

Abstract
Selective Alzheimer disease indicator-1 (seladin-1) is a broadly expressed oxidoreductase and is related to Alzheimer disease, cholesterol metabolism and carcinogenesis. The effect of lipopolysaccharide (LPS) on the expression of seladin-1 was examined using RAW 264.7 macrophage-like cells and murine peritoneal macrophages. Lipopolysaccharide induced the expression of seladin-1 protein and messenger RNA in those macrophages. The seladin-1 expression was also augmented by a series of Toll-like receptor ligands. The LPS augmented the expression of seladin-1 via reactive oxygen species generation and p38 activation. Seladin-1 inhibited LPS-induced activation of p38 but not nuclear factor-κB and inhibited the production of tumour necrosis factor-α in response to LPS. Moreover, seladin-1 inhibited LPS-induced osteoclast formation and enhanced LPS-induced alkaline phosphatase activity. Therefore, it was suggested that seladin-1 might be an LPS-responsible gene product and regulate the LPS-induced inflammatory response negatively.
Keywords: lipopolysaccharide, p38, selective Alzheimer disease indicator-1 (Seladin-1), toll-like receptor, tumour necrosis factor-α
Introduction
Seladin-1 is a human homologue of the plant DIMINUTO/DWARF1 gene, primarily described in Arabidopsis thaliana, located in the endoplasmic reticulum and, to a lesser extent, in the Golgi apparatus and expressed in all types of cells.1–5 The expression of seladin-1 in brain areas is reduced in Alzheimer's disease, hence it's name – seladin-1 (selective Alzheimer disease indicator-1).4,5 Seladin-1 is involved in cell protection from oxidative stress as a hydrogen peroxide (H2O2) scavenger.6 Seladin-1 expression is enhanced under acute stress and the augmented expression confers the resistance to H2O2-induced toxicity, which is mediated by increased cellular cholesterol biosynthesis.7 On the other hand, chronic exposure to oxidative stress diminishes seladin-1 expression, but the protective effect is still maintained through elevated p53 ubiquitination.7 Seladin-1 is also identical to the human DHCR24 complementary DNA, which is a broadly expressed oxidoreductase and catalyses the reduction of the Δ24 double bond in desmosterol to produce cholesterol.8 The mutation of seladin-1 causes desmosterolosis, a rare autosomal recessive disorder, accompanied by severe anomalies such as generalized osteosclerosis, multiple congenital malformations and neuropsychological alterations.8–10
Lipopolysaccharide (LPS) is a component of the Gram-negative bacterial cell wall and provokes a variety of inflammatory responses via excessive release of pro-inflammatory mediators.11 LPS is recognized by Toll-like receptor 4 (TLR-4) and triggers the activation of a series of signal pathways including nuclear factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs) and phosphoinositide 3 kinase.11 Subsequently LPS triggers the production of a number of pro-inflammatory mediators, such as tumour necrosis factor-α (TNF-α), interleukins and nitric oxide in macrophages.12 Lipopolysaccharide also leads to the generation of reactive oxygen species (ROS).13 We have reported that LPS induces osteoclast formation in macrophages via TNF-α production.14,15 Cross-talk between seladin-1 and LPS might therefore be presumed, although there is no report of a relationship between seladin-1 and LPS. In the present study, we examined the effect of LPS on seladin-1 expression, and the involvement of seladin-1 in LPS-induced TNF-α production and bone resorption in macrophages. We report that seladin-1 is an LPS-responsible gene product that negatively regulates LPS-induced inflammatory bone resorption.
Materials and methods
Reagents
The LPS from Escherichia coli O111, cholesterol and N-acetyl-l cysteine (NAC) were obtained from Sigma (St Louis, MO). Antibodies to p38, phosphorylated p38 (pp38), stress-activated protein kinase (SAPK/JNK), phosphorylated SAPK, extracellular signal regulated kinase (ERK 1/2), phosphorylated ERK 1/2, p53, AKT and rabbit immunoglobulin G were purchased from Cell Signalling Technology (Beverly, MA). Rabbit anti-DHCR24 (alternative symbol seladin-1) antibody was obtained from Protein Tech Group (Chicago, IL). BAY 11-7082, SB203508 and SP600125 were obtained from Calbiochem (La Jolla, CA) and nutlin-3 was purchased from Cayman Chemical (Ann Arbor, MI). Recombinant murine receptor activator of NF-κΒ ligand (RANKL) was purchased from PeproTech Inc. (Rocky Hill, NJ). Recombinant mouse macrophage colony-stimulating factor (M-CSF) was purchased from R&D Systems (Minneapolis, MN) and recombinant mouse TNF-α and H2O2 were supplied by Wako (Osaka, Japan).
