


Abstract
The distribution of optimal local alignment scores of random sequences plays a vital role in evaluating the statistical significance of sequence alignments. These scores can be well described by an extreme-value distribution. The distribution’s parameters depend upon the scoring system employed and the random letter frequencies; in general they cannot be derived analytically, but must be estimated by curve fitting. For obtaining accurate parameter estimates, a form of the recently described ‘island’ method has several advantages. We describe this method in detail, and use it to investigate the functional dependence of these parameters on finite-length edge effects.
INTRODUCTION
Local sequence alignment is perhaps the most widely used tool in computational molecular biology, with most protein and DNA database search programs (1–4) implementing heuristic versions of local alignment algorithms (5,6). These algorithms seek the highest-scoring alignment of segments from the two sequences being compared. An alignment’s score is calculated by adding substitution scores, defined for each aligned pair of letters, and gap scores for each run of letters in one segment aligned with null characters inserted into the other.
A key question is what alignment scores may be expected to occur purely by chance. This question is generally addressed by analyzing the distribution of optimal alignment scores from random or real but unrelated sequences. We confine attention to random sequences, defined as strings of independent letters chosen with fixed background probabilities, because they are easier to control and study. Depending upon the details of the alignment scoring system and the background letter probabilities, the optimal score for the alignment of two random sequences of length n tends to grow proportionally either to n or to log(n) (7–10). The linear scoring regime corresponds to optimal alignments that tend to involve virtually the entire sequences; the logarithmic regime, with substitution and gap scores that are on average more negative, corresponds to optimal alignments that are relatively short. Many alignments representing true biological relationships involve only segments of the sequences compared, but these will tend to be outscored by long ‘random alignments’ when a scoring system in the linear regime is employed. Therefore, attention has focused primarily on scoring systems in the logarithmic regime, and we deal here exclusively with such scores.
In the asymptotic limit of long sequences, optimal local alignment scores follow an extreme-value distribution (11), described by two parameters λ and K. For the type of scoring system in most general use, these parameters cannot be calculated but must instead be estimated by random simulation. Most directly, one may generate optimal alignment scores for a large number of random sequence pairs, and fit an extreme-value distribution to these scores. Recently, an alternative approach has been described; it uses scores for local alignment ‘islands’ generated by a slight modification of the Smith–Waterman algorithm (12). We will discuss in detail the implementation and application of the island parameter estimation method, and compare it to the direct method in several ways. The island method has a number of useful features. (i) It renders explicit a tradeoff between parameter estimate bias and stochastic error, and allows this tradeoff to be easily controlled. (ii) It estimates accurately the tail behavior of score distributions for small-length comparisons. (iii) It allows parameter estimates to be obtained for arbitrary length sequence comparisons, including the infinite-length limit. In some circumstances, the first two of these features can be transferred advantageously to the direct method, appropriately modified. For asymptotic parameter estimation, however, the island method has a clear speed advantage.
THE DIRECT ESTIMATION OF STATISTICAL PARAMETERS
An asymptotic theory for local alignment scores has been developed for the case in which no gaps are permitted. In brief, for the comparison of random sequences of sufficient lengths m and n, the number of distinct local alignments with score at least x is approximately Poisson distributed, with mean
E(x) ≈ Kmne–λx, 1
where λ and K are easily calculated parameters (13,14). This implies that the optimal alignment score S′ approximately follows an extreme-value distribution (11), with
Prob (S′ ≥ x) ≈ 1 – exp(–Kmne–λx). 2
For local alignments that allow gaps, no asymptotic score distribution
has been established analytically. However, computational experiments
strongly suggest that equations 1 and 2 apply
to this type of alignment as well (12,15–22).
The key to using equations 1 and 2 is
the accurate estimation of the statistical parameters λ and K. Perhaps the most direct approach to estimating
these parameters for a fixed scoring system and set of background
letter frequencies is to generate a large number of pairs of random
sequences of equal length n, and find the optimal
local alignment score for each pair. From these scores one may calculate
maximum-likelihood estimates ![]() and
and ![]() for the
statistical parameters in equation 2 (23). If R scores are generated,
the ratio
for the
statistical parameters in equation 2 (23). If R scores are generated,
the ratio ![]() /λ is
approximately normally distributed, with mean 1 and standard error
0.78/√R (23). Note that the estimates developed
by Lawless (23) assume continuous data, whereas
alignment scores are almost always discrete. If the scale parameter λ times
the lattice spacing of possible scores is small, the error introduced
by assuming continuous scores is minor. One may, however, derive
maximum-likelihood estimates
/λ is
approximately normally distributed, with mean 1 and standard error
0.78/√R (23). Note that the estimates developed
by Lawless (23) assume continuous data, whereas
alignment scores are almost always discrete. If the scale parameter λ times
the lattice spacing of possible scores is small, the error introduced
by assuming continuous scores is minor. One may, however, derive
maximum-likelihood estimates ![]() and
and ![]() that
explicitly assume discrete scores (Appendix).
that
explicitly assume discrete scores (Appendix).
