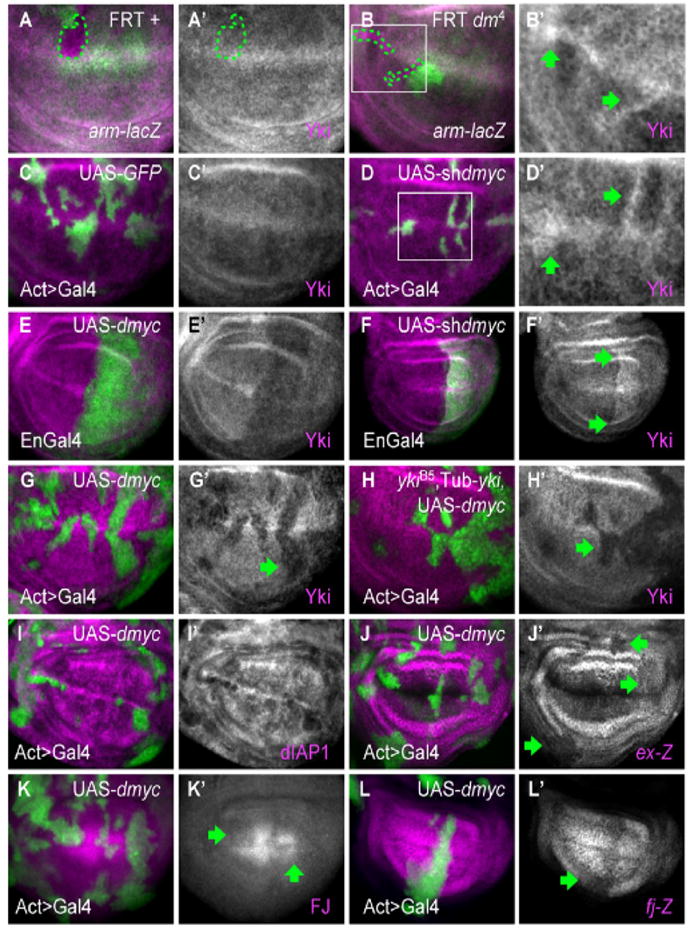
Panels A–H show Yki immunostaining (magenta) in wing discs. (A, A′) Control wing disc clones; (B, B′) and dm4 mutant clones. Clones are marked by the absence of green (arm-lacZ, dotted line indicates clone boundaries). B′, high magnification of area boxed in B. dm4 clones have elevated Yki expression (arrows). (C–D) Control ActGal4>UAS-GFP clones (green) (C, C′) or clones expressing UAS-shdmyc (D, D′). D′ is a higher magnification of area boxed in D. Moderate reduction of dMyc is sufficient to increase Yki protein (arrows). (E–E′) Wing discs expressing UAS-dmyc (E, E′) or UAS-shdmyc (F, F′) in posterior cells (green) under EnGal4 control. Yki expression is decreased in high dMyc cells throughout the disc, and increased in low dMyc cells within the wing pouch (upper and lower boundaries are marked by arrows in F′). See also Figure S5. (G, G′) Clones expressing dmyc, marked with UAS-GFP expression (green). Yki is decreased in clones expressing dmyc (arrow). (H–H′) Clones expressing dmyc in a ykiB5 animal rescued with a Tub-yki transgene. All Yki expression is from the transgene. UAS-dmyc represses Tub-yki in the clones (arrows), indicating that dMyc also regulates Yki expression post-transcriptionally. dmyc expression also reduces yki mRNA 30% below wildtype levels (see Figure S3). (I–L) Expression of Yki targets is diminished by UAS-dmyc expression. dIAP1 protein (I), expanded-lacZ (J, J′), Four Jointed (K, K′), and fj-lacZ (L, L′) are all downregulated (arrows) in clones expressing UAS-dmyc.
Figure 6. Wildtype levels of dMyc regulate Yki.
