

Abstract
Loss of the PTEN tumor suppressor gene occurs frequently in non-small-cell lung carcinoma (NSCLC), although neither genetic alterations nor epigenetic silencing are significant predictors of PTEN protein levels. Since recent reports implicated neural precursor cell expressed, developmentally down-regulated 4-1 (NEDD4-1) as the E3 ubiquitin ligase that regulates PTEN stability, we investigated the role of NEDD4-1 in the regulation of PTEN expression in cases of NSCLC. Our findings indicate that NEDD4-1 plays a critical role in the development of NSCLC and provides novel insight on the mechanisms that contribute to inactivate PTEN in lung cancer. Immunohistochemical analysis on tissue microarrays containing 103 NSCLC resections revealed NEDD4-1 overexpression in 80% of tumors, which correlated with the loss of PTEN protein (n = 98; P < 0.001). Accordingly, adoptive NEDD4-1 expression in NSCLC cells decreased PTEN protein stability, whereas knock-down of NEDD4-1 expression decreased PTEN ubiquitylation and increased PTEN protein levels. In 25% of cases, NEDD4-1 overexpression was due to gene amplification at 15q21. In addition, manipulation of NEDD4-1 expression in different lung cell systems demonstrated that suppression of NEDD4-1 expression significantly reduced proliferation of NSCLC cells in vitro and tumor growth in vivo, whereas NEDD4-1 overexpression facilitated anchorage-dependent and independent growth in vitro of nontransformed lung epithelial cells that lack pRB and TP53 (BEAS-2B). NEDD4-1 overexpression also augmented the tumorigenicity of lung cancer cells that have an intact PTEN gene (NCI-H460 cells).
Lung cancer, the commonest cause of cancer mortality worldwide, comprises two different groups, small cell lung cancer and non-small-cell lung cancer (NSCLC), the latter comprising three major histological subtypes: adenocarcinoma (ADC), squamous cell carcinoma (SCC), and large cell lung cancer.1,2,3
At the molecular level, the activation of the phosphatidylinositol 3-kinase (PI3K)/PTEN/AKT pathway plays a critical role both in the initiation and progression of NSCLC.4,5,6 Accordingly, amplification and/or activating mutations in PI3K or AKT1 genes or the loss of PTEN that lead to the constitutive activation of PI3K have been reported in NSCLC.7,8,9
The PTEN gene, located at 10q23.3, is a tumor suppressor that encodes a lipid phosphatase that antagonizes the action of phosphatidylinositol 3-kinase by dephosphorylating the second messenger phosphatidylinositol 3,4,5-triphosphate.10,11 Loss of PTEN expression occurs frequently in lung cancer and represents an independent poor prognostic factor for patients with NSCLC.12,13 PTEN expression is reduced or lost in a high fraction of NSCLC (55% to 74%), though neither genetic alterations nor epigenetic silencing are significant predictors of PTEN protein expression in NSCLC.14,15 Mutations of the PTEN gene in patients with NSCLC have been reported in 8% to 17% of NSCLC,16,17,18,19 suggesting that PTEN is infrequently targeted at the genetic level during the development of lung cancer. In addition, loss of heterozygosity at microsatellites surrounding and intragenic to the PTEN locus on chromosome 10q23 occurs in ∼20% of informative tumors,15 and epigenetic phenomena like promoter methylation occur at low frequency (up to 26%).14 These considerations leave open the possibility that poorly defined posttranslational mechanisms may take part in PTEN inactivation in NSCLC.
Previous studies indicated that PTEN undergoes multiple posttranslational modifications20,21 and is degraded by the ubiquitin-pathways.22,23 Recently, neural precursor cell expressed, developmentally down-regulated 4-1 (NEDD4-1) was identified as the E3-ubiquitin ligase that promotes ubiquitin-mediated PTEN degradation.24 NEDD4-1 is the prototypical protein in a family of E3 ubiquitin ligases that have a catalytic C-terminal HECT domain and N-terminal C2 and WW domains responsible for substrate recognition.25 It was shown that NEDD4-1 cooperates with K-RAS in transforming mouse embryonic fibroblasts in a PTEN-dependent manner.24 However, recent work failed to find a role for NEDD4-1 in the regulation of PTEN stability in mouse tissues.26
The aim of the present study was to investigate the role of NEDD4-1 in lung cancer with regard to its effects on the PI3K/AKT/PTEN pathway. The data reported here demonstrate that NEDD4-1 is strongly implicated in the onset/progression of lung cancer since it is overexpressed in a significant fraction of NSCLC, promotes PTEN degradation, and increases malignant properties of lung epithelial cells, providing new insights in the molecular pathogenesis of lung carcinomas and identifying NEDD4-1 as a potential therapeutic target for NSCLC.
Materials and Methods
Clinical Samples
Surgical samples from 103 patients given a diagnosis of NSCLC27 were obtained from Monaldi Hospital (Naples, Italy) and INT Fondazione Pascale (Naples, Italy). Median age was 64 years old (range, 28 to 82); there were 20 women and 83 men. Stage was known for 92 patients: 71 patients had stage I to II disease and 21 had stage III to IV disease. Grade was known for 94 patients: seven were G1, 32 were G2, and 55 were G3. The study was approved by the Internal Ethical Committee.
Tissue Microarray
Tissue microarrays (TMAs) were constructed in collaboration with the Unit of Immunostaining at the Centro Nacional de Investigaciones Oncologicas (Madrid, Spain). Microarray recipient block containing paraffin-embedded NSCLC tissue samples from 103 archival patient specimens was constructed according to established methods.28 From every archival paraffin block, two 0.6-mm cylinders were taken from representative areas and transferred to a paraffin recipient block by using a Tissue Arrayer (Beecher Instruments, Gene Micro-Array Technologies, Silver Spring, MD). One cylinder of normal tissue adjacent to each tumor was also transferred to the recipient block. Finally, sections of the resulting tumor tissue microarray block (4-μm thick) were transferred to glass slides.
Cell Culture
Lung cancer cell lines were purchased from ATCC-LGC Promochem (South West London, UK): SCC (BEN-1, NCI-H292, NCI-H226, Calu-1); ADC (A549, NCI-H596, NCI-H2009, NCI- H23, NCI-H522); and large cell carcinoma (LCC; NCI-H460). Cells were maintained in RPMI 1640 (Gibco-Invitrogen Cell Culture, Carlsbad, CA), supplemented with 10% of fetal bovine serum and 100 units/ml penicillin-streptomycin (Life Technologies, Inc., Carlsbad, CA). Normal human bronchial epithelial cells and nontumorigenic human lung cells (BEAS-2B)29 were purchased from Cambrex (Milan, Italy) and grown as suggested by the manufacturer.
Quantitative Reverse Transcription Real-Time PCR and Quantitative Real-Time
Total RNA and genomic DNA were prepared as described.30,31 Quantitative RT-PCR and quantitative PCR (Q-PCR) were performed by using the Power SYBR Green PCR Master Mix in an ABI Prism 7300 thermocycler (Applied Biosystems, Foster City, CA) according to the manufacturer’s instruction. Primers used in quantitative RT-PCR were as follows: NEDD4-1 forward: 5′-GGTGGAGGTGTTCGGGCT-3′, NEDD4-1 reverse: 5′-GCAAGGCCTATTCCGGCTA-3′; glyceraldehyde-3-phosphate dehydrogenase forward: 5′-GAGTCAACGGATTTGGTCGT-3′, glyceraldehyde-3-phosphate dehydrogenase reverse: 5′-GACAAGCTTCCCGTTCTCAG-3′. NEDD4-1 expression was normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA content.
