

Abstract
Elevated saturated FFAs including palmitate (C16:0) are a primary trigger for peripheral insulin resistance characterized by impaired glucose uptake/disposal in skeletal muscle, resulting from impaired GLUT4 translocation in response to insulin. We herein demonstrate that palmitate induces down-regulation of sortilin, a sorting receptor implicated in the formation of insulin-responsive GLUT4 vesicles, via mechanisms involving PKCθ and TNF-α-converting enzyme, but not p38, JNK, or mitochondrial reactive oxygen species generation, leading to impaired GLUT4 trafficking in C2C12 myotubes. Intriguingly, unsaturated FFAs such as palmitoleate (C16:1) and oleate (C18:1) had no such detrimental effects, appearing instead to effectively reverse palmitate-induced impairment of insulin-responsive GLUT4 recycling along with restoration of sortilin abundance by preventing aberrant PKCθ activation. On the other hand, shRNA-mediated reduction of sortilin in intact C2C12 myotubes inhibited insulin-induced GLUT4 recycling without dampening Akt phosphorylation. We found that the peroxisome proliferator-activated receptor γ agonist troglitazone prevented the palmitate-induced sortilin reduction and also ameliorated insulin-responsive GLUT4 recycling without altering the palmitate-evoked insults on signaling cascades; neither highly phosphorylated PKCθ states nor impaired insulin-responsive Akt phosphorylation was affected. Taken together, our data provide novel insights into the pathogenesis of PKCθ-dependent insulin resistance with respect to insulin-responsive GLUT4 translocation, which could occur not only through defects of insulin signaling but also via a reduction of sortilin, which directly controls trafficking/sorting of GLUT4 in skeletal muscle cells. In addition, our data suggest the insulin-sensitizing action of peroxisome proliferator-activated receptor γ agonists to be at least partially mediated through the restoration of proper GLUT4 trafficking/sorting events governed by sortilin.
Keywords: Glucose Transport, Insulin Resistance, Membrane Trafficking, Signal Transduction, Skeletal Muscle, FFA, GLUT4, Sortilin
Introduction
Skeletal muscle cells as well as adipocytes exhibit the highest levels of insulin-stimulated glucose uptake, which is achieved by translocation of the insulin-responsive glucose transporter (GLUT4) from intracellular storage compartment(s) to the plasma membrane (1). It has become increasingly apparent that sortilin, a type I transmembrane protein originally identified as a major component of GLUT4-containing vesicles from rat adipocytes (2, 3), plays crucial roles in the development of the insulin-responsive GLUT4 translocation system not only in adipocytes (4, 5) but also in skeletal muscle cells (6). Evidence available thus far indicates sortilin to be directly involved in the biogenesis of insulin-responsive GLUT4 storage vesicles by regulating sorting events of GLUT4 protein between the trans-Golgi network and endosomes (4, 5, 7), and experimental suppression of sortilin in adipocytes has been shown to inhibit insulin-responsive GLUT4 translocation (4).
Intriguingly, a recent report demonstrated sortilin expression to be significantly reduced in skeletal muscle and adipose tissues of obese and diabetic db/db mice and human patients, appearing to be inversely correlated with the expression levels of pro-inflammatory TNF-α in adipose tissue (8). In addition, injecting TNF-α, which can induce insulin resistance, into mice resulted in late onset down-regulation of sortilin mRNA and protein levels in skeletal muscle and adipose tissues (8), suggesting possible involvement of sortilin reduction in the pathogenesis of chronic insulin resistance induced by TNF-α, especially in terms of insulin-responsive GLUT4 translocation.
Insulin resistance is defined as the pathophysiological condition in which the ability of insulin to regulate glucose homeostasis in target cells is reduced, a state commonly associated with obesity (9). Indeed, high fat feeding and increased levels of circulating FFAs progressively led to the induction of peripheral insulin resistance characterized by impaired insulin-responsive GLUT4 translocation in skeletal muscle (10, 11). The deleterious effects of saturated FFAs such as palmitate (C16:0) in skeletal muscle have been attributed to abnormal accumulation of palmitoyl-CoA, diacylglycerol, and/or ceramide, which subsequently leads to aberrant activation of various serine/threonine kinases such as PKCθ (12–14).
PKCθ is a novel PKC isoform abundantly expressed in skeletal muscle, which reportedly may be relevant to FFA-induced muscle insulin resistance (15). One of the established pathogenic effects of PKCθ activity is detrimental phosphorylation of serine residues on insulin receptor substrate (IRS)3 proteins, which in turn reduces the ability of the IRS proteins to activate phosphatidylinositol 3-kinase cascades (13, 16, 17). In addition, PKCθ has the unique ability to activate transcriptional factor NF-κB, and the PKCθ-NF-κB signaling cascade has been directly implicated in the expression of various pro-inflammatory cytokines including TNF-α (14). TNF-α has also been shown to trigger phosphorylation of the critical serine residues of IRS proteins (18, 19). Thus, it is generally accepted that, as a consequence of the impaired insulin signaling competency resulting from at least these two distinct mechanisms involving PKCθ, insulin-responsive glucose uptake/disposal in skeletal muscle is diminished.
In the present study, we treated C2C12 myotubes with various saturated and unsaturated FFAs to study the molecular mechanisms underlying the FFA-induced insulin resistance in skeletal muscle cells as assessed by insulin-responsive GLUT4 recycling. We demonstrated saturated FFAs, especially palmitate (C16:0), but not unsaturated FFAs, to induce down-regulation of sortilin via a mechanism involving PKCθ, leading to impaired GLUT4 trafficking in differentiated C2C12 myotubes. In addition, we demonstrated a crucial role for sortilin in maintaining proper insulin-responsive GLUT4 trafficking even in palmitate-treated cells, because a PPARγ agonist restored sortilin abundance and GLUT4 recycling without improving palmitate-induced impairments of signaling cascades. These findings provide novel insights into the beneficial actions of PPARγ agonists and the pathogenesis of PKCθ-dependent insulin resistance elicited by palmitate, which could occur via a modulation of sortilin, a sorting receptor involved in GLUT4 sorting/trafficking in skeletal muscle cells.
EXPERIMENTAL PROCEDURES
Materials
The Western blot detection kit (West super femto detection reagents) and Immobilon-P were purchased from Pierce and Millipore Corp. (Bedford, MA), respectively. DMEM, penicillin/streptomycin, and trypsin-EDTA were purchased from Sigma. Cell culture equipment was obtained from BD Biosciences (San Jose, CA). Calf serum and FBS were purchased from BioWest (Nuaille, France). FFA-free BSA was purchased from Wako Pure Chemical Industries (Osaka, Japan). Anti-sortilin antibody was purchased from BD Biosciences. Anti-phospho-PKCθ (Thr538), anti-phospho-NF-κB (Ser536), anti-phospho-Akt (Ser473), and anti-phospho-ATF-2 (Thr69/71) antibodies were purchased from Cell Signaling Technology Inc. (Danvers, MA). Anti-β-actin antibody was purchased from Sigma. The ELISA kit for TNF-α was purchased from R & D Systems (Minneapolis, MN). N-(R)-[2-(Hydroxyaminocarbonyl)methyl]-4-methyl-pentanoyl-l-naphthylalanyl-l-alanine, 2-amino-ethyl amide, TNF-α protease inhibitor-1 (TAPI-1) was purchased from Merck. Fatty acids, pyrrolidine dithiocarbamate, SB203580, rottlerin, etomoxir, thenoyltrifluoroacetone, and carbonyl cyanide m-chlorophenylhydrazone were purchased from Sigma. Wy-14643, GW501516, and troglitazone were purchased from Cayman Chemicals (Ann Arbor, MI), Alexis Biochemicals (Lausen, Switzerland), and Sigma, respectively. The shRNA lentiviral particles targeting mouse PKCθ, sortilin, or scramble control were purchased from Santa Cruz Biotechnology. Other chemicals were purchased from Sigma or Wako.