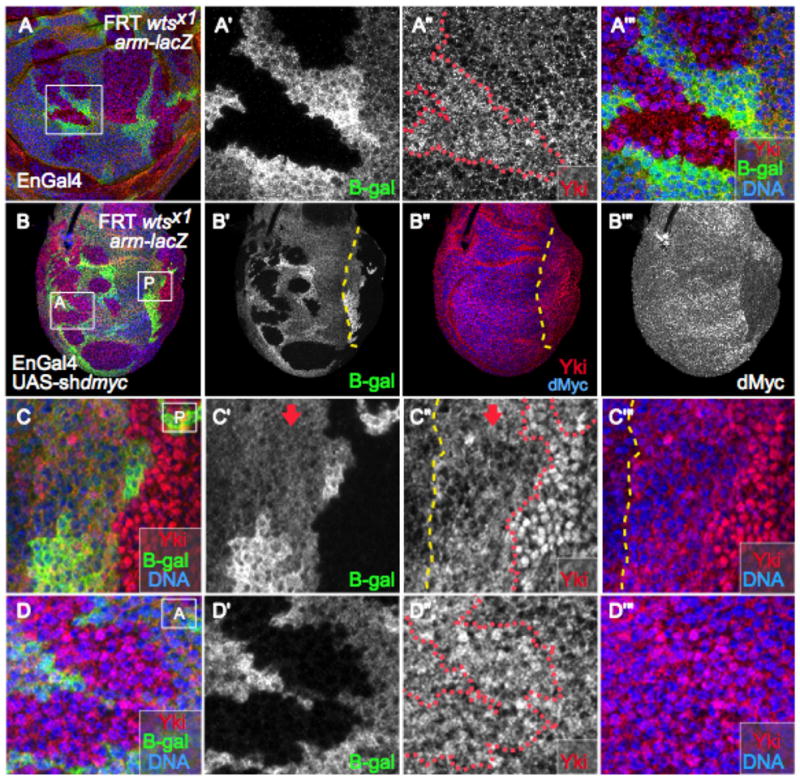
(A) Wing disc with wtsx1mutant clones immunostained for Yki (red). (A′) Close-up view of the region boxed in (A). wtsx1 mutant cells are marked by absence of β-gal. (A″) Yki expression. In the wts mutant clone Yki is somewhat diffuse, with both nuclear and cytoplasmic localization. (A‴) Close-up of merged channels of box in (A). (B) Yki staining (red) in wing disc with wts mutant clones and expressing UAS-shdmyc in posterior cells under EnGal4 control. wts mutant cells are marked by lack of β-gal (B′). Posterior cells are located to the right of the dashed yellow line (B′, B″) and show reduced levels of dMyc (B‴). Box A is located in the anterior compartment and box P in the posterior. (C–C‴) Close up view of the region inside box P. The red dotted line marks the wtsx1clone boundary, mutant cells lack β-gal. Cells to the right of the yellow dashed line express UAS-shdmyc; the arrow points to non-mutant tissue (β-gal-positive). Note the substantial accumulation of Yki in the nucleus of wtsx1 cells that have reduced dMyc levels due to expression of UAS-shdmyc. (D–D″) Close up view of the region inside box A. wtsx1 mutant clone (red line in D) located in the anterior compartment is marked by the absence of β-gal (D′). As in A″, Yki staining is more diffuse, with some nuclear localization (D″), but substantially less than in wtsx1 clones located in the posterior compartment, which express lower levels of dMyc (compare to C″).
Quantitative PCR experiments showed that dMyc-expressing cells had approximately 30% less yki mRNA than controls, suggesting that the reduction in Yki expression by high dMyc is partially under transcriptional control (Supp. Fig. 3C). To test for post-transcriptional regulation of Yki by dMyc, we used the ykiB5 null allele to eliminate endogenous yki expression but provided it through a transgene driven by the tubulin promoter (Tub-yki), which rescues the homozygous mutants to viability (Huang et al., 2005). In ykiB5animals carrying Tub-yki, all Yki protein is expressed from the rescuing transgene and is thus not subject to transcriptional control by dMyc. Under these conditions, Yki protein was still strongly decreased in cells with high dMyc (Fig. 5H, H′), indicating that much of the regulation of Yki by dMyc occurs via a post-transcriptional mechanism. These results demonstrate that dMyc’s regulation of Yki occurs though both transcriptional and post-transcriptional mechanisms, and suggest that the relative level of dMyc in a cell regulates the cellular quantity of Yki.
Negative feedback from dMyc controls Yki activity
Our experiments suggest that a circuit of negative feedback exists between dMyc and Yki, such that expression of dMyc is dependent upon Yki activity. In turn, cellular Yki levels are influenced by the quantity of dMyc within a cell. Yki activity requires nuclear localization (Huang et al., 2005), but should also be affected by its overall level in the cell. If so, changes in Yki expression should have functional consequences by influencing expression of its target genes. This view is supported by the observation that the Sd-dependent transcriptional response in the wing disc is sensitive to the dose of Yki (Goulev et al., 2008). To test this idea we expressed UAS-dmyc in flp-out Gal4 clones to reduce Yki levels and examined the expression of Yki target genes in those cells. As predicted, expression of three Yki targets - diap1, expanded (ex), and four-jointed (fj) - was significantly decreased in UAS-dmyc-expressing cell clones (Fig. 5I–L). The activity of transcriptional reporters of each gene also dropped sharply in response to UAS-dmyc expression, demonstrating that transcription of each target was reduced (Fig. 5J, L and data not shown). High levels of dMyc that reduce Yki expression therefore also diminish the transcriptional activity of Yki on its target genes.