For gene dosage analysis, the following primers were designed to amplify 98 bp spanning exon 5 and intron 5 of NEDD4-1 gene and subsequently normalized by β-actin expression. NEDD4-1 forward: 5′-ATGGCAACATTCAACTGCAA-3′, NEDD4-1 reverse 5′-GGCCTGGTTGCTATACATGG-3′; β-actin forward: 5′-TGCGTGACATTAAGGAGAAG-3′, β-actin reverse: 5′-GCTCGTAGCTCTTCTCC-3′. Each sample was run three times and each PCR experiment included two nontemplate control wells. The relative amounts of mRNA or DNA were calculated by the comparative cycle threshold method by Livak and Schmittgen.32
Fluorescence in Situ Hybridization
Fluorescence in situ hybridization (FISH) analysis was performed on formalin-fixed paraffin-embedded samples TMAs. TMA slides were deparaffinized, heated in a pressure cooker with 1 mmol/L EDTA, pH 8.0 for 10 minutes, and incubated with pepsin at 37°C for 30 minutes. The slides were then dehydrated in increasing ethanol concentrations, and then air-dried. The probes were denatured at 96°C for 5 minutes, and a hybridization solution was applied on each slide and incubated at 75°C for 1 minute. After overnight incubation at 37°C in a humid chamber, slides were washed with 0.4 × standard saline citrate and 0.3% NP40 for 2 minutes at 75°C, air-dried in darkness, counterstained with 4′,6-diamidino-2-phenylindole, and a coverslip was applied.
Copy number of the NEDD4-1 locus was determined by using two different probes: two BAC clones covering the gene (RP11-291H24; RP11-81G24) that were labeled with dUTP-Sprectrum Orange (Vysis Inc., Downers Grove, IL) and a green control probe (Kreatech Diagnostics, LG Amsterdam, The Netherlands; Poseidon FISH probes SE15 KBI-20015G D15Z) covering chromosome 15 centromeric region. The BAC clones used were designed according to the Ensembl database (http://www.ensembl.org, last accessed on April 30, 2010).
FISH evaluation was performed by two independent investigators with no previous knowledge of the genetic, clinical, or immunohistochemistry (IHC) results. Scoring of fluorescence signals was carried out in each sample by counting the number of single copy gene and control probe signals in an average of 130 (60 to 210) well-defined nuclei.
For evaluation of copy number of NEDD4-1 gene, NSCLC cases were divided into classes: disomy, polysomy (three or more copies of chromosome 15 in ≥50% of cells), and gene amplification (ratio of the NEDD4-1 gene to chromosome 15 of ≥ two copies of NEDD4-1/cell or presence of small or nonenumerable clusters of the NEDD4-1 signal in ≥50% of cells). Patients were considered FISH-negative for NEDD4-1 (disomy and amplification in <50% of cells) and FISH-positive for NEDD4-1 (amplification in ≥50% of cells).
Immunohistochemistry
Immunostaining was performed by using the avidin-biotin-peroxidase method (LSAB kit; Dako, Glostrup, Denmark) on formalin-fixed, paraffin-embedded tissues as described previously.8 All sections were counterstained with hematoxylin. Two pathologists reviewed all slides (R.F. and G.V.). Immunostaining for PTEN and NEDD4-1 was scored counting at least 500 cancer cells in 10 high-power fields. The antibody used in immunostaining of TMAs for PTEN (number 9559, Cell Signaling Technology, Beverly, MA) had been described extensively in several previous studies.33,34 The antibody used in immunostaining of TMAs for NEDD4-1 (number 07–049, Upstate Biotechnology, Lake Placid, NY) had been developed by immunizing rabbit with a synthetic peptide of the NEDD4-1WW domain and its specificity was described in a previous study on colon and gastric cancer.35 The specificity of the antibodies used in our study was assessed by three control experiments: absence of consistent immunostaining of cells when the anti-NEDD4-1 or anti-PTEN antibodies were replaced with normal rabbit serum at the same dilution; progressive reduction of signal intensity as dilution of the antibody was increased; and abrogation of signal after pre-incubation with the immunizing peptide.
PTEN expression was scored as positive (staining in >50% of cells) or negative (staining in <50% of cells) in agreement with literature.33,34
As a control for PTEN IHC staining, we performed immunohistochemical analysis of paraffin-embedded PTEN positive (MCF-7 breast carcinoma) and PTEN-negative (MDA-468 breast carcinoma) cell pellets as indicated by the manufacturer (Cell Signaling Technology).
For NEDD4-1 IHC staining, as a positive control we used a paraffin-embedded, NEDD4-1 positive, human kidney cancer cell pellet (Caki). Additionally, the same antibody used for IHC was able to recognize NEDD4-1 by Western blotting in NEDD4-1-transfected HEK293 cells. As a negative control for NEDD4-1 IHC staining, we used either a paraffin-embedded pellet of Caki cells short hairpin RNA-interfered for NEDD4-1, or a TMA slide treated with replacement of primary antibody with nonimmune serum.
NEDD4-1 expression was scored as high (>25% of positive tumor cells), moderate (10% to 25%), or negative (0% to 10% of positive tumor cells). The cut-off of 10% of stained cells to classify tumors negative or positive was as described.35
Antibodies and Reagents
Anti-PTEN (sc-6818 or sc-6974), anti NEDD4-1 (sc-25508), anti-β-tubulin (sc-80011), anti-AKT1 (sc-81434), anti-gelsolin (sc-6406), and anti-c-MYC (sc-42) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); anti-phospho-Akt (S473) antibody used was number 4060 (Cell Signaling Technology). For IHC, we used anti-NEDD4-1 number 07–049 (Upstate Biotechnology) and anti-PTEN number 9559 (Cell Signaling Technology). Cycloheximide (Sigma-Aldrich, St. Louis, MO) was used at 25 μg/ml; MG132 (carbobenzoxy-L-leucyl-L-leucyl-L-leucine) was from EMD Biosciences (Darmastadt, Germany) and used at 25 μmol/L for 4 hours.
Western Blot and Immunoprecipitation
Whole cell extracts were prepared by homogenizing cells in NP-40 lysis buffer (10 mmol/L Tris–HCl [pH 7.5], 150 mmol/L NaCl, 1% NP-40) containing protease inhibitors. Lysates were cleared by centrifugation, and proteins were separated by SDS-polyacrylamide gel electrophoresis. Western blot analysis was carried out by standard methods. Immunoprecipitation was performed by incubating lysates from 5 × 106 cells with 1 μg of antibody and 30 μl of protein A/G-conjugated agarose beads (Santa Cruz Biotechnology) at 4°C overnight. The precipitates were resuspended in 2× Laemmli buffer and resolved by SDS-polyacrylamide gel electrophoresis followed by immunoblotting.
Virus Generation and Infection
The human NEDD4-1 (NM_006154) mission shRNA set (Sigma) and the mission nontarget control transduction virus (SHC002V; Sigma) were used to generate lentiviral particles in HEK 293T packaging cells. Subconfluent HEK293T cells were co-transfected with 13 μg of NEDD4-1 mission shRNA set, 18 μg of pCMV-deltaR8.91, and 12 μg of pCMV-VSVG.36 After transfection, supernatants were collected at 8-hour intervals, filtered, and used for three rounds of transduction of NCI-H292 cells in the presence of 8 μg/ml of polybrene (Sigma).
To generate NEDD4-1-encoding retrovirus, HEK293T cells were co-transfected with 13 μg of pBABEpuro-NEDD4-1, 12 μg of pCMV-VSVG, and 18 μg of PEQ-PAM and collected and used to transduce NCI-H460 or BEAS-2B cells. Transduced cells underwent three rounds of infection (8 hours each round) and were selected in medium containing 1 μg/ml puromycin.