Cell Culture
Mouse skeletal muscle cell lines, C2C12 myoblasts, were maintained in DMEM supplemented with 10% FBS, 30 μg/ml penicillin, and 100 μg/ml streptomycin (growth medium) at 37 °C under a 5% CO2 atmosphere. For biochemical study, the cells were grown on six-well plates (BD Biosciences) at a density of 3 × 104 cells/well in 3 ml of growth medium. Three days after plating, the cells had reached ∼80–90% confluence (day 0). Differentiation was then induced by replacing the growth medium with DMEM (4.5 g/liter glucose) supplemented with 2% calf serum, 1 nm insulin, 30 μg/ml penicillin, and 100 μg/ml streptomycin (differentiation medium) (20). The differentiation medium was changed every 24 h, and the differentiated cells (at days 4 and 5) were used for subsequent experiments. FFA-containing media were prepared by preincubation of FFA with DMEM supplemented with 2% FFA-free BSA as described previously (20). Briefly, FFAs were dissolved in ethanol and diluted 1:150 in DMEM containing 2% (w/v) FFA-free BSA and 2% calf serum, and the lipid-containing media were incubated for 1 h at 37 °C before administration to the culture. Under these conditions, the molar ratio of FFA (0–1.0 mm) to 2% BSA (calculated molecular weight of 665,000) would be ∼0–3.3. The inhibitors were prepared in dimethyl sulfoxide and then added to the media at 0.1% (v/v).
Quantitative Real Time PCR
Total RNA was prepared using the TRIzol reagent (Invitrogen) following the manufacturer's instructions. Quantitative real time PCR analysis was performed using the Light Cycler instrument and SYBR Green detection kit according to the manufacturer's instructions (Roche Applied Science). PCR primers for sortilin were: 5′-CCT CTG TGA CTT TGG CTA CTT C-3′ and 5′-ATC CCA CCT TGG CAT TTG T-3′. PCR primers for β-actin as a control were: 5′-CGT TGA CAT CCG TAA AGA CCT C-3′ and 5′-AGC CAC CGA TCC ACA CAG A-3′.
Western Blot Analysis
Cell lysates were prepared using lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1 μg/ml pepstatin, 5 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, 6500 IU/ml aprotinin, phosphatase inhibitor mixture-1; Sigma), and the protein concentrations of cell lysates were then measured using the BCA protein assay kit (Pierce). Proteins (20 μg) were subjected to 10% SDS-PAGE and then transferred to a PVDF membrane (Immobilon-P; Millipore), and the membranes were then blocked for 2 h at room temperature with 5% BSA in TBS containing 0.1% Tween 20. The membranes were next immunoblotted with primary antibodies at dilutions of 1:500 to 1:1000. Specific total or phospho-proteins were visualized after subsequent incubation with a 1:10000 dilution of anti-mouse or rabbit IgG conjugated to horseradish peroxidase and the SuperSignal Chemiluminescence detection procedure (Pierce). Three independent experiments were performed for each condition.
Detection of Secreted Fragment of Sortilin in Conditioned Media
Conditioned media were collected and concentrated using Amicon Ultra-15/30K (Millipore Corp., Billerica, MA), followed by immunoprecipitation using anti-sortilin antibody. Immunoprecipitated materials were then subjected to Western blot analysis.
ELISA for TNF-α
TNF-α levels in media were evaluated using the mouse TNF-α assay kit (R & D Systems).
Caspase-3 Assay
Cellular caspase-3 activity was evaluated using the EnzChek Caspase-3 assay kit (Invitrogen) according to the manufacturer's instructions. Briefly, the cell lysates were incubated with a caspase-3 substrate, Z-DEVD-AMC, which upon cleavaged by active caspase-3, generates fluorescent products. Fluorescent intensity was measured using a SpectraMax M5 (excitation, 342 nm; emission 441 nm) and subtracted from that of the cell lysate-free sample, followed by normalization using the corresponding protein amount levels.
Anti-c-Myc Antibody Uptake Assay
GLUT4 recycling was analyzed as described previously (21). Briefly, C2C12 myotubes expressing Myc-GLUT4-ECFP were serum-starved, washed three times with Krebs-Ringer/phosphate/HEPES (KRPH) buffer, and then placed in a CO2 incubator with 2 ml of KRPH buffer. After 10 min of incubation, 4 μg/ml of the anti-c-Myc antibody were added to the buffer, and the cells were stimulated with or without 100 nm insulin for 30 min. After incubation for 30 min with the anti-Myc antibody, the cells were placed on ice to stop the reaction, and washed five times with PBS. The cells were harvested using 1× Laemmli buffer and subjected to Western blot analysis using anti-mouse IgG and anti-c-Myc antibodies. In the same experiments, we also measured amounts of Alexa 555-conjugated anti-c-Myc antibody bound to cell surface-exposed Myc-GLUT4-ECFP at 4 °C using a laser-induced fluorescence scanner (PharosFX Plus; Bio-Rad) following SDS-PAGE of the whole cell lysates.
Live Cell Imaging of Intracellular Trafficking and Data Analysis
C2C12 myoblasts were maintained and differentiated into myotubes on poly-l-lysine-coated glass-bottomed recording chamber (thickness, 0.15–0.18 mm; Matsunami Glass, Osaka, Japan). The differentiated myotubes treated with fatty acids were stained with 50 nm LysoTracker Red DND-99 (Invitrogen) according to the manufacturer's instructions. Then the cells were washed with imaging medium (150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, and 10 mm HEPES-NaOH, pH 7.4) containing 5.5 mm d-glucose. The images were acquired every 1 s for 100 s with an inverted microscope (IX81; Olympus, Tokyo, Japan) equipped with a laser scanner (FV1000; Olympus) and an oil-immersion objective lens (PlanApo 60×, numerical aperture of 1.40). The fluorescence was excited at 543 nm and detected at >560 nm. The movement of stained acidic compartments was tracked with G-Track software with a centroid fitting mode (G-angstrom; Sendai). We tracked each object that was successfully fitted with 6 × 6-pixel regions-of-interest for at least 30 consecutive frames. We showed the movement speed in two ways: individual and effective speed (22). The former was calculated based on displacement between two consecutive frames, and the latter was determined by dividing the distance between the origin and the end of the trace by the trace duration. The movement speed calculated as above inevitably contains a factor resulting from instrumental noise. Therefore, we corrected the “apparent” speeds by subtracting the value obtained from cells fixed with 1% paraformaldehyde after staining with LysoTracker.
Statistical Analysis
The data are expressed as the means ± S.E. Statistical significance was determined by analysis of variance followed by the Williams' test unless otherwise indicated. The differences were considered significant when the p values were less than 0.05.
RESULTS
Saturated, but Not Unsaturated, FFAs Suppress Sortilin in C2C12 Myotubes
We examined the effects of FFAs on the expression of sortilin mRNA in differentiated C2C12 myotubes (Fig. 1A). Treatment with palmitate (C16:0) or stearate (C18:0) for 16 h significantly reduced sortilin mRNA expression in a dose-dependent manner, whereas unsaturated FFAs (palmitoleate (C16:1) and oleate (C18:1)) did not reduce the sortilin mRNA level. Consistent with these mRNA data, Western blot analysis demonstrated that only saturated FFAs, palmitate (C16:0) and stearate (C18:0), decreased sortilin protein (Fig. 1B) in a dose-dependent manner (Fig. 1C). Time course experiments demonstrated the reduction of sortilin protein to be induced after 16 h of palmitate treatment (Fig. 1D).
FIGURE 1.

Effects of saturated and unsaturated fatty acids on sortilin expression in C2C12 myotubes. A, C2C12 myotubes were treated with FFAs for 16 h, and total RNA was then prepared. The levels of sortilin and β-actin (control) mRNA were quantified by real time PCR. FFAs were expressed as follows: C16:0, palmitate; C18:0, stearate; C16:1, palmitoleate; and C18:1, oleate. The data are expressed as the means ± S.E. of three independent experiments. *, p < 0.05 versus untreated control. B, C2C12 myotubes were treated with 1 mm FFA for 16 h, and the cell lysates were then subjected to Western blot analysis. C, C2C12 myotubes were treated with different doses of palmitate for 16 h, and the cell lysates were then subjected to Western blot analysis. D, C2C12 myotubes were treated with 1 mm palmitate for 2–20 h, and the cell lysates were then subjected to Western blot analysis.