Since high dMyc reduces Yki target gene expression, it might be expected that reducing dMyc levels would upregulate Yki targets. However, this was not the case: cells with reduced dMyc did not show obvious changes in Yki target gene expression (data not shown). Since previous work suggested that a threshold level of Yki is necessary to overcome regulation of its nuclear localization by Wts (Dong et al., 2007), the increased Yki in cells with low dMyc levels might not have reached this threshold. Consistent with this idea, we were unable to clearly detect nuclear Yki in the cells with reduced dMyc. This led us to consider the possibility that, in wildtype cells, endogenous Yki is constrained by limits on its cellular accumulation (e.g., by dMyc) as well as by its subcellular localization (e.g., by Wts). This type of mechanism could help to explain why in wtsx1 mutant cells, the distribution of Yki is both nuclear (transcriptionally active) and cytosolic (transcriptionally inactive) (Dong et al., 2007). Based on this idea, we hypothesized that nuclear, active Yki upregulates dMyc, which then decreases Yki levels, thereby reducing the amount of Yki available for nuclear localization. A prediction of this hypothesis is that if the negative input on Yki from endogenous dMyc was relaxed, more Yki should enter the nucleus in wts mutant clones. To test this, we decreased dMyc levels in posterior cells by expressing UAS-shdmyc with EnGal4, while at the same time relieving a cytosolic constraint on Yki by making clones of wtsx1 mutant cells. wtsx1 mutant clones were generated throughout the disc, allowing the anterior clones to provide an internal control for the behavior of Yki in wtsx1 cells with wildtype dMyc levels. The anterior wtsx1 mutant cells possessed both cytosolic and nuclear Yki, and the clones contained an average of only 8.5% of nuclei with complete or near-complete nuclear Yki staining (±6.3, n=5; Fig. 6B, D). Posterior cells with wildtype Wts had increased Yki expression due to the reduced dMyc expression, but it was primarily cytosolic (Fig. 6C–C‴, arrow). In striking contrast, 47.8% (±6.5, n=5) of posterior, UAS-shdmyc-expressing cells that were also wtsx1 mutant had nuclear Yki staining that completely covered the nucleus (Fig. 6B–C). Together, these results provide compelling evidence that, at wildtype levels, dMyc functions to limit Yki expression and thereby restrains its subcellular location. These data are consistent with the idea that the cellular level of dMyc contributes to regulation of Yki activity.
A model for stabilization of tissue growth through feedback between dMyc and Yki
Based on our results, we propose the following model of how the growth-promoting properties of dMyc and Yki are integrated to stabilize growth (Fig. 7D). Under normal growth conditions, Hpo activity is relatively low, allowing a pool of nuclear, active Yki to promote expression of target genes that regulate cell survival and growth, including dMyc. The accumulation of dMyc in the cell represses Yki expression, thereby setting a limit on how much Yki is able to translocate into the nucleus and activate target genes. This classic negative feedback relationship would calibrate the expression of each factor and hence their respective growth promoting activities, and at the same time integrate the output of the two pathways. Tissue growth would be promoted while at the same time kept in check, thus the mechanism is potentially homeostatic (Fig. 7D).
Figure 7. A model for a homeostatic feedback mechanism that prevents runaway growth.
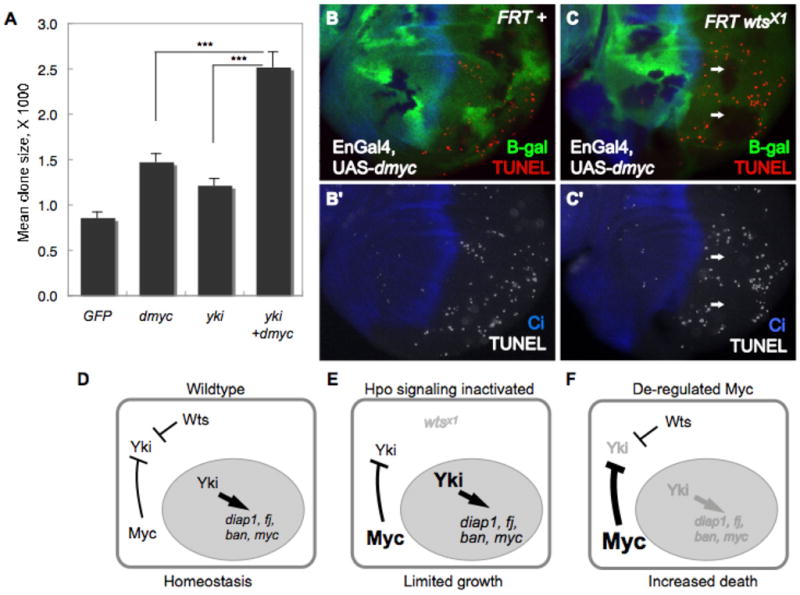
(A) Size of Act>Gal4 cell clones expressing UAS-GFP, UAS-dmyc, UAS-yki or co-expressing UAS-yki and UAS-dmyc. Simultaneous expression of Yki and dMyc drives signficantly more growth than expression of either factor alone. *** p < 0.001. (B–C) Cell death, assessed by TUNEL staining (red), in wing discs with wildtype (B) or wtsx1 mutant clones (C) where UAS-dmyc was expressed in the posterior compartment. wtsx1 cells are marked by absence of β-gal (green) and the posterior compartment is marked by absence of Ci, a marker of anterior cells (blue). wtsx1 mutant tissue is protected from cell death induced by de-regulated dMyc (arrows). (D–F) A model of co-dependent regulation between Yki and dMyc. In wt cells (D), nuclear (active) Yki activates expression of dmyc and other Hpo target genes (diap1, bantam, fj). Negative feedback between dMyc activity and Yki expression keeps the activity of each in balance. (E) In wts mutant cells, Hpo signaling is inactivated; more active Yki increases dMyc expression. High dMyc expression and activity will decrease Yki expression, which in turn will curb dMyc expression, resetting a balance and limiting over-growth. (F) Cells expressing deregulated levels of dMyc will decrease Yki expression, resulting in loss of Yki pro-survival targets and cell death. In the model, deregulation of both dMyc and Yki expression will abolish the negative feedback loop and promote overgrowth; in mammals, this could lead to cancer.