Cell culture
The murine macrophage-like cell line RAW 264.7 was obtained from Riken Cell Bank (Tsukuba, Japan) and maintained in RPMI-1640 medium containing 10% heat-inactivated fetal calf serum (FCS; Gibco-BRL, Gaithersburg, MD) and antibiotics at 37° in 5% CO2. Peritoneal cells were collected from BALB/c mice (Japan SLC, Hamamatsu, Japan) 3 days after an intraperitoneal injection of 1 ml sterile 10% thioglycollate (Remel, Kansas City, MO). Thioglycollate-elicited peritoneal cells were obtained by washing out the peritoneal cavity with RPMI-1640 medium (Gibco-BRL, Gaithersburg, MD) containing 5% FCS. The cells were suspended in RPMI-1640 medium containing 5% FCS without antibiotics and incubated in a plastic dish for 5 hr. The adherent cells as peritoneal macrophages were incubated with RPMI-1640 medium containing 5% FCS and antibiotics at 37° in a humidified 5% CO2 incubator for 24 hr. The animal experiments were carried out following the Guide for Care and Use of Laboratory Animals, Aichi Medical University.
Transfection of small interfering (si) RNA
Seladin-1-specific siGENOME 1 targets plus SMART pool and a non-targeting siRNA were obtained from Dharmacon (Chicago, IL). RAW 264.7 cells were seeded at a concentration of 2 × 105 cells/well in a 48-well plate in growth medium without antibiotics. Cationic lipid complexes were prepared by incubating 200 or 400 nm siRNA with 3 μl Hiperfect transfection reagent (Qiagen, Hilden, Germany) in 50 μl medium without FCS and were then added to the cells. After 8 hr of incubation, the cells were further cultured with fresh growth medium (200 μl) without antibiotics for 2 days. The efficiency of the seladin-1 silencing was evaluated by immunoblotting and real-time polymerase chain reaction (PCR).
Immunoblotting
Immunoblotting was performed as described elsewhere.16 In brief, the cell lysates were extracted by lysis buffer and were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis using a 10% gel under reducing conditions. The proteins were electrically transferred to a membrane and the membranes were treated with various antibodies, followed by horseradish peroxidase-conjugated secondary antibody. The protein bands were visualized using a chemiluminescence reagent (Pierce, Rockford, IL). For re-probing, the membranes were stripped with the solution containing 2% sodium dodecyl sulphate, 62·5 mm Tris–HCl at pH 6·8, 100 mm 2-mercaptoethanol at 50° for 30 min and treated with corresponding antibodies. The molecular sizes of the antigens were determined by comparison with a pre-stained protein size marker kit (Invitrogen, Carlsbad, CA).
NF-κB activation
Binding of NF-κB p65 subunit to the NF-κB binding consensus sequence 5′-GGGACTTTCC-3′ was determined with the enzyme-linked immunosorbent assay (ELISA) -based Trans AM NF-κB p65 kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. Briefly, nuclear extracts were prepared using a nuclear extract kit (Active Motif) and applied to the Trans-AM kit 96-well microplate coated with an oligonucleotide containing the NF-κB binding consensus sequence. The active form of the NF-κB p65 subunit was detected using the manufacturer's instruction that it is accessible only when the subunit is activated and bound to its target DNA. Results are expressed as activation value of p65, which was determined with the absorbance at 450 nm. The phosphorylation of NF-κB p65 was also analysed by immunoblotting.
Determination of TNF-α and interleukin-6 production
RAW 264.7 cells were cultured with LPS at 100 ng/ml for 6 and 24 hr. The concentration of TNF-α in the culture supernatant was determined by ELISA (R&D Systems, Minneapolis, MN). In some experiments, the cells were pre-treated with cholesterol (10 μg/ml) for 6 hr or nutlin-3 (2·5 μm) for 1 hr before LPS stimulation. The concentration of interleukin-6 (IL-6) in the culture supernatant was also determined using ELISA (BioSource, Camarillo, CA).
Real-time PCR
Quantitative real-time PCR was performed as described elsewhere.16 RNA was extracted from cells using the RNeasy mini kit (Qiagen, Chatsworth, CA). RNA was reverse-transcribed in ReverTra Ace (Toyobo, Osaka, Japan) with a three-step incubation according to the manufacturer's instructions, and quantitative PCR was carried out using the SYBR green real-time PCR master mix (Toyobo) following the manufacturer's instructions. The primers were designed as follows: seladin-1 (sense 5′-CACAGGCATCGAGTCATCGT-3′, anti-sense 5′-GGCACGGCATAGAACAGGTC-3′), TNF-α (sense: 5′-TGTTGCCTCCTCTTTTGCTT-3′ anti-sense: 5′-TGGTCACCAA-AATCAGCGTTA-3′); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (sense: 5′-TGAAGCAGGCATCTGAGGG-3′, anti-sense: 5′-CGAAGGTGGAAGAGTGGGAG-3′) (Invitrogen). The PCR was performed with an ABI PRISM 7700 sequence detection system (Applied Biosystems, Hamilton, New Zealand) and the PCR conditions were as follows: 95° for 10 min and 40 cycles at 95° for 30 seconds, 60° for 1 min. Relative quantitative values of seladin-1 and TNF-α expression were normalized using the expression levels of the reference gene GAPDH.