As previously reported (14), treatment with palmitate (C16:0) dose-dependently augmented secretion of TNF-α (Fig. 2A), which might be involved in the reduction of sortilin expression (8). However, administration of a neutralizing antibody against TNF-α failed to prevent the palmitate-induced reduction of sortilin protein, at least under these culture conditions (Fig. 2B). Moreover, a very high concentration of TNF-α (100 ng/ml), i.e. 3000-fold higher than that secreted by palmitate-treated C2C12 myotubes (∼35 pg/ml), was required to reduce sortilin protein (Fig. 2C).
FIGURE 2.
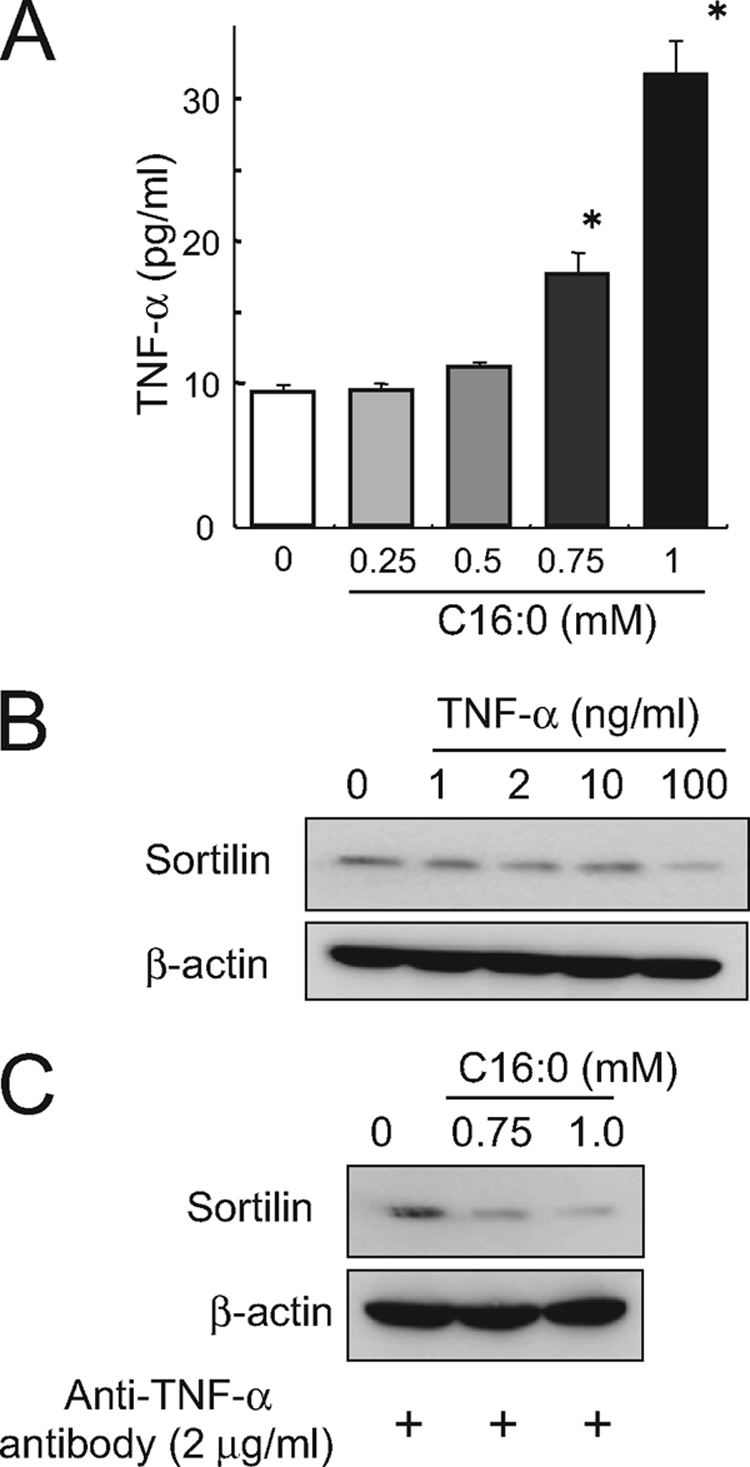
Palmitate-induced sortilin reduction is not mediated through autocrine secreted TNF-α. A, C2C12 myotubes were treated with 0.25–1 mm palmitate for 16 h, and the conditioned medium was then subjected to ELISA for TNF-α concentration measurement. B, C2C12 myotubes were treated with 1–100 ng/ml of TNF-α, and the cell lysates were subjected to Western blot analysis. C, C2C12 myotubes were treated with the indicated concentrations of palmitate for 16 h in the absence or presence of anti-TNF-α neutralizing antibody, and the cell lysates were then subjected to Western blot analysis. Three independent experiments were performed, and representative results were obtained.
It should be noted that high concentrations of saturated FFAs and TNF-α (100 ng/ml), but not unsaturated FFAs, induced significant increases in caspase-3 activity (data not shown), indicating that myotubes were undergoing apoptotic responses in response to either saturated FFAs or TNF-α under these culture conditions, as reported previously (23, 24).
PKCθ and TACE/ADAM17, but Neither p38 MAPK nor NF-κB Are Involved in Palmitate-induced Reduction of Sortilin in C2C12 Myotubes
Because treatment with palmitate resulted in aberrant activations of a wide variety of intracellular signaling cascades including p38 MAPK, JNK, and PKCθ in C2C12 myotubes as we previously reported (20), we next attempted to define intracellular signaling cascades involved in the palmitate-induced reduction of sortilin expression. Pharmacological inhibition of PKCθ by rottlerin abolished the effect of palmitate, and the sortilin amount was restored when the cells were treated with palmitate in the presence of 30 μm rottlerin (Fig. 3, A and B). Involvement of PKCθ in the palmitate-induced sortilin reduction was also confirmed by shRNA-mediated knockdown experiments, and the effect of palmitate was attenuated in C2C12 myotubes treated with PKCθ-specific, but not scramble control, shRNA lentiviral particles (Fig. 3C).
FIGURE 3.

Involvement of PKCθ in palmitate-induced sortilin reduction. A and B, C2C12 myotubes were treated with or without 1 mm palmitate (C16:0) in the presence of 3–30 μm of rottlerin (a novel PKC-specific inhibitor) for 16 h, and the cell lysates were then subjected to Western blot analysis. Specific bands in the Western blots were quantified using a densitometer. The data are expressed as the means ± S.E. of three independent experiments. *, p < 0.05 versus control. C and D, differentiated myotubes derived from either PKCθ-specific or scrambled shRNA lentiviral particle-treated C2C12 myoblasts were cultured with 0.25–1 mm of palmitate for 16 h, and the cell lysates were then subjected to Western blot analysis. Specific bands in the Western blots were quantified using a densitometer. The data are expressed as the means ± S.E. of three independent experiments. *, p < 0.05 versus control.
To further investigate the underlying mechanism of the PKCθ-dependent sortilin suppression elicited by palmitate treatment, we examined the effect of TAPI-1, a selective inhibitor for TACE (also known as ADAM17) because sortilin reportedly undergoes shedding of its luminal domain with TACE (25), which could be induced by phorbol 12-myristate 13-acetate, a PKC activator (26). Palmitate-induced sortilin reduction was significantly blunted by TAPI-1 in a dose-dependent manner, although 30 μm of TAPI-1 could not reverse the sortilin amount to the control levels (Fig. 4). Consistent with these findings, the secreted fragment of sortilin was detected in the conditioned media after palmitate treatment (16 h) but then disappeared with the addition of TAPI-1 in a dose-dependent manner (Fig. 4).