Since inactivating the Hpo pathway or over expression of Yki leads to growth that is greater than normal (e.g., Fig. 2A–C), our model predicts that this growth would still be limited by negative feedback from endogenous dMyc (Fig. 7E). However, eliminating the feedback loop between dMyc and Yki should prevent integration of the two pathways’ outputs and allow each factor to promote growth independently, thereby greatly enhancing the extent of growth. We tested this idea by comparing the extent of growth induced by heterologous expression of dMyc, Yki, or both, under Gal4 control. These conditions should override both the constraints from the Hpo pathway and some of those imposed by dMyc. Even though dMyc’s post-transcriptional control of Yki would not be prevented, this should be offset by the continuous expression of each factor under Gal4 control. The results of this experiment show that during the same growth period, the growth of clones of cells expressing both UAS-dmyc and UAS-yki was enhanced, and grew significantly larger than clones expressing either factor alone (Fig. 7A).
A second prediction arising from our model follows from the observation that high dMyc cells have reduced expression of Yki target genes. Thus, lowered expression of Yki’s effector of cell survival, dIAP1, should result in increased apoptosis of cells expressing high levels of dMyc. We tested this by expressing dMyc in posterior wing disc cells under EnGal4 control, and re-supplying Yki activity in some cells by making wtsx1 mutant clones in this background. As observed previously, expression of UAS-dmyc induced apoptosis in many posterior cells of these discs (Fig. 7B) (de la Cova et al., 2004; Montero et al., 2008). However, in this background, the resulting increase in Yki activity and consequently in dIAP1 expression in wts mutant clones (Huang et al., 2005; Nolo et al., 2006; Thompson and Cohen, 2006; Wu et al., 2003) attenuated the effect of high dMyc, so that the clones formed islands of cells protected from death (Fig. 7C). Thus, although de-regulated high-level expression of dMyc has the potential to fuel aberrant growth, its feedback regulation of Yki expression provides a contingency mechanism that prevents the cells from surviving. These data provide evidence that endogenous dMyc expression provides a critical layer of regulation of Yki activity. Taken as a whole, our data suggest that the relationship between dMyc and Yki integrates and coordinates the activities of both and provides a mechanism that sets limits on tissue growth.
Discussion
The data we have presented here provide evidence for a mechanism that integrates the expression and functions of two well-conserved gene regulatory factors - dMyc and Yki - that play critical roles in Drosophila organ size control. In a mouse hepatic carcinoma cancer model induced by overexpression of the Yki homolog Yap, c-Myc was upregulated in microarray experiments, but the functional relationship between Yap and cMyc was not explored (Dong et al., 2007). Our results demonstrated that Yki regulates expression of dMyc and also requires dMyc to drive growth. In addition, we showed that endogenous Yki directly binds specific DNA sequences in the Drosophila myc gene, dm, and we identified three P-element insertions into the locus in the vicinity of these binding sites that respond robustly to Yki expression, suggesting that Yki might directly transcriptionally activate expression of dMyc. In addition, our data showed that the Yki-associating DNA binding partner, Sd binds to the dm locus at the same regions occupied by Yki, and is required for dMyc expression in the wing disc. These data provide strong suggestive evidence that Yki/Sd transcriptional complexes regulate the activity of the dm downstream of Hpo signaling. However, our results do not rule out the possibility that additional transcription factors participate in dm regulation by Yki. In eye discs, Yki associates with the Hox co-factor Hth to regulate the bantam locus (Peng et al., 2009). We think it unlikely that Hth functions with Yki to transactivate dMyc since loss of hth does not affect dMyc expression (data not shown). Still, Yap and its paralog, Taz utilize numerous transcription factors as transcriptional co-activators (Wang et al., 2009), and it is quite plausible that this is true for Yki as well. Given the significant evolutionary conservation of both Myc and the Hpo pathway functions, the relationship we uncovered here may be an important regulatory mechanism of growth in many animals.
Feedback between dMyc and Yki promotes stable growth during wing development
A key finding of our study is that in addition to its role as a Yki target gene and growth effector, dMyc exerts control over Yki expression. This provides Yki with another layer of regulation over its well-described regulation by subcellular localization. Interestingly, our experiments demonstrate that dMyc’s regulation of Yki occurs through both transcriptional and post-transcriptional mechanisms. By regulating Yki, dMyc indirectly regulates its own expression and that of other Hpo pathway targets, and thus balances its growth-promoting function with the growth-suppressing function of the Hpo pathway.