Because λ enters equations 1 and 2 exponentially, accurate estimates of λ are
particularly important. Marginally significant alignments from current
database searches typically have a scaled score λx > 25, for which even a 4% error
in λ leads to an estimated E-value in
error by greater than a factor of 2.7. Thus, standard errors of <2%,
or even 1%, in ![]() may be desirable.
may be desirable.
THE ISLAND METHOD
Recently, Olsen et al. (12) proposed the island method for estimating λ and K; it is a variant of ideas introduced by Waterman and Vingron (18,19) that translates into a very efficient algorithm. Rather than finding optimal alignment scores for pairs of random sequences, they propose generating scores for each island (as defined below) in a path graph. To generate sufficiently many scores for accurate parameter estimation, a single large or multiple smaller pairwise comparisons may be used.
Briefly, the Smith–Waterman algorithm generates a score for each cell C in a path graph, corresponding to the highest-scoring local alignment ending at C (5). This local alignment starts at a specific anchoring cell, and an island consists of all cells with identical anchors (Fig. 1). The score assigned to an island is the maximum score of the cells it contains. A simple modification of the Smith–Waterman algorithm, involving only a fixed amount of extra computation per cell, allows one to record which island each cell belongs to, and to keep track of each island’s score. Note also that as one moves row by row through a path graph with n columns, there can be at most O(n) islands represented on any given row. This allows one to tabulate all island scores generated by an m × n path graph in O(mn) time, and using only O(n) space.
Figure 1.
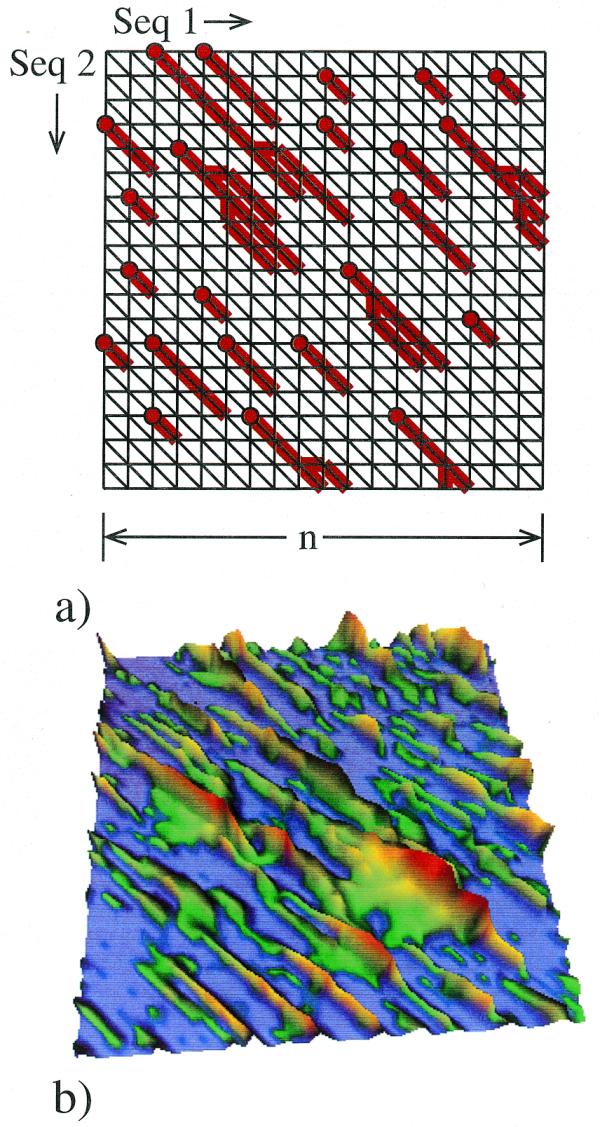
Islands in a local alignment path graph. (a) Schematic representation of the path graph. In every cell C the red line recalls the choice made by the optimization procedure of the Smith–Waterman algorithm. By these lines, all the cells with non-zero scores are partitioned into islands according to which anchoring points (circles) they are connected to. (b) Score landscape on a 50 × 50 path graph. The score at every cell of the path graph is represented by its height above the surface and color-coded with zero scores corresponding to blue areas and increasingly red colors for higher scores. The example shown is generated with a BLOSUM-62 scoring matrix, and a score –(11 + k) for each gap of length k. The islands are easily seen.