PTEN Localization
NCI-H292 cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS for 15 minutes at room temperature. Cells were incubated for 60 minutes at room temperature with an anti-PTEN antibody (Santa Cruz Biotechnology) diluted at 1:100 by PBS-containing 3% bovine serum albumin. After washing, cells were stained by a fluorescein isothiocyanate-conjugated secondary antibody (Santa Cruz Biotechnology), followed by staining with 4′,6-diamidino-2-phenylindole (Sigma). Cells were visualized with a Leica TCS-SP2 Confocal Laser Scanning microscopy using a 63× objective (Leica Microsystems, Wetzlar, Germany). Cellular lysates were fractionated by NE-PER Nuclear and Cytoplasmic Extraction Reagent (Pierce, Rockford, IL). Anti-Sp1 (Calbiochem, San Diego, CA) and anti β-tubulin (Santa Cruz Biotechnology) were used as loading controls.
In Vitro Proliferation Assay
Cells were plated at a density of 2.5 × 105 cells/well in triplicate and were harvested, diluted 1:1 with 0.4% Trypan Blue solution (Sigma), and counted daily up to 72 or 96 hours. This experiment was repeated thrice.
Soft Agar Assay
NCI-H460 and BEAS-2B cells (5 × 103 and 3 × 103, respectively) were resuspended in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum or Bronchial Epithelial cell Basal Medium (BEBM) medium (Cambrex), containing 0.35% low-melting agarose (type VII; Sigma) and seeded onto 0.5% low-melting agarose in Dulbecco’s modified Eagle’s medium-containing 10% fetal bovine serum. Colonies were scored 3 weeks after preparation and stained with 0.1% crystal violet (Sigma). Quantification and SD were obtained from results of three independent experiments.
Tumorigenic Assays
Cells (1 × 106) were suspended in 100 μl 10% fetal bovine serum and 100 μl Matrigel (BD Biosciences) and subcutaneously injected into the right flank of 6-week-old athymic nude mice (Charles River, West Germany) in triplicates. Every 7 days tumor size was measured with a caliper.
Statistical Analysis
Statistical significance of differences was determined by using the Student’s t-test and, for IHC-analysis, the χ2 test with Yates’ correction or the two-tailed Fisher’s exact test as appropriate. Data were analyzed with standard statistical software (SPSS version 9; SPSS, Chicago, IL). A probability value <0.05 was considered statistically significant.
Results
Expression of NEDD4-1 in NSCLC Cell Lines
To determine the role of NEDD4-1 in human cancer, we analyzed expression of NEDD4-1 protein in 10 cell lines derived from human NSCLC (Figure 1A). Primary human normal bronchial epithelial cells were used as control of normal lung. Western blot analysis demonstrated that NEDD4-1 expression was increased in most ADC-derived and SCC-derived cell lines. Subsequent mRNA analysis of NEDD4-1 expression by quantitative RT-PCR (Figure 1B) showed a good correlation between levels of NEDD4-1 mRNA and protein in most cell lines analyzed: cells with low levels of NEDD4-1 protein (ie, BEN and NCI-H23) also presented low amount of mRNA, and cells with high NEDD4-1 protein levels showed enhanced amount of NEDD4-1 mRNA (ie, NCI-H292, NCI-H226, and NCI-H596).
Figure 1.

NEDD4-1 expression in human NSCLC cell lines. A: Immunoblot analysis of NEDD4-1 in human NSCLC-derived cell lines. NHBE, normal human lung epithelial cells; ADC-derived cell lines: A549, NCI-H522, NCI-H2009, NCI-H23, NCI-H460, and NCI-H596; SCC-derived cell lines: NCI-H292, NCI-H226, CALU1, and BEN1. B: NEDD4-1 relative mRNA expression by quantitative RT-PCR. C: NEDD4-1 relative gene number by Q-PCR. DNA copy number from normal PBLs was set arbitrarily to 1.
Previous cytogenetic analysis of NSCLC cells has shown that the chromosome 15q, where the NEDD4-1 gene has been assigned (15q21.3), is frequently gained.37 Thus we examined the amplification status of the NEDD4-1 gene in some NSCLC cell lines analyzed (Figure 1C). Genomic DNA extracted from 10 NSCLC cell lines, from normal human bronchial epithelial cells, and from control normal human lymphocytes (Figure 1C, peripheral blood lymphocytes [PBLs]) was analyzed by Q-PCR. DNA copy number of NEDD4-1 gene was considered increased when values obtained (after normalization with actin) were greater than twofold the value obtained with normal diploid human PBLs. Using these parameters, our results indicated that at least three cell lines presented increased copy number of the NEDD4-1 gene (NCI-H460, NCI-H522, and NCI-H2009) in comparison to genomic DNA from normal human bronchial epithelial cells or from other cell lines examined. The three cell lines that presented amplification of the NEDD4-1 gene expressed high levels of the corresponding mRNA and protein.
Expression of NEDD4-1 in Primary NSCLC
The study of NEDD4-1 expression was extended to primary NSCLC. We performed immunostaining for NEDD4-1 on TMAs containing 41 SCCs, 46 ADCs, four carcinoids, seven adenosquamous carcinomas (ADSs), and five LCCs. The summary of NEDD4-1 expression in NSCLC is shown in Table 1. We found that NEDD4-1 is not detectable in the epithelial cells from normal alveolar epithelium and from upper airways (23/28) where it is expressed only in few samples with <10% focally distributed cytoplasmic-positive cells. In contrast, NEDD4-1 expression is increased in a total of 82/103 (80%) of NSCLC analyzed (Figure 2). Among NSCLC positive for NEDD4-1 staining, expression was classified as negative when the staining for NEDD4-1 was detected in 0% to 10% of cells (21/103), moderate when staining was found in 10% to 25% of cells (26/103), and high when tumors showed strong diffuse cytoplasmic staining in more than 25% of cells per high power field (56/103). Positive NEDD4-1 expression was significantly higher in the carcinoma samples than either normal alveolar or bronchial epithelium (P < 0.001; Fisher’s exact test). Among the different types of NSCLC, NEDD4-1 expression was moderate in 10/46 and highly positive in 24/46 ADCs; moderate in 11/41 and highly positive in 25/41 SCCs. ADS and LCC histotypes presented high NEDD4-1 expression in 5/7 and 1/5 cases, respectively (Table 1). Patients’ characteristics are shown in Supplemental Table S1 (available at http://ajp.amjpathol.org). We found no significant association between NEDD4-1 expression and tumor size, node involvement, grade, or stage; conversely, NEDD4-1 overexpression was more frequently represented in male patients (P = 0.03; see Supplemental Table S2 at http://ajp.amjpathol.org), though the significance of this association deserves further investigation.
Table 1.
Expression of NEDD4-1 in NSCLC
| Histology | Sample no. | NEDD4-1 expression*
|
||
|---|---|---|---|---|
| Negative (0–10%) | Moderate (10–25%) | High (>25%) | ||
| Normal | 28 | 23 | 3 | 2 |
| ADC | 46 | 12 | 10 | 24 |
| SCC | 41 | 5 | 11 | 25 |
| ADS | 7 | 1 | 1 | 5 |
| LCC | 5 | 2 | 2 | 1 |
| CAR | 4 | 1 | 2 | 1 |
| Total tumors | 103 | 21 | 26 | 56 |
CAR, carcinoid.
The expression of NEDD4-1 was scored as negative (<10% of the tumor cells with weak, focal immunopositivity, or absence of staining), moderate (>10% and <25% of positive tumor cells), and high (>25% of tumor cells with strong or diffuse immunopositivity).
Figure 2.

NEDD4-1 expression in human primary NSCLC by TMA immunostaining. Expression of NEDD4-1 in NSCLC: representative immunostaining of NEDD4-1 in normal and pathological lung tissues. Serial formalin-fixed paraffin-embedded sections from normal lung (number 11520), SCC (number 2252), or ADC (number 3044) were analyzed for NEDD4-1 expression. Epithelial cells from normal lung tissue are negative for NEDD4-1 expression (upper panels); cancer cells show intense NEDD4-1 expression in SCC and ADC. Original magnification: left column, ×10; right column, ×40.