FIGURE 4.
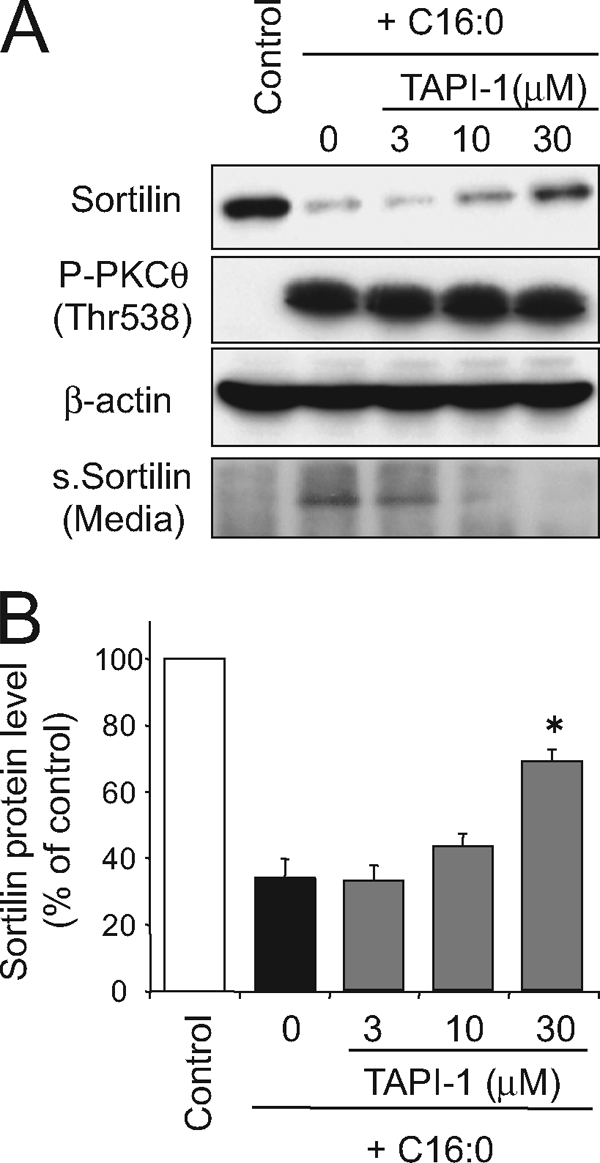
Effects of pharmacological inhibition of TACE/ADAM17 on palmitate-induced sortilin down-regulation. A and B, C2C12 myotubes were treated with or without 1 mm palmitate (C16:0) in the presence of 3–30 μm of TAP-1 (the TACE/ADAM17 selective inhibitor) for 16 h, and the cell lysates were then subjected to Western blot analysis. Conditioned media were collected and immunoprecipitated using anti-sortilin antibody, and the immunoprecipitated materials were then subjected to Western blot analysis. Specific bands in the Western blots were quantified using a densitometer. The data are expressed as the means ± S.E. of three independent experiments. *, p < 0.05 versus control. s.Sortilin, secreted fragment of sortilin.
The effect of palmitate on sortilin reduction was not inhibited by pyrrolidine dithiocarbamate (a NF-κB cascade inhibitor), SB203580 (a p38 MAP kinase inhibitor), or SP600125 (a JNK inhibitor) (Fig. 5, A–C), whereas these inhibitors potently dampened palmitate-induced phosphorylation of NF-κB or ATF2 (a p38 substrate) (Fig. 5, A and B).
FIGURE 5.

Effects of pharmacological inhibition of NF-κB, p38 MAPK, or fatty acid oxidation on palmitate-induced sortilin down-regulation. A–D, C2C12 myotubes were treated with or without 1 mm palmitate (C16:0) in the presence of several concentrations of pyrrolidine dithiocarbamate (PDTC; NF-κB inhibitor), SB203580 (p38 inhibitor), etomoxir (200, 500, and 1000 μm; carnitine palmitoyl transferase-1 inhibitor), thenoyltrifluoroacetone (TTFA; 10, 30, and 100 μm; electron transport complex II inhibitor), or carbonyl cyanide m-chlorophenylhydrazone (CCCP; 0.5, 3, and 10 μm; uncoupler) for 16 h, and the cell lysates were then subjected to Western blot analysis. Representative immunoblots obtained from three independent experiments are shown.
We also examined the possible involvement of mitochondria-derived reactive oxygen species by using pharmacological inhibitors for enzymes related to fatty acid oxidation. Etomoxir (carnitine palmitoyltransferase-1 inhibitor), thenoyltrifluoroacetone (electron transport complex II inhibitor), and carbonyl cyanide m-chlorophenylhydrazone (an uncoupler of oxidative phosphorylation) all failed to exert any inhibitory effect on palmitate-induced sortilin reduction (Fig. 5D), whereas these inhibitors completely abolished reactive oxygen species production (data not shown). We also found that administration of ceramide, a possible mediator of the actions of palmitate (27, 28), did not mimic palmitate-induced sortilin down-regulation at least with 16 h of C2-ceramide (1–100 μm) treatment (data not shown).
Unsaturated FFAs Reverse Palmitate-induced Sortilin Reduction
As reported previously, unsaturated FFAs such as oleate (C18:1) and palmitoleate (C16:1) potently reverse palmitate-induced detrimental effects including secretion of inflammatory cytokines (e.g. IL-6 and TNF-α), cyclooxyganase-2 expression, and insulin resistance (14, 20, 29–31). We therefore investigated the impacts of unsaturated FFAs on palmitate-induced sortilin reduction (Fig. 6). Both oleate (C18:1) and palmitoleate (C16:1) exhibited dose-dependent inhibitory effects on palmitate-induced sortilin suppression concomitantly with diminished PKCθ phosphorylation, and a sufficient concentration of each unsaturated FFA completely restored sortilin protein to levels comparable with those under intact conditions (Fig. 6). It is noteworthy that potencies against palmitate-induced sortilin reduction differed significantly, with oleate (C18:1) clearly being more potent than palmitoleate (C16:1) as we previously observed in their protective effects on palmitate-induced COX-2 expression in C2C12 myotubes (20).
FIGURE 6.

Effects of unsaturated FFAs on palmitate-induced sortilin down-regulation and PKCθ phosphorylation status. A, B, C2C12 myotubes were treated with 1 mm palmitate (C16:0) alone or in combination with palmitoleate (C16:1) (closed triangle) or oleate (C18:1) (open square) for 16 h, and the cell lysates were then subjected to Western blot analysis. B, specific bands in the Western blots were quantified using a densitometer. The data are expressed as the means ± S.E. of three independent experiments. *, p < 0.05 versus C16:0 alone.
PPARγ, but Neither PPARα nor PPARβ/δ, Is Involved in the Protective Action of Unsaturated FFAs against Palmitate-induced Sortilin Reduction
We also examined the possible involvement of PPARs in the protective actions of unsaturated FFAs on palmitate-induced events, because unsaturated FFAs have been shown to serve as potent ligands for these PPARs (31, 32). The palmitate-induced sortilin reduction was not reversed by either Wy-14643 (PPARα agonist) or GW501516 (PPARβ/δ agonist) (33, 34). However, troglitazone (PPARγ agonist) (35) displayed a remarkable dose-dependent inhibitory effect on palmitate-induced sortilin reduction with no obvious alterations in palmitate-induced PKCθ phosphorylation (Fig. 7). Although relatively higher concentrations of troglitazone were required for its protective effect against palmitate-induced sortilin down-regulation to be exerted, this is perhaps due to supplementation with BSA, which reportedly binds troglitazone and affects its potency (36, 37).
FIGURE 7.

Effects of PPAR activators on palmitate-induced sortilin down-regulation and PKCθ phosphorylation status. A, B, C2C12 myotubes were treated with 1 mm palmitate (C16:0) alone or in combination with 300 μm oleate (C18:1) and several concentrations of Wy-14643 (PPARα activator), GW50156 (PPARβ/δ activator), or troglitazone (PPARγ activator) for 16 h. A, the cell lysates were subjected to Western blot analysis. Three independent experiments were performed, and representative results were obtained. B, specific bands in the Western blots were quantified using a densitometer. The data are expressed as the means ± S.E. of three independent experiments. *, p < 0.05 versus C16:0 alone.