Our experiments demonstrated that when under Gal 4 control, co-overexpression of dMyc and Yki causes significantly more growth than either factor alone. On its own, this result is consistent with the interpretation that these two growth-promoting factors are independent and function in parallel to enhance growth when co-expressed. Indeed, each regulator does appear to carry out independent functions. For example, over-expression of dMyc increases cell size and nucleolar size in a manner that does not depend upon Yki. Whereas Yki promotes “balanced growth”, in which cells divide at the same rate as they increase in mass, yielding cells of normal size (Huang et al., 2005; Pantalacci et al., 2003; Tapon et al., 2002), growth promoted by dMyc is “unbalanced”: it promotes cellular growth by enhancing ribosome biogenesis but is unable to promote cell division in the wing disc, resulting in large cells with large nucleoli (Johnston et al., 1999). However, our results also indicate that the activities of dMyc and Yki are not completely independent. dMyc is positively regulated by Yki, and thus is dependent upon Yki for its expression, and Yki expression is negatively regulated by, and therefore dependent upon, dMyc. These data provide evidence for a co-dependent relationship between dMyc and Yki that integrates their expression and function. We therefore favor the interpretation that the additional growth driven by the combined overexpression of dMyc and Yki occurs because continuous Gal 4-driven expression of both factors bypasses Wts regulation and dampens dMyc’s ability to repress Yki expression, thereby allowing the potent growth promoting properties of both factors to prevail (Fig. 7D–E).
Implications of the regulatory relationship between dMyc and Yki
Does this regulatory relationship between dMyc and Yki have functional consequences for growth? We suggest that it does. The indirect influence dMyc has on Yki target gene expression would limit the expression and activities of both, and thereby could limit the overall growth of the tissue. It could also ensure the death of cells with de-regulated Myc activity, and as a result prevent expansion of cells with dangerously high growth potential. Indeed, negative feedback on Yki from dMyc provides an explanation for Myc’s well-known propensity for sensitizing cells to apoptosis when it is highly expressed (de la Cova et al., 2004; Montero et al., 2008; Pelengaris et al., 2002).
Our experiments suggest that the dMyc-Yki regulatory relationship is important for regulating the extent of tissue growth during the disc growth period. However, complete abrogation of the negative feedback loop is not possible without an understanding of the post-transcriptional mechanism by which dMyc controls Yki. Yki is anchored in the cytoplasm via phosphorylation by the Wts kinase and 14-3-3 binding (Huang et al., 2005), and also through direct binding of various Hpo pathway members by phosphorylation-independent processes (Badouel et al., 2009a; Oh et al., 2009). Both of these mechanisms prevent its nuclear localization and activity. Interestingly, the stability of Yap1 stability is regulated by interactions with β-TRCP, a component of the SCF E3 ubiquitin ligase complex, in cultured mammalian cells. However, the relevant phosphodegron is not conserved in Yki (Zhao et al., 2010), suggesting that other mechanisms underlie post-transcriptional control of Yki by dMyc. Nevertheless, other observations may shed light on how the regulatory relationship between dMyc and Yki affects organ growth. For example, loss of dmyc by mitotic recombination is generally lethal to cells due to the block to cellular growth and to cell competition (Johnston et al, 1999). However, loss of dmyc from all cells of the wing disc midway through larval development allows the formation of adult wings, albeit rudimentary in size (Wu and Johnston, 2010). This might be explained by our finding that dMyc negatively regulates Yki expression and activity: because competition between cells is eliminated and Yki is no longer subject to negative regulation by dMyc, a limited amount of growth may be possible because of increased cell survival and a few cell divisions. Increased negative feedback from dMyc to Yki may also explain the observation that adult wings are only 15% larger when dMyc is expressed at 70% over wildtype levels under heterologous control, much less than expected given the high level of dMyc expression (Wu and Johnston, 2010). Conversely, loss of Yki-regulated transcription of dMyc could affect their functional coordination and reduce the overall growth rate; consistent with this possibility, constitutive expression of dmyc at 80% of wildtype levels in an otherwise null dmyc mutant slows wing disc growth and leads to adult wings that are 33% smaller than wildtype (Wu and Johnston, 2010).
Although dMyc receives input from the disc patterning system, evidence suggests it is indirect (Herranz et al., 2008; Johnston et al., 1999; Wu and Johnston, 2010). Yki’s control of dMyc expression effectively couples the patterning system to regulation of dMyc, since the Hpo pathway is controlled by Dpp, a major organizer of pattern in the wing disc (Rogulja et al., 2008). In addition, Yki has a role in propagating expression of the wing selector gene, vestigial (vg), which drives expansion of the presumptive wing region of the wing disc (Zecca and Struhl, 2010). Early in wing disc development dMyc is expressed in all cells while Vg expression is restricted to cells at the dorsal-ventral boundary. This raises the possibility that Yki’s regulation of dMyc expression provides fuel for the proliferation of cells that are recruited by Vg to form the wing blade. Moreover, as later dMyc expression becomes similar to Vg, dMyc could also provide restraint to the recruitment process through its feedback on Yki expression.