Subsequently, to investigate the molecular mechanisms that lead to overexpression of NEDD4-1 in NSCLC, we performed quantitative RT-PCR analysis on RNA extracted from five normal lung tissues and 16 representative NSCLC (nine SCCs, five ADCs, and two ADSs). The average mRNA level of NEDD4-1 in normal lung tissue (n = 5) was set arbitrarily as 1. The mRNA expression of NEDD4-1 from NSCLC tissues was expressed as relative values compared with the corresponding normal tissues (fold increase). We found that on average the levels of NEDD4-1 mRNA (five ADCs, nine SCCs, and two ADSs) were considerably higher compared with those observed in normal lung tissue (P < 0.05). See Figure 3A for a representative experiment. As in cell lines, we found a significant relationship between NEDD4-1 mRNA and protein levels also in primary NSCLC (not shown).
Figure 3.

NEDD4-1 expression in human primary NSCLC by quantitative RT-PCR. A: NEDD4-1 relative mRNA expression by quantitative RT-PCR in primary NSCLC. The mean NEDD4-1 expression of normal lung tissues (n = 5) was set to 1. B: FISH analysis of primary NSCLC: normal diploid cells (upper row, left picture), tumors with five and seven signals of NEDD4-1 gene (middle and lower row left picture, respectively). Red, NEDD4-1 gene; green, chromosome 15 centromere. Nuclei were visualized under an Olympus BX61 microscope using a 60× objective; an enlargement of the inset is shown in the row right picture. C: NEDD4-1 relative gene copy number by Q-PCR. DNA copy number from normal PBLs was set arbitrarily to 1.
Finally, to determine whether the observed increased expression of NEDD4-1 could be ascribed to gene amplification at 15q21.3, we examined the amplification status of the NEDD4-1 gene in the same samples analyzed for protein and mRNA expression by FISH analysis performed on NSCLC TMAs. Altogether we were able to analyze 20 normal tissues and 57 tumors. We found that 19 of 20 normal lung tissues were disomic at the 15q21.3 locus with one showing low-level amplification. Conversely, 21 of 57 samples showed consistent alterations of the NEDD4-1 locus at 15q21.3. Interestingly, all of the cytogenetics alterations we observed were ascribed to gene amplification (a ratio of red-to-green signals >2), whereas there was no evident case of trisomy and/or polysomy. In seven cases (five SCCs and two ADCs), we found NEDD4-1 gene amplification in <50% of cells (20% to 30% on average) and thus were not considered FISH-positive, whereas in 14 cases (eight SCCs, four ADCs, one ADS, and one LCC) NEDD4-1 gene amplification was observed in more than 50% of cells and were considered FISH-positive (P < 0.05, for tumors with amplified NEDD4-1 gene compared with normal lung tissues; see Figure 3B for a representative picture). FISH data were confirmed by evaluation of copy number by Q-PCR on genomic DNA extracted from eight representative NSCLCs (four ADCs and four SCCs). Normal human diploid PBLs were used as controls. DNA copy number of NEDD4-1 gene was considered increased when values obtained by Q-PCR were greater than twofold the value obtained with PBLs. Using these criteria, we found complete concordance with the FISH data: two FISH-positive ADCs and four FISH-positive SCCs showed increased copy number of NEDD4-1 gene by Q-PCR; on the other hand, two FISH-negative ADCs did not show any copy number variation (Figure 3C). These results suggest that gene amplification of the NEDD4-1 locus at 15q21.3 may represent a common event in the pathogenesis of NSCLC and may account for increased expression of NEDD4-1 mRNA and protein observed in ∼25% of NSCLC. Accordingly, 14 of 14 cases with clear-cut NEDD4-1 gene amplification by FISH were positive for NEDD4-1 expression by immunostaining analysis.
NEDD4-1 has been reported to negatively regulate PTEN stability and nuclear localization by catalyzing PTEN polyubiquitination.24 To determine the role of NEDD4-1-mediated PTEN degradation in lung cancer, we first investigated the existence of an inverse correlation between NEDD4-1 and PTEN proteins in 98 NSCLC cases of which the expression of both proteins was available. PTEN immunostaining in NSCLC TMAs was performed with anti-PTEN monoclonal antibody. Results are summarized in Table 2. For this association we considered positive for NEDD4-1 expression NSCLC samples with moderate (10% to 25% of positive cells) and high (>25% of positive cells) staining (see previous paragraph). Conversely, NSCLC samples were classified as positive (staining in >50% of cells) or negative (staining in <50% of cells) for PTEN as described previously.15 NSCLC showed three major patterns of NEDD4-1/PTEN expression. Fifty-six tumors (57%) were characterized by low PTEN and high NEDD4-1 levels; 15 tumors (15%) expressed low NEDD4-1 and high PTEN levels; in nine tumors (9%) both low NEDD4-1 and PTEN levels were observed. Only 18 tumors with high NEDD4-1 expression retained high levels of PTEN protein. The χ2 test indicated that the percentage of high NEDD4-1 expressors was significantly greater in the low PTEN group than in the high PTEN expression group (P < 0.001). Representative immunostaining that shows inverse expression of NEDD4-1 and PTEN in NSCLC is reported in Figure 4, A and B. The presence of several tumors (n = 9) with low NEDD4-1 levels that showed loss of PTEN protein could be explained by the multiplicity of the mechanisms that contribute to inactivate PTEN mutations, loss of heterozygosity, and methylation also in the absence of NEDD4.
Table 2.
Inverse Correlation between NEDD4-1 and PTEN Expression in NSCLC (n = 98; P < 0.001)
| PTEN positive† | PTEN negative† | Total | |
|---|---|---|---|
| NEDD4-1 Positive* | 18 | 56 | 74 |
| NEDD4-1 Negative* | 15 | 9 | 24 |
| Total | 33 | 65 | 98 |
NSCLC samples were considered positive for NEDD4-1 expression when scored as weak and/or high staining and negative when scored low. The expression of NEDD4-1 was graded as high (>25% of tumor cells showed strong or diffuse immunopositivity), moderate (10–25%), or negative (0–10% of the tumor cells showed weak, focal immunopositivity, or absence of staining).
PTEN was scored by observing 500 cancer cells in at least 10 high-power fields and were classified as positive (staining in >50% of cells) or negative (staining in <50% of cells) as described previously.
Figure 4.

Inverse correlation between NEDD4-1 and PTEN expression in NSCLC. A: Immunostaining of NEDD4-1 and PTEN in a SCC (number 335) with high NEDD4-1 and low PTEN expression. B: Immunostaining of NEDD4-1 and PTEN in a SCC (number 4510) that shows low NEDD4-1 and high PTEN expression. Original magnification: left column, ×10; right column, ×40.
The correlation data disaggregated by histological types (ADC and SCC) are shown in Supplemental Tables S3 and S4 (available at http://ajp.amjpathol.org). The results shown indicate that the association between high NEDD4-1 expression and low PTEN levels was significant in SCC (n = 41; P < 0.001) but not in ADC (n = 46; P > 0.05). These results indicate that NEDD4-1 ubiquitin ligase may contribute to onset and/or progression of NSCLC, and especially in the case of SCC, by regulating PTEN expression.
Regulation of PTEN Protein Stability by NEDD4-1 in NSCLC Cells
Subsequently, we investigated the effects induced by the manipulation of NEDD4-1 in NSCLC cells on PTEN protein levels and localization. First, we adoptively expressed HA-tagged NEDD4-1 into NCI-H460 cells by retroviral transduction. We found that NCI-H460 cells overexpressing NEDD4-1 (NCI-H460-N4 cells) showed reduced PTEN levels and increased AKT activation (Figure 5A); moreover, overexpression of NEDD4-1 leads to an increase of the ubiquitylated form of PTEN, as assessed in cell lysates from NCI-H460-N4 or control cells treated with proteasome inhibitor MG132 (25 μmol/L) for 4 hours and then immunoprecipitated with anti-PTEN and analyzed by immunoblot with anti-ubiquitin and anti-PTEN antibodies, respectively (Figure 5B). Analysis of PTEN protein stability by cycloheximide treatment of vector or NEDD4-1-transduced NCI-H460 cells demonstrated that NEDD4-1 overexpression decreased PTEN half-life from >72 hours to slightly <48 hours (Figure 5C).