Involvement of Sortilin Reduction in Palmitate-induced Impairment of Insulin-responsive GLUT4 Recycling in C2C12 Myotubes
Finally, we examined the involvement of sortilin reduction in the generation of insulin resistance in C2C12 myotubes, because sortilin plays important roles in establishing the insulin-responsive glucose transport system in skeletal muscle cells (6). To address this possibility, we employed a Myc-GLUT4 recycling assay using anti-Myc antibody as we reported previously (21). Consistent with a previous report (20), palmitate treatment impaired insulin-responsive GLUT4 recycling, concomitantly with the sortilin reduction, with impaired insulin responsiveness being partially attributable to the increased GLUT4 recycling even under basal conditions (Fig. 8). The palmitate-induced impairment of insulin-responsive GLUT4 recycling was reversed by adding concentrations of palmitoleate (C16:1), oleate (C18:1), or troglitazone sufficient to restore sortilin expression levels (Fig. 8). The impairment of insulin-induced Akt phosphorylation was reversed by unsaturated FFAs, but not by troglitazone. Insulin-induced GLUT4 translocation was significantly inhibited by shRNA-mediated sortilin knockdown in C2C12 myotubes without influencing Akt phosphorylation (Fig. 8C).
FIGURE 8.

Effect of sortilin down-regulation either by palmitate treatment or shRNA lentiviral infection and impaired GLUT4 recycling. A and B, C2C12 myotubes expressing Myc-GLUT4-ECFP were treated with 1 mm palmitate (C16:0) alone or in combination with palmitoleate (C16:1, 1000 μm), oleate (C18:1, 300 μm), and troglitazone (Tro, 100 μm) for 16 h. A, the cells were serum-starved and then treated with 100 nm of insulin for 30 min in the presence of 4 μg/ml anti-Myc antibody. The cells were then washed five times with PBS and analyzed by Western blotting with the use of anti-mouse IgG HRP conjugate antibody (for evaluation of the translocated GLUT4 amounts) and anti-EGFP antibody (for evaluation of total Myc-GLUT4-ECFP amounts). To evaluate Akt phosphorylation status, the serum-starved cells were treated with 100 nm insulin for 5 min. A, the cell lysates were subjected to Western blot analysis using anti-phospho Akt (Ser473) and anti-Akt antibodies. B, the results from A, uptake of anti-Myc antibody in response to insulin, were subjected to densitometric analysis for quantification. C, differentiated C2C12 myotubes expressing Myc-GLUT4-ECFP (day 3) were treated with or without lentiviral particles containing PKCθ-specific shRNA or scramble control shRNA and then further cultured for 72 h. The Myc-GLUT4 recycling assay and Western blot analysis were performed as described above for A and B. The data are expressed as the means ± S.E. of four independent experiments. *, p < 0.05 versus each basal conditions. Representative immunoblots obtained from two to four independent experiments are shown.
Palmitate Impairs Lysosome Motility
To examine whether palmitate affects intracellular trafficking events other than GLUT4 translocation, we analyzed the movement of acidic compartments including lysosomes in C2C12 myotubes by staining with LysoTracker Red (Fig. 9A). The dye-positive structures showed highly dynamic movement in the cells (Fig. 9B), as in other types of cells (38, 39). We quantitatively analyzed the movement by calculating the movement speed based on displacement between two consecutive frames or between the origin and the end of the trace (Fig. 9, C–E; see “Experimental Procedures”). Interestingly, treatment with palmitate for 16 h significantly reduced the movement of intracellular acidic compartments (Fig. 9, C–E). These effects of palmitate were dose- and time-dependent (Fig. 9, F and G). Importantly, oleate significantly inhibited the deleterious effects of palmitate and completely restored the movement of acidic compartments to levels comparable with those under intact conditions (Fig. 9, D and E). These observations indicate that palmitate affects not only GLUT4 trafficking but also intracellular trafficking more widely and that oleate has a restorative effect on palmitate-induced derangements of intracellular trafficking in C2C12 myotubes.
FIGURE 9.

Effects of palmitate and oleate on movement of intracellular acidic compartments. A, fluorescent image of C2C12 myotubes stained with LysoTracker Red. The area between the white lines represents a single myotube. B, typical traces of acidic compartment movement. C, distributions of apparent individual speed of acidic compartment movements when treated without (white) or with (black) 1 mm palmitate for 16 h. D and E, mean individual (D) and effective (E) speeds of acidic compartment movement in control (Cont, white), 1 mm palmitate-treated (Pal, 16 h, black), or 1 mm palmitate + 0.3 mm oleate-treated (Pal + Ole, 16 h, gray) C2C12 myotubes. The apparent speed in fixed cells (individual speed,0.028 μm s−1; effective speed, 0.002 μm s−1) was subtracted from the values of the cells treated as indicated (see “Experimental Procedures”). Statistical analyses were performed with Tukey's multiple comparison test. *, p < 0.05; **, p < 0.01. F and G, dose response (F) and time course (G) of the effects of palmitate on acidic compartment movement. The data were corrected with the apparent movement speed in fixed cells, as in D. Statistical analyses were performed with Dunnett's multiple comparison test versus 0 mm (D) or 0 h (E). *, p < 0.05; **, p < 0.01; ***, p < 0.001. The data were obtained from four to nine cells.
DISCUSSION
The role of PKCθ activity elicited by the palmitate-induced accumulation of DAG and acyl-CoA has been well established in generating pathogenic insulin resistance (15), which is generally explained by impaired insulin signaling cascades resulting from detrimental PKCθ-induced phosphorylation of serine residues on IRS proteins (13, 17, 40) and PDK1 (41). In the present study, we demonstrated that palmitate-induced PKCθ activation is also attributable to the down-regulation of sortilin (Fig. 3), a sorting receptor protein implicated in generating insulin-responsive GLUT4 vesicles. Importantly, unsaturated FFAs including palmitoleate (C16:1) and oleate (C18:1) effectively reversed palmitate-induced impairment of insulin-responsive GLUT4 recycling along with restoration of sortilin protein abundance by preventing aberrant PKCθ phosphorylation (Fig. 6). On the other hand, shRNA-mediated reduction of sortilin in intact C2C12 myotubes slightly but significantly inhibited insulin-induced GLUT4 recycling without dampening Akt phosphorylation (Fig. 8). In addition, palmitate treatment appeared to significantly impair lysosome motility, which was again effectively restored by oleate administration (Fig. 9). Although the degree to which detrimental PKCθ actions on the generation of insulin signaling defects or the down-regulation of sortilin expression contributes to the development of impaired GLUT4 recycling remains to be determined, our findings shed novel mechanistic insights into the pathogenic roles of PKCθ in generating insulin resistance in skeletal muscle cells (Fig. 10).
FIGURE 10.

Schematic model for palmitate-induced sortilin down-regulation and its possible consequences for the impaired insulin-responsive GLUT4 translocation in myotubes. Palmitate-induced activation of PKCθ, but not mitochondrial reactive oxygen species, p38, JNK, or NF-κB, contributes to sortilin down-regulation resulting from actions at both transcriptional (mRNA expression) and post-transcriptional (TACE-mediated shedding) levels. Unsaturated FFA such as oleate effectively prevented aberrant PKCθ activation elicited by palmitate and thereby restored sortilin contents as well as insulin-responsive GLUT4 translocation. Thus, palmitate-induced deleterious PKCθ activation contributes to not only dampening of insulin receptor signals (signaling defects), but also the induction of sortilin down-regulation, possibly resulting in derangements in GLUT4 sorting/targeting (trafficking defects). In addition to these PKCθ-dependent events, palmitate also induces significant impairments in lysosome motility, indicating that various intracellular trafficking events are severely deranged under the insulin-resistant conditions elicited by palmitate.