Tartrate-resistant acid phosphatase staining
Tartrate-resistant acid phosphatase (TRAP) staining was carried out to identify osteoclasts. Briefly, the cells were washed with phosphate-buffered saline and treated with a fixation solution at room temperature for 5 min. The cells were washed with distilled water and stained with TRAP reagent (Wako, Osaka, Japan) at 37° for 60 min. After washing with distilled water, the cells were observed under a microscope. The images were taken with a digital camera attached to a microscope. More than 150 cells were counted microscopically for the frequency of TRAP-positive cells. In some experiments, cells were pre-incubated with recombinant TNF-α (25 ng/ml) for 1 hr before LPS treatment.
Alkaline phosphatase assay
The alkaline phosphatase (ALP) activity was determined as described elsewhere.15 In brief, RAW 264.7 cells were stimulated with 100 ng/ml LPS in a 96-well plate for 2 days. An enzyme assay solution (200 μl) containing 8 mmp-nitrophenyl phosphate, 12 mm MgCl2 and 0·1 mm ZnCl2 in 0·1 m glycine–NaOH buffer at pH 10·5 were added to each well and the plate was incubated at 37° for 10 min. The enzyme reaction was terminated by 0·5 m NaOH and the amount of p-nitrophenol released was determined with the absorbance at 405 nm.
Statistical analysis
All the experiments were performed at least three times independently. Experimental data are expressed as the mean of triplicates ± standard deviation in at least three independent experiments. Statistical analysis based on Student's t-test was carried out for comparisons between two experiments. A value of P < 0·01 was considered statistically significant.
Results
LPS induces the expression of seladin-1 in RAW264.7 cells and mouse peritoneal macrophages
The effect of LPS on the expression of seladin-1 in RAW264.7 cells was examined. The RAW 264.7 cells were stimulated with various concentrations of LPS for 4 hr and the expression of seladin-1 protein was determined by immunoblotting (Fig. 1a). Seladin-1 was only marginally expressed in non-treated control cells. LPS at 1 and 10 ng/ml slightly augmented the seladin-1 expression and LPS at 100 ng/ml induced the highest expression of seladin-1. On the other hand, LPS at 1000 ng/ml reduced the seladin-1 expression. RAW264.7 cells were stimulated with LPS (100 ng/ml) for various hours and the time–course of the seladin-1 expression was followed (Fig. 1b). Seladin-1 expression increased 1 hr after LPS stimulation, reached a peak at 4 hr, and then declined gradually. The LPS-induced seladin-1 messenger RNA (mRNA) expression was examined by real-time PCR (Fig. 1c). The expression of seladin-1 mRNA was significantly augmented 1 hr after LPS stimulation and increased up to 4 hr. Further, the effect of LPS on the expression of seladin-1 was examined using mouse peritoneal macrophages (Fig. 2). Administration of LPS at 100 ng/ml induced the highest expression of seladin-1 in peritoneal macrophages as well as RAW 264.7 cells (Fig. 2a). The seladin-1 expression was augmented 2 and 4 hr after the stimulation with LPS (Fig. 2b).
Figure 1.
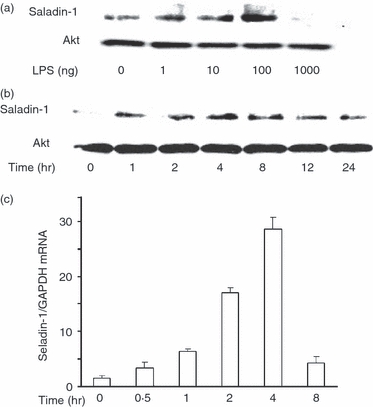
Induction of seladin-1 expression by lipopolysaccharide (LPS). (a) RAW 264.7 cells were stimulated with various concentrations of LPS for 4 hr. (b) RAW 264.7 cells were stimulated with LPS (100 ng/ml) for various lengths of time. The expression of seladin-1 protein was determined with immunoblotting. (c) RAW 264.7 cells were stimulated with LPS (100 ng/ml) for 4 hr. The expression of seladin-1 messenger RNA was determined with real-time polymerase chain reaction. A typical experiment of three independent experiments is shown.
Figure 2.