Figure 5.

NEDD4-1 induces PTEN degradation and increased ubiquitination in NSCLC (NCI-H460) cells. A: Immunoblot analysis of HA-tagged NEDD4-1, PTEN, and phosphorylated S473 (pAKT) in vector-transfected and NEDD4-1-transfected NCI-H460 cells. B: Effects of NEDD4-1 on PTEN ubiquitylation in NSCLC cells (NCI-H460 cells). Lysates from vector- or NEDD4-1-HA-transduced NCI-H460 were immunoprecipitated with PTEN antibodies (except for lane 1, in which control normal goat serum was used) and then probed by immunoblot with anti-ubiquitin antibody (upper panel). C: Vector or NEDD4-1-HA-transduced NCI-H460 cells were incubated with 25 μg/ml cycloheximide for the indicated time, and cells were harvested, lysed, and analyzed by immunoblotting. Half-life of PTEN protein is shown in the graph. *P < 0.005, compared with control at 72 hours.
As a second approach, we transduced NCI-H292 cells with lentiviruses encoding three different shRNAs to NEDD4-1 (shNEDD4-1 numbers 1, 2, and 3) or a scrambled control sequence (SCR). Cells were selected in puromycin (1 μg/ml for 3 to 5 days) and analyzed by immunoblot for NEDD4-1, PTEN, and pAKT (S473). NEDD4-1-interfered NCI-H292 cells showed reduced amount of NEDD4-1 (65% to 90%), higher levels of PTEN, and reduced AKT phosphorylation at S473 compared with control cells (Figure 6A). In addition, NCI-H292 cells interfered for NEDD4-1 presented consistently higher levels of nuclear PTEN, as demonstrated by indirect immunofluorescence and biochemical fractionation (Figure 6, B and C).
Figure 6.

Block of NEDD4-1 expression reduces PTEN ubiquitylation, increases PTEN nuclear expression, and suppresses AKT activation in human NSCLC (NCI-H292) cells. A: Immunoblot analysis of NEDD4-1, PTEN, phosphorylated S473 (pAKT), total AKT, and β-tubulin in NCI-H292 cells transduced with scrambled oligos (SCR) or with three different NEDD4-1-specific shRNA lentiviruses (shNEDD4-1). B: Indirect immunofluorescence for PTEN in NCI-control H292 cells (SCR) or transduced with shRNA to NEDD4-1 (shNEDD4-1). PTEN-positive cells were evidentiated by using fluorescein isothiocyanate-conjugated secondary antibody. Cell nuclei were identified by 4′,6-diamidino-2-phenylindole staining. Fluorescence was visualized under a Leica TCS-SP2 confocal microscopy with a 63× magnification. C: Immunoblot analysis of PTEN localization in NCI-H292 cells (SCR or shNEDD4-1) subjected to nuclear-cytoplasmic fractionation. Sp1 was used as loading control of nuclear proteins. D: SCR or shNEDD4-1-transduced NCI-H292 cells were incubated with 25 μg/ml cycloheximide for the indicated time, to determine the half-life of PTEN protein. Bars indicate SDs of a representative experiment carried out with two different shRNA to NEDD4-1. *P = 0.005, compared with control at 48 hours. E: Effects of NEDD4-1 on PTEN ubiquitylation in NCI-H292 cells. Lysates from SCR or shNEDD4-1-transduced NCI-H292 cells were immunoprecipitated with PTEN antibodies (except for lane 1, in which control normal goat serum was used) and then probed by immunoblot with anti-ubiquitin antibody (upper panel).
Finally, the knock-down of NEDD4-1 augmented PTEN half-life from ∼30 hours to >48 hours (Figure 6D). To confirm that the proteasome pathway was involved in the degradation of PTEN protein in NSCLC cells, scrambled or NEDD4-1-interfered NCI-H292 cells were treated with solvent or with proteasome inhibitor MG132 (25 μmol/L) for 4 hours. Cells were lysed and PTEN was immunoprecipitated and analyzed by immunoblot with anti-ubiquitin and anti-PTEN antibodies. We found that, as compared with untreated (or dimethyl sulfoxide-treated) cells, ubiquitylated PTEN accumulated in NCI-H292 cells treated with MG132. However, the level of PTEN ubiquitylation decreased steadily in NCI-H292 cells knocked down for NEDD4-1 (Figure 6E).
NEDD4-1 Contributes to Cell Proliferation and Tumor Formation
We determined the role of NEDD4-1 on the proliferative potential of human lung epithelial cells. To this aim we analyzed NEDD4-1-interfered NSCLC NCI-H292 cells by trypan blue exclusion assay. Control and NEDD4-1-interfered cells were plated and counted every day for 3 days (Figure 7A). In parallel with the decrease in NEDD4-1 expression, we found that the growth rate of NSCLC cells interfered with anti-NEDD4-1 shRNA lentivirus was markedly reduced compared with parental cells (not shown) or cells transduced with control scrambled lentivirus. This is consistent with the increased expression of the cyclin dependent-kinase inhibitor p21, gelsolin, and decreased c-MYC expression observed in interfered cells (Figure 7B) and points to a prominent role of NEDD4-1 in the proliferative pathways of NSCLC cells. These changes are consistent with the induction of a more mature, slower-growing phenotype.
Figure 7.

Block of NEDD4-1 expression decreases growth and tumorigenesis of human NSCLC (NCI-H292) cells. A: Trypan blue exclusion assay of SCR and three different shNEDD4-1 transduced NCI-H292 cells. Cells (2.5 × 105) were seeded in 6-well plates, and the number of viable cells excluding trypan blue was measured every day for three days. *P < 0.005 for shNEDD4-1 cells, compared with control at 72 hours. B: Immunoblot analysis of NEDD4-1, p21, c-MYC, gelsolin, and β-tubulin in SCR- and shNEDD4-1-transduced NCI-H292 cells. A representative blot relative to SCR-control cells and cells transduced with one of the three shNEDD4-1 is shown. C: SCR and shNEDD4-1-transduced NCI-H292 cells were subcutaneously injected into the flank of athymic nude mice, and the growth of xenotransplated tumor was measured as described in Material and Methods. A representative image of a SCR- and a shNEDD4-1-NCI-H292 xenotransplanted tumor is shown in the inset. **P < 0.005, compared with control. D: IHC-analysis of PTEN localization in NCI-H292 tumor xenografts 8 weeks after the injection (original magnification, ×40). E: Immunoblot analysis of NEDD4-1, PTEN, and phosphorylated S473 (pAKT) in lysates obtained from two different NCI-H292 tumor xenografts eight weeks after injection.
Finally, we determined the effect of suppressing NEDD4-1 expression on the tumorigenic potential of NSCLC cells. Control, scrambled-transduced or NEDD4-1-interfered NCI-H292 cells (1 × 106) were injected subcutaneously into immunodeficient athymic mice (n = 6). Mice were monitored for tumor appearance. We found that the suppression of NEDD4-1 expression in NCI-H292 cells markedly reduced in vivo growth of NSCLC cells (P = 0.002; Figure 7C). Notably, we found that the tumors explanted by xenografted mice had retained shRNA-dependent interference of NEDD4-1 expression, increased levels of nuclear PTEN as determined by IHC analysis (Figure 7D), and reduced AKT activation as assessed by immunoblotting analysis of lysates from tumors (Figure 7E). Above all, these shRNA experiments indicated that NEDD4-1 plays a significant role in the malignant behavior of NSCLC cells.