Involvement of PKCθ and TACE/ADAM17 in Palmitate-induced Sortilin Down-regulation
It was recently reported that exogenous administration of TNF-α, an established inducer of insulin resistance (42), resulted in decreased sortilin expression in both adipose and skeletal muscle tissues in vivo and that TNF-α-induced sortilin suppression was also observed in cultured adipocytes (8). However, we demonstrated the contribution of autocrine TNF-α to the palmitate-induced sortilin reduction in C2C12 myotubes to be minimal at least under the culture conditions of the present study, because the anti-TNF-α neutralizing antibody had no impact (Fig. 2), with palmitate instead directly inducing sortilin down-regulation through a mechanism involving PKCθ as an intracellular signaling intermediate (Fig. 3). In agreement with this observation, pharmacological inhibition of NF-κB cascades by pyrrolidine dithiocarbamate, which completely abolished palmitate-induced TNF-α secretion (data not shown), failed to interfere with the sortilin reduction (Fig. 5A). Because high concentrations of TNF-α suppressed sortilin expression in C2C12 myotubes (Fig. 2), similar to observations in cultured adipocytes (8), sortilin expression in skeletal muscle is perhaps regulated by various different and/or combined insults such as saturated FFAs and circulating TNF-α triggering insulin resistance in vivo.
We provide compelling evidence that PKCθ activity is predominantly involved in palmitate-induced sortilin down-regulation by showing that inhibition of PKCθ activity and phosphorylation by either the pharmacological inhibitor rottlerin (Fig. 3) or unsaturated FFAs (Fig. 6) completely prevent this palmitate-induced event. However, shRNA-mediated reduction of PKCθ only partially prevented the decrease in sortilin abundance by palmitate treatment (Fig. 3D). We also found that palmitate-induced suppression of sortilin mRNA expression was obvious at concentrations higher than 0.75 mm, whereas sortilin protein was significantly reduced at 0.5 mm palmitate (Fig. 1). Taken together, these results suggest that palmitate-induced sortilin down-regulation is complex, being induced at both the transcriptional and the post-transcriptional level, possibly involving multiple harmful intermediates evoked by palmitate in addition to PKCθ activity.
In this regard, we found that palmitate-induced sortilin reduction is significantly inhibited by TAPI-1, the selective TACE/ADAM17 inhibitor (Fig. 4), indicating PKCθ-dependent sortilin reduction to be mediated at least partially through the post-translational modification (shedding) involving TACE activity (25), which could be activated by a PKC activator, TPA (26). Intriguingly, several recent studies have demonstrated that TACE activity is increased in the skeletal muscles of obese type 2 diabetes patients and in lipid-induced insulin resistance (43, 44). In addition, increased TACE activity has been directly implicated in the pathogenesis of insulin resistance, which is generally explained by the augmented secretion of TNF-α (43, 45, 46). Thus, our findings provide an important connection between TACE activity and PKCθ-dependent sortilin reduction in skeletal muscle cells under palmitate-induced insulin-resistant conditions, which appeared to be directly involved in the pathogenesis of impaired GLUT4 trafficking as discussed below.
At present, we have not yet defined the molecular mechanism underlying the palmitate-induced sortilin mRNA down-regulation. PKCθ has been shown to modulate the activities of many transcription factors including nuclear factor of activated T cell c2, NF-κB, and activator protein-1 (47), and several lines of evidence indicate PKCθ to also be involved in transcriptional repression by modulating nuclear receptor-corepressor and/or -coactivator interaction in T lymphoid cells (48) (49). Because significant increases in caspase-3 activity were detected in C2C12 myotubes exposed to saturated FFAs but not unsaturated FFAs, the suppression of sortilin mRNA expression observed herein might be a consequence of apoptotic responses to some degree. Further studies are needed to clarify this important issue.
Palmitate-induced Sortilin Down-regulation and Impaired GLUT4 Trafficking
In our previous study designed to elucidate the role of sortilin in the development of insulin-responsive GLUT4 translocation in C2C12 myocytes, we observed that sortilin serves as an important factor in generating the insulin-responsive glucose transport system (6), essentially as seen in the 3T3L1 adipogenic cell line (4). In addition, we reported sortilin to also be directly involved in the early phase of myogenic differentiation processes by cooperatively functioning with p75 neurotrophin receptor and pro-form of NGF of nerve growth factor in C2C12 myocytes (6). Because siRNA-mediated sortilin knockdown in C2C12 myoblasts severely inhibited their myogenic differentiation (data not shown) (6), we utilized shRNA lentiviral technology to knock down sortilin expression in differentiated C2C12 myotubes in the present study (Fig. 8). Similar to observations in the 3T3L1 adipogenic cell line (4), our data clearly demonstrated that experimental suppression of sortilin abundance in differentiated C2C12 myotubes resulted in impaired insulin-responsive GLUT4 recycling, which is partially attributable to increased GLUT4 recycling even in the basal state, without affecting insulin-induced Akt phosphorylation (Fig. 8).
Supporting an essential role for sortilin in the formation of insulin-responsive GLUT4 vesicles (4–7), our data (shown in Fig. 8) strongly suggest that sortilin down-regulation induced by palmitate treatment is involved in the pathogenesis of impaired GLUT4 trafficking and, therefore, that restoration of sortilin abundance either by unsaturated FFAs or troglitazone is crucial for maintaining GLUT4 trafficking in response to insulin stimulation in C2C12 myotubes. These findings are consistent with previous studies showing that unsaturated FFAs such as palmiteoleate (C16:1) and oleate (C18:1) have protective effects on palmitate-induced insulin resistance at the level of glucose uptake in L6 myotubes (50, 51). At present we have not yet defined in detail the mechanism(s) underlying the impacts of sortilin reduction on impaired GLUT4 trafficking. However, because our data indicate the palmitate-induced sortilin reduction to be detectable after 16 h of treatment with relatively high levels of palmitate (Fig. 1), this palmitate-induced late onset event perhaps contributes to generating the chronic, but not the acute, phase of insulin resistance (52–54), possibly resulting from impairments in the formation of insulin-responsive GLUT4 storage vesicles. The most plausible explanation for the impaired GLUT4 trafficking elicited by prolonged palmitate treatment is the derangement in sorting/targeting events of GLUT4 governed by sortilin, possibly contributing to the higher basal GLUT4 localization at the plasma membrane with a subsequent reduction in insulin-responsive recycling as we observed herein (Fig. 8). Given that the formation of insulin-responsive GLUT4 storage vesicles is a prerequisite for skeletal muscle cells to properly respond to the intracellular signals evoked by insulin stimulation (6), future studies are warranted to clarify the mechanism(s) underlying the impacts of sortilin reduction on impaired GLUT4 trafficking, especially on GLUT4 storage vesicle formation.
An interesting observation made in the present study was that troglitazone, a PPARγ agonist (35), but neither α nor β/δ agonist, prevented the palmitate-induced sortilin reduction and also ameliorated insulin-responsive GLUT4 recycling impairment (Fig. 8) without altering the palmitate-evoked insults on signaling cascades; neither highly phosphorylated PKCθ states (Fig. 7) nor impaired insulin-responsive Akt phosphorylation was affected (Fig. 7). These data strongly suggest that sortilin restoration, rather than signaling competency involving Akt phosphorylation, is crucial for maintaining the insulin-responsive GLUT4 recycling in skeletal muscle cells at least under the present culture conditions. In excellent agreement with this idea, it has become increasingly apparent that the most deleterious defects of skeletal muscle insulin resistance including those evoked by palmitate/high fat occur independently of the IRS signaling cascades (53, 55–57). Moreover, several lines of evidence have also demonstrated that despite its therapeutic benefits, rosiglitazone, a PPARγ agonist, failed to ameliorate the IRS signaling cascade defects in the skeletal muscles of FFA-induced insulin resistant rodents (58) as well as that in human patients with type 2 diabetes (59). On the other hand, as mentioned above, Kaddai et al. (8) recently reported that sortilin expression was significantly down-regulated in adipose and muscle tissues in obese mice and humans. Importantly, they also found that rosiglitazone prevented the sortilin reduction triggered by TNF-α treatment in cultured adipocytes (8). Taken together with the results of our present study, these compelling lines of evidence have now set the stage for the hypothesis that insulin resistance, especially as regards insulin-responsive GLUT4 translocation, could occur not only through insulin signaling defects but also via malfunction/reduction of protein(s) such as sortilin that directly controls trafficking and/or sorting of GLUT4. In addition, our data provide novel insights into the beneficial actions of PPARγ agonists, which are highly likely to exert their insulin-sensitizing actions in skeletal muscle cells via effects on multiple cellular functions (60), including sortilin restoration, perhaps in concert with the improvements in insulin signaling cascades in skeletal muscles under conditions of insulin resistance (61–63).