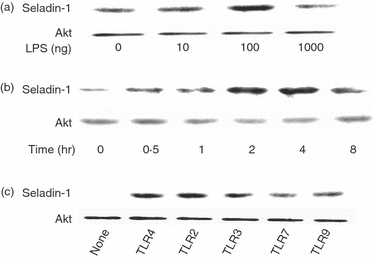
Induction of seladin-1 by lipopolysaccharide (LPS) and various Toll-like receptor (TLR) ligands. (a) Murine peritoneal macrophages were stimulated with various concentrations of LPS for 4 hr. (b) Murine peritoneal macrophages were stimulated with LPS (100 ng/ml) for various lengths of time. (c) RAW 264.7 cells were stimulated with LPS (100 ng/ml), Pam3CysSK4 (10 μg/ml), poly I : C (10 μg/ml), imiquimod R837 (10 μg/ml) and CpG DNA (10 μm) as TLR-4, -2 -3, -7 and -9 ligands, respectively, for 4 hr. The expression of seladin-1 protein was determined with immunoblotting. A typical experiment of three independent experiments is shown.
In addition, the effect of other TLR ligands on the seladin-1 expression was examined (Fig. 2c). RAW 264.7 cells were stimulated with LPS (100 ng/ml), Pam3CysSK4 (10 μg/ml), poly I : C (10 μg/ml), imiquimod R837 (10 μg/ml) and CpG DNA (10 μm) as TLR-4, -2, -3, -7 and -9 ligands, respectively, for 4 hr and the seladin-1 expression was determined by immunoblotting. The LPS and Pam3CysSK4 induced higher expression of seladin-1 than poly I: C, imiquimod R837 and CpG DNA.
LPS triggers the expression of seladin-1 through ROS generation and p38 activation
To clarify the signals triggering LPS-induced seladin-1 expression, the effect of NAC as a ROS scavenger17 or SB203580 as a p38 pharmacological inhibitor on LPS-induced seladin-1 expression was examined. RAW 264.7 cells were stimulated with LPS (100 ng/ml) for 2 hr in the presence or absence of NAC (15 mm) or SB203580 (10 μm). The seladin-1 expression was determined by immunoblotting. The addition of NAC or SB203580 completely abolished LPS-induced seladin-1 expression (Fig. 3a). Although the effect of a series of signal inhibitors on LPS-induced seladin-1 expression was examined, none of them, including BAY 11-7082 (10 μm) as an NF-κB inhibitor and SP600125 (10 μm) as an SAPK/JNK inhibitor exhibited an inhibitory action on it (data not shown). Moreover, RAW 264.7 cells were pre-incubated with 25 μm H2O2 for 1 hr and then treated with LPS 100 ng/ml for 2 hr. Treatment with H2O2 significantly augmented the seladin-1 expression in the presence or absence of LPS (Fig. 3b), suggesting that ROS as well as p38 augments the seladin-1 expression.
Figure 3.
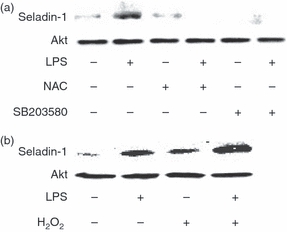
Involvement of reactive oxygen species (ROS) and p38 in lipopolysaccharide (LPS)-induced seladin-1 expression. (a) RAW 264.7 cells were pretreated with N-acetyl-l cysteine (NAC; 15 mm) as a ROS inhibitor or SB203580 (10 μm) as a p38 inhibitor for 2 hr and stimulated with LPS (100 ng/ml) for 4 hr. (b) RAW 264.7 cells were pretreated with H2O2 (25 μm) for 1 hr and stimulated with LPS (100 ng/ml) for 2 hr. The expression of seladin-1 protein was determined with immunoblotting. A typical experiment of three independent experiments is shown.
Seladin-1 inhibits LPS-induced p38 activation
In the preceding section we demonstrated that LPS augmented the seladin-1 expression via ROS generation and p38 activation. On the other hand, seladin-1 is reported to augment H2O2-induced p38 and SAPK/JNK activation via an unknown mechanism.6 It is possible that seladin-1 might affect the activation of a series of MAPKs in response to LPS as well as H2O2. RAW 264.7 cells were transfected with seladin-1 siRNA and stimulated with LPS (100 ng/ml) for 1 hr and the phosphorylation of p38, SAPK/JNK and ERK1/2 was determined by immunoblotting. First, we confirmed the silencing of the seladin-1 protein expression by seladin-1 siRNA (Fig. 4a). By using the effective seladin-1 siRNA, the effect of seladin-1 siRNA on the phosphorylation of p38, SAPK/JNK and ERK1/2 was examined in LPS-stimulated RAW 264.7 cells (Fig. 4b). Introduction of seladin-1 siRNA augmented the phosphorylation of p38 but not SAPK/JNK and ERK1/2 in response to LPS (Fig. 4b). On the other hand, the negative control siRNA failed to do this.