To further investigate the role played by NEDD4-1 in the control of cell cycle progression in lung cells, we transduced three different human lung cells: normal human primary bronchial epithelial cells, nontumorigenic human bronchial epithelial cells immortalized with Adenovirus (BEAS-2B),29 and a low-tumorigenic human lung cancer cell line that retains PTEN expression (NCI-H460). Primary human normal bronchial epithelial cells were transduced with retroviruses carrying the empty vector or the cDNA encoding HA-tagged NEDD4-1. However, in four distinct experiments, we were not able to select clones overexpressing NEDD4-1. Therefore, we decided to use BEAS-2B cells, human bronchial epithelial cells immortalized by Adenovirus infection and thus with inactivation of pRB and TP53.
After retroviral-transduction, cells were selected in puromycin-containing medium and several control BEAS-2B and BEAS-2B-N4 cells were isolated and expanded. The presence of the exogenous NEDD4-1 protein in transduced cells was detected by immunoblot (Figure 8A). BEAS-2B-N4 cells from two different transduction experiments were selected for further biological studies. We investigated whether the constitutive expression of NEDD4-1 modified cellular parameters of normal human lung cells including proliferation rate, anchorage-independent growth, and tumorigenicity. We found that, compared with control mock transduced cells, BEAS-2B-N4 cells proliferated with a slight accelerated rate in monolayer (anchorage-dependent growth) and formed consistently more colonies (∼40% more) in soft-agar (anchorage-independent growth; Figure 8, B and C, respectively). However, when assayed for tumorigenicity by subcutaneous injection into athymic mice, BEAS-2B-N4 cells were unable to promote tumor growth in vivo (n = 10) up to 3 months after the injection (not shown).
Figure 8.

NEDD4-1 overexpression increases in vitro growth rate of human nontumorigenic lung cells (BEAS-2B). A: Immunoblot analysis of HA-tagged NEDD4-1 in vector-transduced and NEDD4-1-transduced BEAS-2B cells. B: Trypan blue exclusion assay of NEDD4-1-HA-transduced BEAS-2B cells. Cells (2.5 × 105) were seeded in 6-well plates, and the number of viable cells excluding trypan blue was measured every day for four days. *P < 0.05, compared with control at 72 hours. C: Colony formation assay of vector- or NEDD4-1-HA-transduced BEAS-2B cells. Experiments were carried out in triplicate, and colonies were visualized with 0.1% crystal violet staining. **P < 0.005, compared with control.
However, to determine whether NEDD4-1 was able to confer more malignant characteristics on lung tumor cells, we used a cellular model of human adenocarcinoma (NCI-H460). NCI-H460 cells were transduced with a retrovirus carrying the cDNA encoding NEDD4-1. Transduced cells were selected in puromycin-containing medium and several control NCI-H460 and NCI-H460-N4 cells were isolated and expanded. The presence of the exogenous NEDD4-1 protein, PTEN, and AKT phosphorylation in transduced cells was detected by immunoblot (Figure 5A). NCI-H460-N4 cells from three different transduction experiments were selected for further biological studies. We investigated whether the constitutive expression of NEDD4-1 modified proliferation rate, anchorage-dependent and -independent growth, and tumorigenicity of NCI-H460 cells. We found that NCI-H460-N4 cells proliferated with an accelerated rate as a monolayer in culture dishes (anchorage-dependent growth) and formed more colonies (∼30% more) in soft-agar (anchorage-independent growth) than control mock transduced cells (Figure 9, A and B, respectively). In addition, when assayed for tumorigenicity by subcutaneous injection into athymic mice, NCI-H460-N4 cells gave rise to bigger tumors (n = 6; Figure 9C), that showed reduced PTEN levels and increased activation of AKT, as assessed by immunoblotting analysis of lysates from xenotransplanted tumors (Figure 9D).
Figure 9.
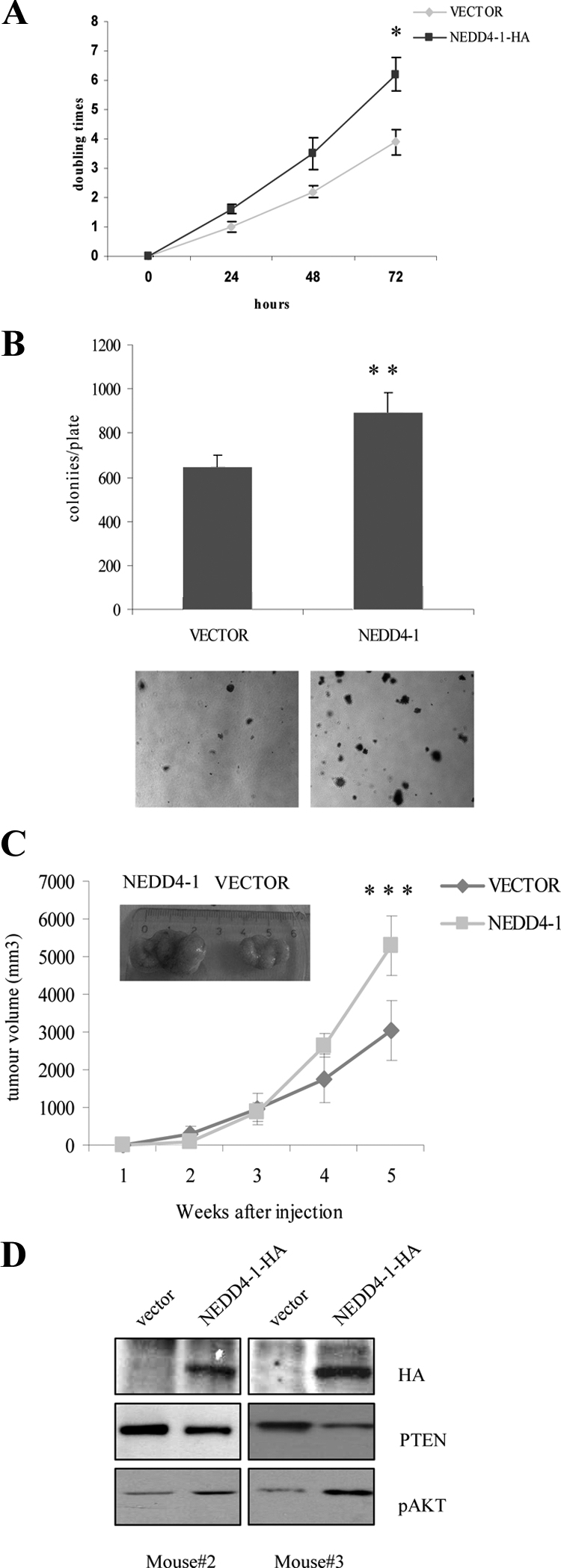
NEDD4-1 overexpression increases growth rate and tumorigenesis of human non-NSCLC cells (NCI-H460). A: Trypan blue exclusion assay of vector transduced or NEDD4-1-HA-transduced NCI-H460 cells. Cells (2.5 × 105) were seeded in 6-well plates, and the number of viable cells excluding trypan blue was measured every day for three days. *P < 0.05, compared with control at 72 hours. B: Colony formation assay of vector-transduced or NEDD4-1-HA-transduced NCI-H460 cells. Experiments were carried out in triplicate, and colonies were visualized with 0.1% crystal violet staining. **P < 0.05, compared with control. C: Control vector and NEDD4-1-HA-transduced NCI-H460 cells were subcutaneously injected into the flank of athymic nude mice, and the growth of xenotransplated tumor was measured as described in Material and Methods. A representative image of a vector- and a NEDD4-1-NCI-H460 xenotransplanted tumor is shown in the inset. ***P < 0.005, compared with control. D: Immunoblot analysis of NEDD4-1, PTEN, and phosphorylated S473 (pAKT) in lysates obtained from two different NCI-H460 tumor xenografts five weeks after injection.