Finally, our findings that palmitate treatment resulted in a significant impairment in lysosomal motility strongly suggest multiple intracellular trafficking events in addition to GLUT4 trafficking to also be deranged under insulin-resistant conditions. Although the underlying mechanisms and pathogenic relevance to development of the impaired insulin-responsive GLUT4 translocation remain to be clarified, our novel findings raise the possibility that insulin-resistant states could also be characterized as intracellular trafficking defects (65) in addition to the pathophysiological conditions characterized as insulin receptor signaling defects (Fig. 10).
Given that muscle insulin resistance has been implicated as one of the earliest defects in type 2 diabetes development, characterized by decreased glucose transport and metabolism (66, 67), it is clear that further understanding of the events underlying palmitate-induced events, both sortilin down-regulation and trafficking defects, could have major implications not only for our knowledge and understanding of glucose homeostasis but also for the development of new treatment strategies and therapies for type 2 diabetes.
Acknowledgment
We thank Akito Kadotani for preliminary experiments.
This work was supported in part by grants from the Ministry of Education, Science, Sports and Culture of Japan and the New Energy and Industrial Technology Development Organization.
- IRS
- insulin receptor substrate
- PPAR
- peroxisome proliferator-activated receptor
- ADAM
- a disintegrin and metalloproteinase
- TACE
- TNF-α-converting enzyme
- TAPI-1
- TNF-α protease inhibitor-1
- FFA
- free fatty acid.
REFERENCES
- 1.Watson R. T., Kanzaki M., Pessin J. E. (2004) Endocr. Rev. 25, 177–204 [DOI] [PubMed] [Google Scholar]
- 2.Lin B. Z., Pilch P. F., Kandror K. V. (1997) J. Biol. Chem. 272, 24145–24147 [DOI] [PubMed] [Google Scholar]
- 3.Morris N. J., Ross S. A., Lane W. S., Moestrup S. K., Petersen C. M., Keller S. R., Lienhard G. E. (1998) J. Biol. Chem. 273, 3582–3587 [DOI] [PubMed] [Google Scholar]
- 4.Shi J., Kandror K. V. (2005) Dev. Cell. 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 5.Shi J., Kandror K. V. (2007) J. Biol. Chem. 282, 9008–9016 [DOI] [PubMed] [Google Scholar]
- 6.Ariga M., Nedachi T., Katagiri H., Kanzaki M. (2008) J. Biol. Chem. 283, 10208–10220 [DOI] [PubMed] [Google Scholar]
- 7.Shi J., Huang G., Kandror K. V. (2008) J. Biol. Chem. 283, 30311–30321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaddai V., Jager J., Gonzalez T., Najem-Lendom R., Bonnafous S., Tran A., Le Marchand-Brustel Y., Gual P., Tanti J. F., Cormont M. (2009) Diabetologia 52, 932–940 [DOI] [PubMed] [Google Scholar]
- 9.Felber J. P., Golay A. (2002) Int. J. Obes. Relat. Metab. Disord. 26, (Suppl. 2) S39–S45 [DOI] [PubMed] [Google Scholar]
- 10.Roden M., Price T. B., Perseghin G., Petersen K. F., Rothman D. L., Cline G. W., Shulman G. I. (1996) J. Clin. Invest. 97, 2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boden G. (2006) Curr. Diab. Rep. 6, 177–181 [DOI] [PubMed] [Google Scholar]
- 12.Schmitz-Peiffer C., Browne C. L., Oakes N. D., Watkinson A., Chisholm D. J., Kraegen E. W., Biden T. J. (1997) Diabetes 46, 169–178 [DOI] [PubMed] [Google Scholar]
- 13.Griffin M. E., Marcucci M. J., Cline G. W., Bell K., Barucci N., Lee D., Goodyear L. J., Kraegen E. W., White M. F., Shulman G. I. (1999) Diabetes 48, 1270–1274 [DOI] [PubMed] [Google Scholar]
- 14.Jové M., Planavila A., Sánchez R. M., Merlos M., Laguna J. C., Vázquez-Carrera M. (2006) Endocrinology 147, 552–561 [DOI] [PubMed] [Google Scholar]
- 15.Kim J. K., Fillmore J. J., Sunshine M. J., Albrecht B., Higashimori T., Kim D. W., Liu Z. X., Soos T. J., Cline G. W., O'Brien W. R., Littman D. R., Shulman G. I. (2004) J. Clin. Invest. 114, 823–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C., Chen Y., Cline G. W., Zhang D., Zong H., Wang Y., Bergeron R., Kim J. K., Cushman S. W., Cooney G. J., Atcheson B., White M. F., Kraegen E. W., Shulman G. I. (2002) J. Biol. Chem. 277, 50230–50236 [DOI] [PubMed] [Google Scholar]
- 17.Li Y., Soos T. J., Li X., Wu J., Degennaro M., Sun X., Littman D. R., Birnbaum M. J., Polakiewicz R. D. (2004) J. Biol. Chem. 279, 45304–45307 [DOI] [PubMed] [Google Scholar]
- 18.Hotamisligil G. S., Murray D. L., Choy L. N., Spiegelman B. M. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 4854–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rui L., Aguirre V., Kim J. K., Shulman G. I., Lee A., Corbould A., Dunaif A., White M. F. (2001) J. Clin. Invest. 107, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kadotani A., Tsuchiya Y., Hatakeyama H., Katagiri H., Kanzaki M. (2009) Am. J. Physiol. Endocrinol. Metab. 297, E1291–E1303 [DOI] [PubMed] [Google Scholar]
- 21.Nedachi T., Kanzaki M. (2006) Am. J. Physiol. Endocrinol. Metab. 291, E817–E828 [DOI] [PubMed] [Google Scholar]
- 22.Westphal V., Rizzoli S. O., Lauterbach M. A., Kamin D., Jahn R., Hell S. W. (2008) Science 320, 246–249 [DOI] [PubMed] [Google Scholar]
- 23.Peterson J. M., Wang Y., Bryner R. W., Williamson D. L., Alway S. E. (2008) Am. J. Physiol. Endocrinol. Metab. 295, E1307–E1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tolosa L., Morlá M., Iglesias A., Busquets X., Lladó J., Olmos G. (2005) Cell. Signal. 17, 1333–1342 [DOI] [PubMed] [Google Scholar]
- 25.Hermey G., Sjøgaard S. S., Petersen C. M., Nykjaer A., Gliemann J. (2006) Biochem. J. 395, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navarro V., Vincent J. P., Mazella J. (2002) Biochem. Biophys. Res. Commun. 298, 760–764 [DOI] [PubMed] [Google Scholar]
- 27.Schmitz-Peiffer C., Craig D. L., Biden T. J. (1999) J. Biol. Chem. 274, 24202–24210 [DOI] [PubMed] [Google Scholar]
- 28.Chavez J. A., Knotts T. A., Wang L. P., Li G., Dobrowsky R. T., Florant G. L., Summers S. A. (2003) J. Biol. Chem. 278, 10297–10303 [DOI] [PubMed] [Google Scholar]
- 29.Jové M., Planavila A., Laguna J. C., Vázquez-Carrera M. (2005) Endocrinology 146, 3087–3095 [DOI] [PubMed] [Google Scholar]