Figure 4.
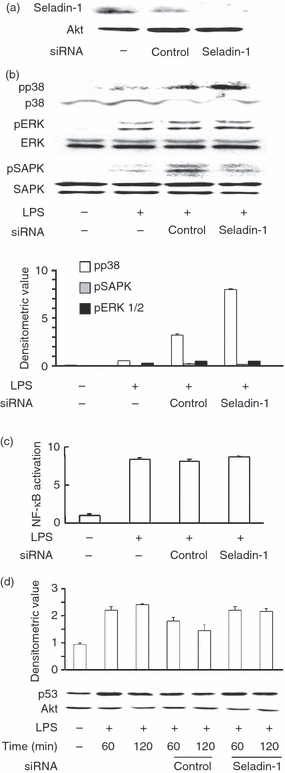
Effect of seladin-1 small interfering (si) RNA on lipopolysaccharide (LPS) -induced p38 activation. RAW 264.7 cells were transfected with seladin-1 siRNA or negative control siRNA for 48 hr and stimulated with LPS (100 ng/ml) for 1 hr. The expression of seladin-1 protein (a) and phosphorylated p38 (pp38), pERK1/2, pSAPK/JNK (b) were determined with immunoblotting. (c) The transfected cells were stimulated with LPS (100 ng/ml) for 30 min. The binding of the activated p65 nuclear factor-κB (NF-κB) subunit to an NF-κB consensus sequence was determined with the NF-κB DNA binding kit. (d) The transfected cells were stimulated with LPS (100 ng/ml) for 60 and 120 min. The expression of p53 protein was determined with immunoblotting. Relative quantification of immunoblots was analysed by densitometry and the result is shown as the mean of three experiments with SD (b,d).
Next, the effect of seladin-1 siRNA on LPS-induced NF-κB activation was examined by the phosphorylation and DNA-binding activity of p65 NF-κB. Seladin-1 siRNA did not alter the phosphorylation (data not shown) and DNA-binding activity of NF-κB (Fig. 4c). Further, seladin-1 is reported to stabilize p53 protein independent of its oxidoreductase activity and to accumulate p53 through displacing the E3 ubiquitin ligase Mdm2 from p53,17 and another recent report suggested that hepatitis C virus-mediated persistent over-expression of seladin-1 suppresses p53 activity.18 Therefore, the effect of seladin-1 siRNA on the expression of p53 was examined. There was no significant difference in LPS-induced p53 accumulation between seladin-1 siRNA and control siRNA (Fig. 4d).
Seladin-1 inhibits the production of TNF-α in LPS-stimulated RAW264.7 cells
As seladin-1 had been found to inhibit LPS-induced p38 activation (Fig. 4b) and because LPS-induced TNF-α production is dependent on the activation of p38 as well as NF-κB,11,12,19 the effect of seladin-1 siRNA on LPS-induced TNF-α production was examined. Seladin-1 or control siRNA-transfected cells were stimulated with LPS (100 ng/ml) for 6 or 24 hr. Introduction of seladin-1 siRNA into the cells augmented the production of TNF-α in response to LPS (Fig. 5a) whereas introduction of control siRNA did not. The TNF-α production was also augmented in seladin-1 siRNA-transfected cells 24 hr after LPS stimulation (Fig. 5b). Subsequently, the effect of seladin-1 siRNA on LPS-induced TNF-α mRNA expression was examined (Fig. 5c). Seladin-1 siRNA significantly augmented the expression of TNF-α mRNA in LPS-stimulated cells, suggesting that seladin-1 inhibited LPS-induced TNF-α production via p38 inactivation. Seladin-1 or control siRNA-transfected cells were pre-incubated with cholesterol or nutlin-3, which inhibits p53-Mdm2 interaction, and then stimulated with LPS (100 ng/ml) for 6 hr. The addition of cholesterol and nutlin-3 failed to inhibit seladin-1-induced TNF-α augmentation, suggesting no involvement of cholesterol and p53 in the augmentation of TNF-α production by seladin-1 (data not shown). The effect of seladin-1 siRNA on LPS-induced IL-6 production was also examined (Fig. 5d). Introduction of seladin-1 siRNA resulted in the augmented production of IL-6 in response to LPS.
Figure 5.