Island scores correspond to distinct locally optimal alignments, and thus the number of islands with score at least x should be well described by equation 1 when x is sufficiently large. The island method generates maximum-likelihood estimates of λ and K from equation 1, while the direct method generates these estimates from equation 2.
The concept of two or more local alignments being distinct is a subtle one, and a variety of definitions have been proposed (6,12,24,25). The differences among these definitions are relevant more for the comparison of real than random sequences. Because using any reasonable definition of distinct alignments should yield equivalent statistical results, the advantage of the ‘island’ (12) over the ‘declumping’ definition (18,19,24,25) for parameter estimation is its algorithmic efficiency.
In general, equation 1 becomes increasingly
accurate for larger values of x, so to obtain a
good estimate for λ one should confine attention to islands
whose score attains at least some threshold value c.
Assume the set Ic of
such islands has cardinality Rc,
and let ![]() be
the mean score in excess of c of these islands:
be
the mean score in excess of c of these islands:
![]() , 3
, 3
where S(i) is the score of island i. Then, assuming island scores are integral, with unit lattice spacing, the maximum-likelihood estimate (Appendix) for λ is
![]() . 4
. 4
The standard error of ![]() /λ is
/λ is
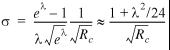 , 5
, 5
where the approximation holds to better than 0.05% for λ < 1. If the island scores
were continuous, the maximum-likelihood estimate ![]() would
instead be simply 1/
would
instead be simply 1/![]() , and the standard error
of
, and the standard error
of ![]() /λ would be 1/√Rc.
/λ would be 1/√Rc.
In conjunction with ![]() , the maximum-likelihood
estimate for K is
, the maximum-likelihood
estimate for K is
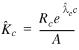 , 6
, 6
where A is the aggregate ‘area’ of the search space from which the collection of islands were drawn. If a single pair of sequences, of lengths m and n, were compared to generate the islands, then A = mn; if B such comparisons were performed, then A = Bmn.
The parameters λ and K of equations 1 and 2 properly apply only in the limit of infinite-length sequences. If one uses either the island or direct method to estimate λ for sequences of finite length, one obtains estimates with an observable finite-length bias. As will be discussed below, this bias can be explained in terms of ‘edge effects’, for which a simple correction can be applied to the lengths m and n in equations 1 and 2. The resulting formulas retain the asymptotic values of λ and K, so it is desirable to avoid any finite-length bias in the estimation of these parameters. We note here that, by eliminating edge effects, the island method can estimate asymptotic values of λ and K directly. This is done by embedding a length n × n sequence comparison within a larger (n + 2b) × (n + 2b) comparison, with a border of length b on each side (Fig. 2). Only islands anchored within the central n × n region are recorded. When b is sufficiently large, edge effects are essentially abolished.
Figure 2.
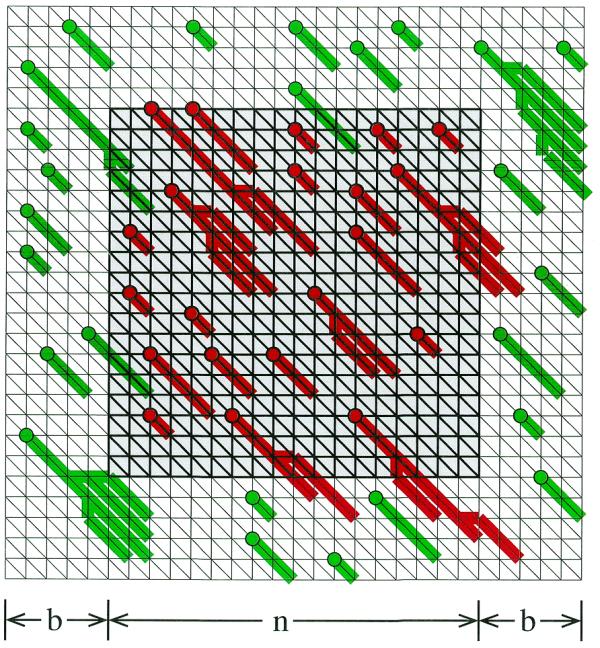
Schematic representation of a path graph used to avoid edge effects in the estimation of λ and K via the island method. The n × n scoring lattice (gray square in the middle) is surrounded by a border of width b. Only islands that are anchored within the central n × n area (shown in dark red) are counted. Islands anchored outside this area (green) are ignored. Note that some of the ignored islands reach into the inner area and some of the accepted islands reach into the border region since the classification of an island depends only on the position of its anchor (circles); borders thus are required on all sides to suppress edge effects properly.