In summary, these data indicate that, although NEDD4-1 overexpression is not able to accelerate the rate of in vivo growth of nontransformed lung epithelial cells that lack pRB and TP53 (BEAS-2B), it facilitates their anchorage-dependent and independent growth in vitro and augments in vivo tumorigenicity of lung cancer cells with an intact PTEN gene (NCI-H460 cells).
Discussion
In this article we have addressed two critical issues in lung cancer: the role of the E3 ubiquitin-ligase NEDD4-1 in NSCLC and its contribution to the inactivation of the tumor suppressor gene PTEN. We report that NEDD4-1 is overexpressed in ∼80% of human NSCLC as compared with normal tissue, frequently as consequence of an increase in the copy number of the NEDD4-1 gene at 15q21 and this apparently represents a major determinant of PTEN down-regulation in NSCLC. In addition, adoptive NEDD4-1 expression in cultured human lung cells promotes anchorage-dependent and independent growth and is required for full tumor growth in xenograft experiments.
So far, little information concerning NEDD4-1 expression in human cancer is available. To our knowledge this is the first thorough study of NEDD4-1 expression in human cancer with a significant correlation with PTEN status: NEDD4-1 overexpression has been reported in limited cohorts of patients with urinary bladder (mRNA) and gastrointestinal (protein) cancers.24,35
The TMA immunostaining studies reported in this article demonstrated that most NSCLC overexpress NEDD4-1 compared with normal lung epithelium, implying that the acquisition of NEDD4-1 expression might represent an oncogenic event during cancer development in the lung. The observation that there is no significant difference in NEDD4-1 levels with respect to histological subtypes, grade, stage, or metastasis, suggests that enhanced NEDD4-1 expression might occur at an early stage of the tumorigenic process.
As to the mechanism whereby NEDD4-1 expression is dysregulated in NSCLC, we observed increased copy number of NEDD4-1 gene in some NSCLC-derived cell lines and in a significant fraction (∼25%) of primary NSCLC, indicating that gene amplification of the NEDD4-1 gene at 15q21 may represent a major mechanism capable of enhancing NEDD4-1 expression in NSCLC. It is of note that our results confirm previous reports indicating that gain of part or of the entire long arm of chromosome 15, where the NEDD4-1 gene maps, are recurrent in NSCLC.37 Yet, because several NSCLC overexpress NEDD4-1 in the absence of gene amplification, other mechanisms must be involved in the dysregulation of NEDD4-1 expression in NSCLC.
NEDD4-1 is the E3 ubiquitin-ligase that regulates stability of PTEN, a lipid phosphatase that suppresses PI3K signaling.5,6 To search the biological link between NEDD4-1 and the PI3K pathway, we investigated the correlation of NEDD4-1 and PTEN in human NSCLC. Several reports have shown that PTEN loss occurs frequently (24% to 74%) in NSCLC.12,14,15 Loss or significant reduction in the cellular levels of PTEN protein was observed in 66% of the cases investigated here. However, previous results indicate that mutation/deletion of the PTEN gene or epigenetic silencing of its promoter do not account for PTEN loss in all NSCLC.14,15 Conversely, our data indicate that NEDD4-1 may contribute to PTEN inactivation in NSCLC by increasing its turnover. On one hand, we found that NEDD4-1 overexpression correlated with decreased levels of PTEN protein in NSCLC (n = 98; P < 0.001): low PTEN and high NEDD4-1 levels were observed in 57% of NSCLC, whereas high PTEN and low NEDD4-1 levels were observed in 15% of NSCLC. On the other hand, manipulation of NEDD4-1 expression in different lung cellular systems (BEAS-2B, NCI-H460, and NCI-H292 cells) demonstrated that NEDD4-1 plays a role in the dysregulation of PTEN in NSCLC cells. Interference with NEDD4-1 expression in NSCLC cells increases PTEN levels as well as its nuclear localization and, conversely, adoptive expression of NEDD4-1 into NSCLC cells induces PTEN down-regulation. However, it must be noted that NSCLC lacking NEDD4-1 expression (approximately 15%) must have developed alternative mechanisms for PTEN inactivation.
As to the role of NEDD4-1 in lung epithelial cells, we found that NEDD4-1 increases in vitro growth of lung epithelial cells and augments the tumorigenicity of lung cancer cells with an intact PTEN gene (NCI-H460). In fact, NEDD4-1 overexpression facilitates anchorage-dependent and independent growth of nontransformed lung epithelial cells that lack pRB and TP53 (BEAS-2B), though xenograft experiments in nude mice indicate that NEDD4-1 is not able to confer full tumorigenicity to BEAS-2B. On the other hand, suppression of NEDD4-1 expression by lentiviral-mediated transduction of shRNA into cultured NCI-H292 cells efficiently reduced proliferation of NSCLC cells in vitro and tumor growth in vivo possibly by inhibiting the degradation of PTEN via the ubiquitin–proteasome pathway. These findings are consistent with the idea that NEDD4-1 is an oncogene that regulates anchorage-independent growth and cooperate with RAS and TP53 in transforming Mouse Embryonic Fibroblasts (MEFs).24 Therefore, it is likely that NEDD4-1 might act in concert with other oncogenic hits to promote malignant transformation of lung cancer cells.
PTEN loss has been reported to predict for an aggressive subset of lung tumors that have a poor prognosis12 and to reduced survival in epidermal growth factor receptor-mutated NSCLC patients treated with gefitinib.38
Conversely, we failed to find any significant correlation between NEDD4-1 overexpression or PTEN loss (Scrima et al, manuscript in preparation) with patients’ clinico-pathological parameters analyzed (tumor size, node involvement, grade, or stage), suggesting the existence of complex relationships among the different members of the PI3K pathway. This idea is further confirmed by the analysis of AKT activation and/or PTEN loss in NSCLC, which has resulted in conflicting results regarding their prognostic significance.
Accordingly, Olaussen et al39 did not find any correlation between PTEN loss and clinico-pathological parameters of the patients analyzed in their study. Notably, in this article, loss of PTEN expression lacks prognostic value in stage I NSCLC. In agreement with these results, the cohort investigated in our study mainly comprised stage I to II patients (n = 67). In addition, the prognostic significance of AKT phosphorylation in NSCLC has resulted in conflicting results as well: in some studies AKT activation as measured by S473 phosphorylation occurs in most NSCLC and predicts for poor prognosis,12 whereas in another report S473 phosphorylated AKT protein expression has apparently no prognostic significance.40 In addition, the finding that there may be different prognostic roles for the various AKT isoforms (AKT1–3)41 or even the two activating AKT residues (T308 and S473, respectively)42 contributes to make the issue more complex.
In conclusion, the results described in this article indicate that NEDD4-1 may play an important role for the development of NSCLC, and that NEDD4-1 overexpression may modulate the malignant phenotype of NSCLC cells, likely by regulating the intracellular levels of PTEN, though our results cannot rule out the possibility that NEDD4-1 may act through other PTEN-independent mechanisms.
Acknowledgments
We thank Dr. Xuejun Jiang (Memorial Sloan Kettering Cancer Center, New York) for providing us with pBABE-NEDD4-1-HA expression vector.
Footnotes
Address reprint requests to Giuseppe Viglietto, Ph.D., Laboratorio di Oncologia Molecolare III, Dipartimento di Medicina Sperimentale e Clinica “G Salvatore,” Facoltà di Medicina e Chirurgia, Università Magna Graecia, Campus Universitario “Salvatore Venuta” Germaneto 88100, Catanzaro, Italy. E-mail: viglietto@unicz.it.
Supported by grants from the Associazione Italiana Ricerca sul Cancro to G.V.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
None of the authors disclosed any relevant financial relationships.