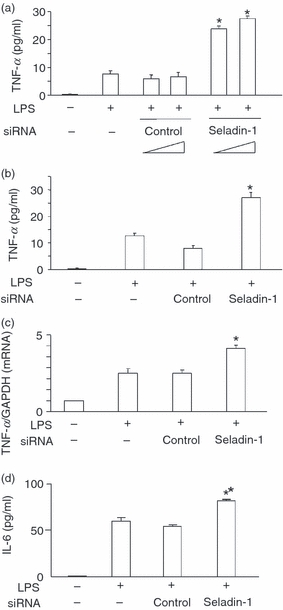
Effect of seladin-1 small interfering (si) RNA on lipopolysaccharide (LPS) -induced tumour necrosis factor (TNF-α) production. RAW 264.7 cells were transfected with seladin-1 siRNA or negative control siRNA for 48 hr and the transfected cells were stimulated with LPS (100 ng/ml) for 6 (a) and 24 hr (b). (c) The level of TNF-α in the culture supernatant was determined by enzyme-linked immunosorbent assay (ELISA). Transfected or control cells were stimulated with LPS (100 ng/ml) for 1 hr. The expression of TNF-α messenger RNA was determined with real-time polymerase chain reaction. (d) Transfected or control cells were stimulated with LPS (100 ng/ml) for 6 h and the level of interleukin-6 (IL-6) in the culture supernatant was determined with ELISA. *P <0·01 versus control siRNA.
Seladin-1 augments LPS-induced osteoclast formation and bone resorption
Lipopolysaccharide-induced osteoclast formation and survival exclusively depend on TNF-α production and TNF-α plays a central role in the pathogenesis of LPS-induced bone resorption.14,15 Since seladin-1 siRNA-transfected cells produced a higher amount of TNF-α in response to LPS (Fig. 5), seladin-1 might affect LPS-induced osteoclast formation. Therefore, the effect of seladin-1 siRNA on LPS-induced osteoclast formation was examined (Fig. 6a). Seladin-1 or control siRNA-transfected cells were stimulated with LPS (100 ng/ml) for 48 hr and the frequency of osteoclast formation was determined with TRAP staining. LPS induced the higher frequency of TRAP+ cells (osteoclasts) in seladin-1 siRNA-transfected cells than control siRNA-transfected cells, suggesting the negative regulation of LPS-induced osteoclast formation by seladin-1. The addition of exogenous TNF-α (25 ng/ml) further increased the frequency of osteoclasts appearing in LPS-induced osteoclast formation (approximately 14%) and the value was almost the same as that of LPS-stimulated seladin-1-siRNA-transfected cells (15%, Fig. 6a).
Figure 6.
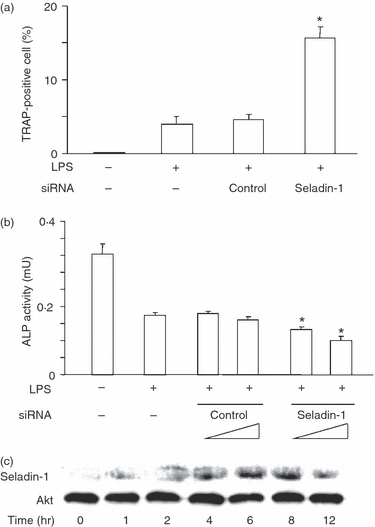
Effect of seladin-1 small interfering (si) RNA on lipopolysaccharide (LPS) -induced osteoclast formation and alkaline phosphatase (ALP) activity. RAW 264.7 cells were transfected with seladin-1 siRNA or negative control siRNA for 48 hr and the transfected cells were stimulated with LPS (100 ng/ml) for 48 hr. (a) The frequency of osteoclasts was determined with tartrate-resistant acid phosphatase staining. (b) ALP activity was measured using an enzyme assay using p-nitrophenol as the substrate. *P <0·01 versus control siRNA. (c) RAW 264.7 cells were stimulated with receptor activator of nuclear factor-κΒ ligand (RANKL; 100 ng/ml) and macrophage colony-stimulating factor (M-CSF; 25 ng/ml) for various lengths of time. The expression of seladin-1 protein was determined with immunoblotting (d). A typical experiment of three independent experiments is shown.
The effect of seladin-1 on the ALP activity as a marker of bone resorption in LPS-simulated RAW 264.7 cells was examined (Fig. 6b). RAW 264.7 cells were transfected with seladin-1 or control siRNA and then stimulated with LPS (100 ng/ml) for 48 hr. The ALP activity in seladin-1 siRNA-transfected cells was lower than in control siRNA-transfected cells.
The effect of RANKL and M-CSF on the seladin-1 expression was examined. RAW 264.7 cells were treated with RANKL (100 ng/ml) and M-CSF (25 ng/ml) for various lengths of time. The seladin-1 expression significantly increased 2 hr after treatment, reached a peak at 6–8 hr and declined gradually (Fig. 6c).
Discussion
In the present study we demonstrate that seladin-1 is an LPS-responsive gene and negatively regulates the TNF-α production and bone resorption in response to LPS. The LPS induces the expression of seladin-1 in macrophages and the augmented seladin-1 expression inhibits LPS-induced inflammatory responses, such as TNF-α and IL-6 production. Therefore, seladin-1 is a negative regulator of LPS signalling. Moreover, other TLR ligands augment the expression of seladin-1 in macrophages. Therefore, seladin-1 is suggested to be a TLR-responsive gene and negatively regulates the TLR-induced inflammatory response. Hence, seladin-1 might play a critical role in the feedback regulation of innate immunity.