We propose that positive regulation of dMyc by Yki and negative feedback from dMyc to Yki moderates the powerful growth stimulatory input to cells, and thus generates an inherently stable system of growth in the developing tissue. In addition, since our experiments demonstrated that changes in Hpo signaling activity locally induce cell competition that specifically mimics competition induced by differences in dMyc, dMyc appears to contribute to organ homeostasis both cell-autonomously, as an effector and regulator of Yki, and non cell-autonomously, as a mediator of cell competition provoked by local changes in the Hpo pathway.
Deregulation of the negative feedback between Myc and Yap: a tumor accelerant?
Myc is a widely conserved transcriptional regulator that controls many aspects of cell biology, and its de-regulation is a key mediator of numerous cancers. Several mammalian components of the Hpo pathway are also linked to cancer, including Mst1/2/Hpo, WW45/Sav, Lats/Wts, Mats, and Yap/Yki (reviewed in (Pan, 2007; Reddy and Irvine, 2008). Amplification of human chromosome 11q22, which contains the Yap and cIAP genes, is observed in glioblastomas and in pancreatic, ovarian, cervical, and lung cancers (Overholtzer et al., 2006). Oncogenomic approaches in mouse transgenic models of breast and liver cancers identified Yap, either alone or in combination with cIAP1, as a tumor accelerant that can synergize with c-Myc de-regulation (Zender et al., 2006). This is a striking parallel to our finding that co-expression of Yki and dMyc significantly enhances clonal growth over that stimulated by Yki or dMyc alone, and suggests broad implications of our findings. Our model of feedback between two growth regulators also raises the possibility that amplification and the contingent de-regulation of Yap is positively selected in nascent tumors to circumvent normal regulation between Yap and Myc. Therefore, an important future challenge is the elucidation of the interactions between these pathways in humans and their potential impact in organ and tissue size control and tumorigenesis.
Methods
Generation of clones and clone size measurements
The Gal4/UAS system was used for misexpression of transgenes (Brand and Perrimon, 1993). Clonal over-expression was achieved using Flp-out Gal4 (Act>y+>Gal4; (Pignoni and Zipursky, 1997)) and MARCM (Lee and Luo, 1999). Mutant clones were generated by Flp/FRT mediated mitotic recombination (Xu and Rubin, 1993). Heat shocks were given at 48h ± 3h AEL and animals dissected for analysis in late third instar (typically between 98h and 110h AEL under our laboratory conditions). MARCM was used to express transgenes in mitotic clones; TubGal4 was used to drive transgene expression, except for the dm4; UAS-yki experiment (Figure 1) where T80Gal4 was used. Heat-shocks were given at 30h ± 2h AEL and animals were dissected at 116h ± 2h AEL. Cell competition was evaluated with MARCM as previously described (de la Cova et al., 2004). Heat shocks were given at 30h ± 2h AEL and animals dissected for analysis at 110h ± 2h AEL. In all cases, clone size was measured in microns and determined by drawing around the perimeter of the marked clones using Axiovision software (Zeiss). 20 clones or more were examined for each experiment. Two-tailed Students t-tests with unequal variance were used to determine significance. The percent of nuclear Yki staining in wtsx1 mutant clones in the presence or absence of WT dMyc was calculated as the percent of cells per anterior clone (control) or posterior clone (expressing UAS-shdmyc) that expressed Yki protein with complete or nearly complete (>3/4 nucleus) overlap with the DNA stain (Hoechst).
Immunocytochemistry
Fixation and immunocytochemistry of imaginal discs were carried out as described (Johnston and Edgar, 1998). RNA in situ hybridizations were carried out using digoxigenin-labeled RNA probes (Johnston and Edgar, 1998). Images were acquired using Apotome software and a Zeiss Axioplan 2 microscope with an Orca-100 CCD camera (Hammatsu) and processed with Photoshop (Adobe) software. Antibodies used were (dilutions and sources in parentheses): guinea-pig anti-dMyc (1:1000, G. Morata), mouse anti-Nubbin (1:100, DSHB), mouse anti-CD2 (1:400, Serotec), rabbit anti-β-galactosidase (1:2000, Cappel), mouse anti-Fibrillarin (1:500, John Aris), rabbit anti-Yki (1:500, D. J. Pan), mouse anti-DIAP1 (1:200, B. Hay), rat anti-Four-Jointed (1:1000, D. Strutt), Rat anti-Ci (1:25, R. Holmgren), anti-mouse Cy3 (1:600, Jackson Laboratories), anti-rat Cy3 (1:600, Jackson Laboratories), anti-rabbit AlexaFluor488 (1:600, Molecular Probes). TUNEL assays were carried out using Apoptag Red (Chemicon International) (Wells et al., 2006).