- 30.Coll T., Jové M., Rodríguez-Calvo R., Eyre E., Palomer X., Sánchez R. M., Merlos M., Laguna J. C., Vázquez-Carrera M. (2006) Diabetes 55, 2779–2787 [DOI] [PubMed] [Google Scholar]
- 31.Coll T., Eyre E., Rodríguez-Calvo R., Palomer X., Sánchez R. M., Merlos M., Laguna J. C., Vázquez-Carrera M. (2008) J. Biol. Chem. 283, 11107–11116 [DOI] [PubMed] [Google Scholar]
- 32.Forman B. M., Chen J., Evans R. M. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kliewer S. A., Forman B. M., Blumberg B., Ong E. S., Borgmeyer U., Mangelsdorf D. J., Umesono K., Evans R. M. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 7355–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliver W. R., Jr., Shenk J. L., Snaith M. R., Russell C. S., Plunket K. D., Bodkin N. L., Lewis M. C., Winegar D. A., Sznaidman M. L., Lambert M. H., Xu H. E., Sternbach D. D., Kliewer S. A., Hansen B. C., Willson T. M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 5306–5311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambe K. G., Tugwood J. D. (1996) Eur. J. Biochem. 239, 1–7 [DOI] [PubMed] [Google Scholar]
- 36.Preininger K., Stingl H., Englisch R., Fürnsinn C., Graf J., Waldhäusl W., Roden M. (1999) Br. J. Pharmacol. 126, 372–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibukawa A., Sawada T., Nakao C., Izumi T., Nakagawa T. (1995) J. Chromatogr. A. 697, 337–343 [DOI] [PubMed] [Google Scholar]
- 38.Sugii S., Lin S., Ohgami N., Ohashi M., Chang C. C., Chang T. Y. (2006) J. Biol. Chem. 281, 23191–23206 [DOI] [PubMed] [Google Scholar]
- 39.Watson P., Stephens D. J. (2006) J. Cell. Sci. 119, 2758–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haasch D., Berg C., Clampit J. E., Pederson T., Frost L., Kroeger P., Rondinone C. M. (2006) Biochem. Biophys. Res. Commun. 343, 361–368 [DOI] [PubMed] [Google Scholar]
- 41.Wang C., Liu M., Riojas R. A., Xin X., Gao Z., Zeng R., Wu J., Dong L. Q., Liu F. (2009) J. Biol. Chem. 284, 2038–2044 [DOI] [PubMed] [Google Scholar]
- 42.Hotamisligil G. S. (1999) Exp. Clin. Endocrinol. Diabetes 107, 119–125 [DOI] [PubMed] [Google Scholar]
- 43.Fiorentino L., Vivanti A., Cavalera M., Marzano V., Ronci M., Fabrizi M., Menini S., Pugliese G., Menghini R., Khokha R., Lauro R., Urbani A., Federici M. (2010) Hepatology 51, 103–110 [DOI] [PubMed] [Google Scholar]
- 44.Monroy A., Kamath S., Chavez A. O., Centonze V. E., Veerasamy M., Barrentine A., Wewer J. J., Coletta D. K., Jenkinson C., Jhingan R. M., Smokler D., Reyna S., Musi N., Khokka R., Federici M., Tripathy D., DeFronzo R. A., Folli F. (2009) Diabetologia 52, 2169–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Federici M., Hribal M. L., Menghini R., Kanno H., Marchetti V., Porzio O., Sunnarborg S. W., Rizza S., Serino M., Cunsolo V., Lauro D., Mauriello A., Smookler D. S., Sbraccia P., Sesti G., Lee D. C., Khokha R., Accili D., Lauro R. (2005) J. Clin. Invest. 115, 3494–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serino M., Menghini R., Fiorentino L., Amoruso R., Mauriello A., Lauro D., Sbraccia P., Hribal M. L., Lauro R., Federici M. (2007) Diabetes 56, 2541–2546 [DOI] [PubMed] [Google Scholar]
- 47.Manicassamy S., Gupta S., Sun Z. (2006) Cell. Mol. Immunol. 3, 263–270 [PubMed] [Google Scholar]
- 48.Ishaq M., DeGray G., Natarajan V. (2003) J. Biol. Chem. 278, 39296–39302 [DOI] [PubMed] [Google Scholar]
- 49.Granja A. G., Perkins N. D., Revilla Y. (2008) J. Immunol. 180, 2429–2442 [DOI] [PubMed] [Google Scholar]
- 50.Dimopoulos N., Watson M., Sakamoto K., Hundal H. S. (2006) Biochem. J. 399, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao D., Griffiths H. R., Bailey C. J. (2009) Br. J. Nutr. 102, 1557–1563 [DOI] [PubMed] [Google Scholar]
- 52.Hirabara S. M., Silveira L. R., Abdulkader F., Carvalho C. R., Procopio J., Curi R. (2007) J. Cell. Physiol. 210, 7–15 [DOI] [PubMed] [Google Scholar]
- 53.Alkhateeb H., Chabowski A., Glatz J. F., Luiken J. F., Bonen A. (2007) Am. J. Physiol. Endocrinol. Metab. 293, E783–E793 [DOI] [PubMed] [Google Scholar]
- 54.Pimenta A. S., Gaidhu M. P., Habib S., So M., Fediuc S., Mirpourian M., Musheev M., Curi R., Ceddia R. B. (2008) J. Cell. Physiol. 217, 478–485 [DOI] [PubMed] [Google Scholar]
- 55.Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., James D. E. (2008) Cell. Metab. 7, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Storgaard H., Jensen C. B., Björnholm M., Song X. M., Madsbad S., Zierath J. R., Vaag A. A. (2004) J. Clin. Endocrinol. Metab. 89, 1301–1311 [DOI] [PubMed] [Google Scholar]
- 57.Hoy A. J., Brandon A. E., Turner N., Watt M. J., Bruce C. R., Cooney G. J., Kraegen E. W. (2009) Am. J. Physiol. Endocrinol. Metab. 297, E67–E75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lessard S. J., Rivas D. A., Chen Z. P., Bonen A., Febbraio M. A., Reeder D. W., Kemp B. E., Yaspelkis B. B., 3rd, Hawley J. A. (2007) Diabetes 56, 1856–1864 [DOI] [PubMed] [Google Scholar]
- 59.Karlsson H. K., Hällsten K., Björnholm M., Tsuchida H., Chibalin A. V., Virtanen K. A., Heinonen O. J., Lönnqvist F., Nuutila P., Zierath J. R. (2005) Diabetes 54, 1459–1467 [DOI] [PubMed] [Google Scholar]
- 60.Konrad D., Rudich A., Bilan P. J., Patel N., Richardson C., Witters L. A., Klip A. (2005) Diabetologia 48, 954–966 [DOI] [PubMed] [Google Scholar]
- 61.Meyer M. M., Levin K., Grimmsmann T., Perwitz N., Eirich A., Beck-Nielsen H., Klein H. H. (2002) Diabetes 51, 2691–2697 [DOI] [PubMed] [Google Scholar]
- 62.Kim Y. B., Ciaraldi T. P., Kong A., Kim D., Chu N., Mohideen P., Mudaliar S., Henry R. R., Kahn B. B. (2002) Diabetes 51, 443–448 [DOI] [PubMed] [Google Scholar]
- 63.Miyazaki Y., He H., Mandarino L. J., DeFronzo R. A. (2003) Diabetes 52, 1943–1950 [DOI] [PubMed] [Google Scholar]
- 64.Deleted in proof
- 65.Fujita H., Hatakeyama H., Watanabe T. M., Sato M., Higuchi H., Kanzaki M. (2010) Mol. Biol. Cell 21, 2721–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saltiel A. R. (2001) Cell 104, 517–529 [DOI] [PubMed] [Google Scholar]
- 67.Petersen K. F., Shulman G. I. (2006) Am. J. Med. 119, S10–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]