Oncogenic ras and oxidative stress (H2O2) are reported to induce the expression of seladin-1 via the ROS signal pathway.17 This was also confirmed by the current study. The ROS signal pathway is at present the only established signal pathway for seladin-1 activation in carcinogenesis.17 On the other hand, the present study demonstrates the involvement of p38 as well as ROS in LPS-induced seladin-1 expression. Lipopolysaccharide causes the ROS generation13 and generated ROS is known to result in the activation of oxidative stress-related p38 signal pathway.20 It is reasonable that LPS-induced seladin-1 expression requires the activation of both ROS and p38 signal pathways. On the other hand, only ROS signalling seems to be required for carcinogenesis.17 The seladin-1 gene expression might require each different signal pathway in response to a variety of stimuli.
Lipopolysaccharide-induced TNF-α production is regulated by p38, SAPK/JNK and ERK1/2 MAPK pathways as well as the NF-κB pathway.21 Blocking one of three MAPK pathways including ERK1/2, SAPK/JNK and p38 is sufficient to inhibit the TNF-α production in response to LPS.19,21 Seladin-1 is shown to inhibit p38 activation in the present study. Therefore, it is reasonable that seladin-1 inhibits LPS-induced TNF-α production via reduced p38 activation. In addition, we demonstrate no involvement of NF-κB, SAPK/JNK and ERK1/2 in the inhibition of LPS-induced TNF-α production by seladin-1. The involvement of p53 is also excluded.
The mutation of seladin-1 protein causes desmosterolosis, a rare autosomal recessive disorder.8–10 Patients with desmosterolosis are characterized by generalized osteosclerosis, abnormal bone formation, multiple congenital malformations and severe neuropsychological alterations.8–10 The present study demonstrates that seladin-1 down-regulates osteoclast formation and bone resorption in LPS-stimulated macrophages and that RANKL and M-CSF augment seladin-1 expression. The LPS-induced osteoclast formation is dependent on TNF-α14,15 and it is further augmented by exogenous TNF-α. Considering that the LPS-induced TNF-α production is negatively regulated by seladin-1, seladin-1 is suggested to inhibit LPS-induced osteoclast formation and bone resorption via reduced TNF-α production. It might be of interest to study the role of TNF-α on the pathogenesis of desmosterolosis, although our experimental system does not necessarily correspond to the pathogenesis of patients with desmosterolosis.
Many proteins control TLR signalling to ensure the appropriate strength and duration of immune responses in innate immunity. The TLR signalling is regulated by signal proteins including IRAKM, ST2, SIGIRR, SOCS1, the tumour suppressor CYLD and A20.22 Cells lacking one of those proteins produce a higher level of pro-inflammatory cytokines on TLR stimulation.22 Aberrant activation of TLR signalling may contribute to abnormal osteoclast formation, rheumatoid arthritis, inflammatory bowel disease and systemic lupus erythematosus.23–25 Although various proteins have been identified as negative regulators of TLR signalling, few mice lacking one of these proteins die shortly after birth.26 However, the mice lacking SOCS1 or A20 have severe immune disorders that lead to premature death.26,27 Seladin-1-deficient mice might undergo multiorgan inflammation and be useful for osteoclast function analysis.
The present study demonstrates that seladin-1 is an LPS-responsible gene and that it is a negative regulator of LPS signalling. Moreover, the seladin-1 might be a general negative regulator of TLR signalling. It may provide a new insight in the prevention of inflammatory responses caused by infection with various micro-organisms. Seladin-1 is likely to become a rational target for therapeutic regimens against inflammatory responses caused by various infectious agents.
Acknowledgments
We are grateful to Kazuko Takahashi and Akiko Morikawa for their technical assistance. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
Glossary
Abbreviations:
- ALP
alkaline phosphatase
- ELISA
enzyme-linked immunosorbent assay
- ERK
extracellular signal-regulated kinase
- FCS
fetal calf serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- H2O2
hydrogen peroxide
- IL-6
interleukin-6
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- M-CSF
macrophage colony-stimulating factor
- mRNA
messenger RNA
- NAC
N-acetyl-l cysteine
- NF-κB
nuclear factor-κB
- PCR
polymerase chain reaction
- RANKL
receptor activator of NF-κΒ ligand
- ROS
reactive oxygen species
- Seladin-1
Selective Alzheimer disease indicator-1
- siRNA
small interfering RNA
- TLR
Toll-like receptor
- TNF-α
tumour necrosis factor-α
- TRAP
tartrate-resistant acid phosphatase
Disclosures
The authors declare no conflict of interest.