Chromatin immunoprecipitation
ChIP assays were based on a previously described protocol (Estella et al., 2008) on wildtype wing, haltere and third leg discs. Mid-third instar (98h AEL) yw; +; + larvae were dissected in cold PBS, and fixed for 25 min. at RT in 1.8% formaldehyde, 50mM HEPES, 1mM EDTA, 0.5mM EGTA, 100mM NaCl. 20 animals were processed at a time and a total of approximately 120 animals were dissected. After quenching (0.125M Glycine, 1xPBS, 0.01% Triton) the tissue was washed twice in Buffer A (10mM HEPES, 10mM EDTA, 0.5mM EGTA, 0.25% Triton, 1mM PMSF, plus complete protease inhibitor cocktail (Roche)), then twice in Buffer B (10mM HEPES, 200mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.01% Triton, 1mM PMSF, plus complete protease inhibitor cocktail (Roche)), for 10 min at 4°C. Wing, third leg and haltere imaginal discs were then dissected from larval cuticle in Buffer B. Discs were sonicated in Buffer C (10mM HEPES, 1mM EDTA, 0.5mM EGTA, 1mM PMSF, plus complete protease inhibitor cocktail (Roche)) on ice. Soluble chromatin was transferred to a new tube after centrifugation and 10% was removed for Input. Fresh chromatin, precleared with protein A/G agarose beads (Santa Cruz), was incubated overnight at 4°C with antibodies in RIPA buffer (140mM NaCl, 10mM HEPES, 1mM EDTA, 1% Glycerol, 1% Triton X-100, 0.1% DOC, 1mM). Antibody-chromatin complexes were pulled down with protein A/G agarose beads for 3 hours at 4°C. Beads were then washed 4-times in RIPA buffer and once in TE. Chromatin was eluted twice in Elution Buffer (1% SDS, 0.1M NaHCO3). To reverse crosslinks, eluted material was incubated at 65°C overnight. 20ug Proteinase K was added and samples incubated for 3 hrs at 55°C. Phenol-chloroform extraction and ethanol precipitation were used to clean up DNA. Real Time PCR analysis was performed on an ABI 7300 instrument using SYBR Green PCR Master Mix (Applied Biosystems) per manufacturer’s instructions. Results were quantified using the delta Ct method, with respect to input samples. Rabbit anti-Yki (D. J. Pan) was used at a final concentration of 1:300, and rabbit anti-GFP used at 1:300 (Ab-Cam); specificity was evaluated by immunoprecipitations performed in parallel with normal rabbit IgG (Santa Cruz). Primer sequences are available on request.
Quantitative RT-PCR
RNA was isolated from wing, haltere and third leg imaginal discs dissected from wandering larvae with large clones expressing UAS-GFP (controls) or UAS-dmyc. Clones were generated with the flp-out Act>Gal4 system with a 15 min. heat shock at 37°C at 72 hr AEL; with this protocol the majority of the disc was GFP-positive. Reverse transcription (RT) to produce single-stranded cDNA was performed using 1 μg total RNA and SuperScript First-Strand Synthesis kit (Invitrogen). Quantitative PCR (Q-PCR) reactions were performed on an ABI 7300 instrument using SYBR Green PCR Master Mix (Applied Biosystems) per manufacturer’s instructions. For each primer set (act5C and yki), a standard curve was produced using serially diluted cDNA made from WT imaginal discs. In analyzing Q-PCRs, primer appropriate standard curves were used to calculate relative cDNA concentration in each control and experimental sample. Primers to act5C were used as loading control and to normalize the data.