References
- Mountain CF. Revisions in the International System for Staging Lung Cancer. Chest. 2007;111:1710–1717. doi: 10.1378/chest.111.6.1710. [DOI] [PubMed] [Google Scholar]
- Gabrielson E. Worldwide trends in lung cancer pathology. Respirology. 2006;11:533–538. doi: 10.1111/j.1440-1843.2006.00909.x. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell. 2002;1:49–52. doi: 10.1016/s1535-6108(02)00027-2. [DOI] [PubMed] [Google Scholar]
- Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184-192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Kawano O, Sasaki H, Endo K, Suzuki E, Haneda H, Yukiue H, Kobayashi Y, Yano M, Fujii Y. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54:209–215. doi: 10.1016/j.lungcan.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, Malara N, Savino R, Rocco G, Chiappetta G, Franco R, Tirino V, Pirozzi G, Viglietto G. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7:665–669. doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- Breuer RH, Postmus PE, Smit EF. Molecular pathology of non-small-cell lung cancer. Respiration. 2005;72:313–330. doi: 10.1159/000085376. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated Akt overexpression and loss of PTEN expression in non-small-cell lung cancer confers poor prognosis. Lung Cancer. 2006;51:181–191. doi: 10.1016/j.lungcan.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Lim WT, Zhang WH, Miller CR, Watters JW, Gao F, Viswanathan A, Govindan R, McLeod HL. PTEN and phosphorylated AKT expression and prognosis in early- and late-stage non-small-cell lung cancer. Oncol Rep. 2007;17:853–857. [PubMed] [Google Scholar]
- Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L, Khuri FR. Lack of PTEN expression in non-small-cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002;8:1178–1184. [PubMed] [Google Scholar]
- Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, Kelsey KT. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005;36:768–776. doi: 10.1016/j.humpath.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Kohno T, Takahashi M, Manda R, Yokota J. Inactivation of the PTEN/MMAC1/TEP1 gene in human lung cancers. Genes Chromosomes Cancer. 1998;22:152–156. doi: 10.1002/(sici)1098-2264(199806)22:2<152::aid-gcc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Yokomizo A, Tindall DJ, Drabkin H, Gemmill R, Franklin W, Yang P, Sugio K, Smith DI, Liu W. PTEN/MMAC1 mutations identified in small cell, but not in non-small-cell lung cancers. Oncogene. 1998;17:475–479. doi: 10.1038/sj.onc.1201956. [DOI] [PubMed] [Google Scholar]
- Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba II, Milchgrub S, Brezinschek R, Virmani A, Gazdar AF, Minna JD. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 2008;17:1557–1565. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]
- Hosoya Y, Gemma A, Seike M, Kurimoto F, Uematsu K, Hibino S, Yoshimura A, Shibuya M, Kudoh S. Alteration of the PTEN/MMAC1 gene locus in primary lung cancer with distant metastasis. Lung Cancer. 1999;25:87–93. doi: 10.1016/s0169-5002(99)00052-5. [DOI] [PubMed] [Google Scholar]
- Adey NB, Huang L, Ormonde PA, Baumgard ML, Pero R, Byreddy DV, Tavtigian SV, Bartel PL. Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding. Cancer Res. 2000;60:35–37. [PubMed] [Google Scholar]
- Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus: implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- Wu W, Wang X, Zhang W, Reed W, Samet JM, Whang YE, Ghio AJ. Zinc-induced PTEN protein degradation through the proteasome pathway in human airway epithelial cells. J Biol Chem. 2003;278:28258–28263. doi: 10.1074/jbc.M303318200. [DOI] [PubMed] [Google Scholar]
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004;23:1972–1984. doi: 10.1038/sj.onc.1207436. [DOI] [PubMed] [Google Scholar]
- Fouladkou F, Landry T, Kawabe H, Neeb A, Lu C, Brose N, Stambolic V, Rotin D. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci USA. 2008;105:8585–8590. doi: 10.1073/pnas.0803233105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh FR, Corrin B, Colby TV. Tumours of the lung. Clinical features and staging. Travis WD, Brambilla E, Muller-Hermelink E, Harris CC, editors. Lyon: IARC Press,; WHO Classification, Tumors of the Lung, Pleura, Thymus and Heart. 2004:pp. 16–19. [Google Scholar]
- Rimm DL, Camp RL, Charette LA, Olsen DA, Provost E. Amplification of tissue by construction of tissue microarrays. Exp Mol Pathol. 2001;70:255–264. doi: 10.1006/exmp.2001.2363. [DOI] [PubMed] [Google Scholar]
- Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris C. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904-1909. [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate–phenol–chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T, editors. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press,; Molecular CloningA Laboratory Manual (2). 1992 [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–ΔΔ C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Saal LH, Gruvberger-Saal SK, Persson C, Lövgren K, Jumppanen M, Staaf J, Jönsson G, Pires MM, Maurer M, Holm K, Koujak S, Subramaniyam S, Vallon-Christersson J, Olsson H, Su T, Memeo L, Ludwig T, Ethier SP, Krogh M, Szabolcs M, Murty VV, Isola J, Hibshoosh H, Parsons R, Borg A. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet. 2008;40:102–107. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lague MN, Paquet M, Fan HY, Kaartinen MJ, Chu S, Jamin SP, Behringer RR, Fuller PJ, Mitchell A, Doré M, Huneault LM, Richards JS, Boerboom D. Synergistic effects of Pten loss and WNT/CTNNB1 signaling pathway activation in ovarian granulosa cell tumor development and progression. Carcinogenesis. 2008;29:2062–2072. doi: 10.1093/carcin/bgn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS, Yoo NJ, Jeong EG, Kim MS, Lee SH. Expression of NEDD4-1, a PTEN regulator, in gastric and colorectal carcinomas. APMIS. 2008;116:779–784. doi: 10.1111/j.1600-0463.2008.00999.x. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- Luk C, Tsao MS, Bayani J, Shepherd F, Squire JA. Molecular cytogenetic analysis of non-small-cell lung carcinoma by spectral karyotyping and comparative genomic hybridization. Cancer Genet Cytogenet. 2001;125:87–99. doi: 10.1016/s0165-4608(00)00363-0. [DOI] [PubMed] [Google Scholar]
- Endoh H, Yatabe Y, Kosaka T, Kuwano H, Mitsudomi T. PTEN and PIK3CA expression is associated with prolonged survival after gefitinib treatment in EGFR-mutated lung cancer patients. J Thorac Oncol. 2006;1:629–634. [PubMed] [Google Scholar]
- Olaussen KA, Soria JC, Morat L, Martin A, Sabatier L, Morere JF, Khayat D, Spano JP. Loss of PTEN expression is not uncommon, but lacks prognostic value in stage I NSCLC. Anticancer Res. 2003;6:4885–4890. [PubMed] [Google Scholar]
- Massion PP, Taflan PM, Shyr Y, Rahman SM, Yildiz P, Shakthour B, Edgerton ME, Ninan M, Andersen JJ, Gonzalez AL. Early involvement of the phosphatidylinositol 3-kinase/Akt pathway in lung cancer progression. Am J Respir Crit Care Med. 2004;170:1088–1094. doi: 10.1164/rccm.200404-487OC. [DOI] [PubMed] [Google Scholar]
- Al-Saad S, Donnem T, Al-Shibli K, Persson M, Bremnes RM, Busund LT. Diverse prognostic roles of Akt isoforms, PTEN and PI3K in tumor epithelial cells and stromal compartment in non-small-cell lung cancer. Anticancer Res. 2009;29:4175–4183. [PubMed] [Google Scholar]
- Tsurutani J, Fukuoka J, Tsurutani H, Shih JH, Hewitt SM, Travis WD, Jen J, Dennis PA. Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. J Clin Oncol. 2006;24:306–314. doi: 10.1200/JCO.2005.02.4133. [DOI] [PubMed] [Google Scholar]