References
- 1.Takahashi T, Gasch A, Nishizawa N, Chua NH. The DIMINUTO gene of Arabidopsis is involved in regulating cell elongation. Genes Dev. 1995;9:97–107. doi: 10.1101/gad.9.1.97. [DOI] [PubMed] [Google Scholar]
- 2.Bishop GJ, Nomura T, Yokota T, et al. The tomato DWARF enzyme catalyses C-6 oxidation in brassinosteroid biosynthesis. Proc Natl Acad Sci USA. 1999;96:1761–6. doi: 10.1073/pnas.96.4.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T, Behl C, Levkau B, Nitsch RM. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer's disease-associated neurodegeneration and oxidative stress. J Neurosci. 2000;20:7345–52. doi: 10.1523/JNEUROSCI.20-19-07345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peri A, Danza G, Serio H. Seladin-1 as a target of estrogen receptor activation in the brain: a new gene for a rather old story? J Endocrinol Invest. 2005;28:285–93. doi: 10.1007/BF03345387. [DOI] [PubMed] [Google Scholar]
- 5.Peri A, Serio M. Neuroprotective effects of the Alzheimer's disease-related gene seladin-1. J Mol Endocrinol. 2008;41:251–61. doi: 10.1677/JME-08-0071. [DOI] [PubMed] [Google Scholar]
- 6.Lu X, Kambe F, Cao X, Kozaki Y, Kaji T, Ishii T, Seo H. DHCR24 is a hydrogen peroxide scavenger, protecting cells from oxidative-stress-induced apoptosis. Endocrinology. 2008;149:3267–73. doi: 10.1210/en.2008-0024. [DOI] [PubMed] [Google Scholar]
- 7.Kuehnle K, Crameri A, Kälin RE, et al. Prosurvival effect of DHCR24/Seladin-1 in acute and chronic responses to oxidative stress. Mol Cell Biol. 2008;28:539–50. doi: 10.1128/MCB.00584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waterham HR, Koster J, Romeijn GJ, et al. Mutations in the 3beta-hydroxysterol delta 24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–94. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.FitzPatrick DR, Keeling JW, Evans MJ, et al. Clinical phenotype of desmosterolosis. Am J Med Genet. 1998;75:145–52. [PubMed] [Google Scholar]
- 10.Clayton P, Mills K, Keeling J, FitzPatrick D. Desmosterolosis: a new inborn error of cholesterol biosynthesis. Lancet. 1996;10:404. doi: 10.1016/s0140-6736(05)65020-9. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. Pathogen recognition with Toll- like receptor. Curr Opin Immunol. 2005;17:338–44. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–9. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Feuillet-Fieux MN, Golub RM, Nguyen AT, Zamfirescu P, Descamps-Latscha B. Effect of LPS on the oxidative metabolism of peritoneal and spleen cells from LPS sensitive and resistant mice. J Clin Lab Immunol. 1984;15:155–61. [PubMed] [Google Scholar]
- 14.Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J Clin Invest. 1997;15:1557–65. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Islam S, Hassan F, Tumurkhuu G, et al. Bacterial lipopolysaccharide induces osteoclast formation in RAW 264.7 macrophage cells. Biochem Biophys Res Commun. 2007;24:346–51. doi: 10.1016/j.bbrc.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 16.Iftakhar-E-Khuda I, Koide N, Hassan F, et al. Novel mechanism of U18666A-induced tumor necrosis factor-alpha production in RAW264.7 macrophage cells. Clin Exp Immunol. 2009;155:552–8. doi: 10.1111/j.1365-2249.2008.03779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxidative stress by seladin-1. Nature. 2004;432:640–5. doi: 10.1038/nature03173. [DOI] [PubMed] [Google Scholar]
- 18.Nishimura T, Kohara M, Izumi K, et al. Hepatic C virus impairs p53 via persistent over-expression 3β-hydroxysterol Δ24-reductase. J Biol Chem. 2009 doi: 10.1074/jbc.M109.043232. doi: 10.1074/jbc.M109.043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1:94–7. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzawa A, Saegusa K, Noguchi T, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–92. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 21.Dumitru CD, Ceci JD, Tsatsanis C, et al. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK dependent pathway. Cell. 2007;103:1071–83. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 22.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 23.Nagasawa T, Kiji M, Yashiro R, et al. Roles of receptor activator of nuclear factor-kappaB ligand (RANKL) and osteoprotegerin in periodontal health and disease. Periodontol 2000. 2007;43:65–84. doi: 10.1111/j.1600-0757.2006.00185.x. [DOI] [PubMed] [Google Scholar]
- 24.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–8. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 25.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–35. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. 1998;95:14395–9. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]