Supplementary Material
Acknowledgments
We thank numerous Drosophila researchers for generously providing fly strains and reagents, G. Karsenty, R. Mann, G. Struhl, and C. Wu for comments on the manuscript, M. Slattery and J. Parker for valuable advice and discussions. Supported by RO1GM78464, RO1HD042770, and the Rita Allen Foundation (to LAJ), and the Fundacao para a Ciencia e a Tecnologia, Portugal (to RMNS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Badouel C, Gardano L, Amin N, Garg A, Rosenfeld R, Le Bihan T, McNeill H. The FERM-domain protein Expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev Cell. 2009a;16:411–420. doi: 10.1016/j.devcel.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Badouel C, Garg A, McNeill H. Herding Hippos: regulating growth in flies and man. Curr Opin Cell Biol. 2009b doi: 10.1016/j.ceb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Bourbon HM, Gonzy-Treboul G, Peronnet F, Alin MF, Ardourel C, Benassayag C, Cribbs D, Deutsch J, Ferrer P, Haenlin M, et al. A P-insertion screen identifying novel X-linked essential genes in Drosophila. Mech Dev. 2002;110:71–83. doi: 10.1016/s0925-4773(01)00566-4. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Lighthouse D, Bachman J, Planck J, Owen S, Skora AD, Nystul TG, Ohlstein B, Allen A, et al. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics. 2007;175:1505–1531. doi: 10.1534/genetics.106.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. Embo J. 2000;19:4543–4554. doi: 10.1093/emboj/19.17.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- de la Cova C, Johnston LA. Myc in model organisms: a view from the flyroom. Semin Cancer Biol. 2006;16:303–312. doi: 10.1016/j.semcancer.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estella C, McKay DJ, Mann RS. Molecular integration of wingless, decapentaplegic, and autoregulatory inputs into Distalless during Drosophila leg development. Dev Cell. 2008;14:86–96. doi: 10.1016/j.devcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulev Y, Fauny JD, Gonzalez-Marti B, Flagiello D, Silber J, Zider A. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr Biol. 2008;18:435–441. doi: 10.1016/j.cub.2008.02.034. [DOI] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- Herranz H, Perez L, Martin FA, Milan M. A Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. Embo J. 2008;27:1633–1645. doi: 10.1038/emboj.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Johnston LA. Competitive interactions between cells: death, growth and geography. Science. 2009;324:1679–1682. doi: 10.1126/science.1163862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Edgar BA. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature. 1998;394:82–84. doi: 10.1038/27925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. Embo J. 1996;15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Liu X, Grammont M, Irvine KD. Roles for scalloped and vestigial in regulating cell affinity and interactions between the wing blade and the wing hinge. Dev Biol. 2000;228:287–303. doi: 10.1006/dbio.2000.9939. [DOI] [PubMed] [Google Scholar]
- Martin FA, Herrera SC, Morata G. Cell competition, growth and size control in the Drosophila wing imaginal disc. Development. 2009;136:3747–3756. doi: 10.1242/dev.038406. [DOI] [PubMed] [Google Scholar]
- Meier P, Silke J, Leevers SJ, Evan GI. The Drosophila caspase DRONC is regulated by DIAP1. Embo J. 2000;19:598–611. doi: 10.1093/emboj/19.4.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero L, Muller N, Gallant P. Induction of apoptosis by Drosophila Myc. Genesis. 2008;46:104–111. doi: 10.1002/dvg.20373. [DOI] [PubMed] [Google Scholar]
- Nolo R, Morrison CM, Tao C, Zhang X, Halder G. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16:1895–1904. doi: 10.1016/j.cub.2006.08.057. [DOI] [PubMed] [Google Scholar]
- Oh H, Reddy BV, Irvine KD. Phosphorylation-independent repression of Yorkie in Fat-Hippo signaling. Dev Biol. 2009;335:188–197. doi: 10.1016/j.ydbio.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103:12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21:886–897. doi: 10.1101/gad.1536007. [DOI] [PubMed] [Google Scholar]
- Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- Peng HW, Slattery M, Mann RS. Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev. 2009;23:2307–2319. doi: 10.1101/gad.1820009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter A, Schottler P, Werner M, Beinert N, Dowe G, Burkert P, Mourkioti F, Dentzer L, He Y, Deak P, et al. Mapping and identification of essential gene functions on the X chromosome of Drosophila. EMBO Rep. 2002;3:34–38. doi: 10.1093/embo-reports/kvf012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignoni F, Zipursky S. Induction of Drosophila eye development by decapentaplegic. Development. 1997;124:271–278. doi: 10.1242/dev.124.2.271. [DOI] [PubMed] [Google Scholar]
- Reddy BV, Irvine KD. The Fat and Warts signaling pathways: new insights into their regulation, mechanism and conservation. Development. 2008;135:2827–2838. doi: 10.1242/dev.020974. [DOI] [PubMed] [Google Scholar]
- Rogulja D, Rauskolb C, Irvine KD. Morphogen control of wing growth through the Fat signaling pathway. Dev Cell. 2008;15:309–321. doi: 10.1016/j.devcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Johnston LA. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc Natl Acad Sci U S A. 2007;104:18543–18548. doi: 10.1073/pnas.0709021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber DA, Hariharan IK. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110:467–478. doi: 10.1016/s0092-8674(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126:767–774. doi: 10.1016/j.cell.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Tyler DM, Li W, Zhuo N, Pellock B, Baker NE. Genes affecting cell competition in Drosophila. Genetics. 2007;175:643–657. doi: 10.1534/genetics.106.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Wang K, Degerny C, Xu M, Yang XJ. YAP, TAZ, and Yorkie: a conserved family of signal-responsive transcriptional coregulators in animal development and human disease. Biochem Cell Biol. 2009;87:77–91. doi: 10.1139/O08-114. [DOI] [PubMed] [Google Scholar]
- Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16:1606–1615. doi: 10.1016/j.cub2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Johnston LA. Control of wing size and proportions by Drosophila myc. Genetics. 2010;184:199–211. doi: 10.1534/genetics.109.110379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Zecca M, Struhl G. A feed-forward circuit linking wingless, fat-dachsous signaling, and the warts-hippo pathway to Drosophila wing growth. PLoS Biol. 2010;8:e1000386. doi: 10.1371/journal.pbio.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell. 2008;13:188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF (beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.