THE TRADEOFF OF SPEED, BIAS AND PRECISION
Because of λ’s exponential role in equations 1 and 2, accurate estimates for λ are far more important than those for K, and we shall therefore focus on the estimation of λ. A key question for applying the island method effectively is how to choose an appropriate threshold parameter c for use in equation 4.
While we believe that the qualitative features presented here are
truly independent of the scoring system used, we will illustrate
below the issues involved in choosing c using a specific
example. To obtain extremely accurate parameter estimates for this
case study, we performed a massive random simulation for a particular
local alignment scoring system. Specifically, we used a set of standard
amino acid frequencies for proteins (26)
to generate over 92 000 pairs of length-7000 ‘random amino
acid sequences’. We compared each pair using the BLOSUM-62
amino acid substitution matrix (27),
in conjunction with affine gap scores (28–31) of –(11 + k)
for gaps of length k. To suppress edge effects,
scores were tabulated only for islands anchored within the central
5000 × 5000 square of each pairwise
comparison; approximately 1012 total island scores were
recorded. Using equations 3–6,
estimates of λ and K were obtained from
these data for a range of cutoff scores c; the
results are summarized in Table 1 and
the values of ![]() are plotted in Figure 3.
are plotted in Figure 3.
Table 1. Island method estimates for λ and K.
|
c |
Rc |
λc |
Kc |
| 20 | 508 087 143 | 0.2790 0.0000 (0.00%) | 0.058 |
| 21 | 382 046 389 | 0.2771 0.0000 (0.01%) | 0.056 |
| 22 | 288 047 946 | 0.2754 0.0000 (0.01%) | 0.053 |
| 23 | 217 666 586 | 0.2739 0.0000 (0.01%) | 0.051 |
| 24 | 164 854 001 | 0.2726 0.0000 (0.01%) | 0.050 |
| 25 | 125 090 432 | 0.2716 0.0000 (0.01%) | 0.048 |
| 26 | 95 080 777 | 0.2707 0.0000 (0.01%) | 0.047 |
| 27 | 72 367 615 | 0.2700 0.0000 (0.01%) | 0.046 |
| 28 | 55 135 823 | 0.2694 0.0000 (0.01%) | 0.045 |
| 29 | 42 040 928 | 0.2689 0.0000 (0.02%) | 0.044 |
| 30 | 32 087 753 | 0.2685 0.0000 (0.02%) | 0.044 |
| 31 | 24 502 349 | 0.2681 0.0001 (0.02%) | 0.043 |
| 32 | 18 721 366 | 0.2678 0.0001 (0.02%) | 0.043 |
| 33 | 14 312 497 | 0.2676 0.0001 (0.03%) | 0.042 |
| 34 | 10 945 852 | 0.2674 0.0001 (0.03%) | 0.042 |
| 35 | 8 372 081 | 0.2672 0.0001 (0.03%) | 0.042 |
| 36 | 6 407 611 | 0.2671 0.0001 (0.04%) | 0.042 |
| 37 | 4 904 102 | 0.2670 0.0001 (0.05%) | 0.041 |
| 38 | 3 755 281 | 0.2671 0.0001 (0.05%) | 0.042 |
| 39 | 2 874 422 | 0.2670 0.0002 (0.06%) | 0.041 |
| 40 | 2 201 167 | 0.2671 0.0002 (0.07%) | 0.042 |
| 41 | 1 684 893 | 0.2670 0.0002 (0.08%) | 0.041 |
| 42 | 1 289 490 | 0.2669 0.0002 (0.09%) | 0.041 |
| 43 | 986 932 | 0.2667 0.0003 (0.10%) | 0.041 |
| 44 | 756 060 | 0.2668 0.0003 (0.12%) | 0.041 |
| 45 | 579 087 | 0.2668 0.0004 (0.13%) | 0.041 |
| 46 | 443 934 | 0.2671 0.0004 (0.15%) | 0.042 |
| 47 | 339 913 | 0.2671 0.0005 (0.17%) | 0.042 |
| 48 | 260 519 | 0.2675 0.0005 (0.20%) | 0.042 |
| 49 | 199 117 | 0.2671 0.0006 (0.22%) | 0.042 |
| 50 | 152 595 | 0.2674 0.0007 (0.26%) | 0.042 |
| 51 | 116 705 | 0.2671 0.0008 (0.29%) | 0.042 |
| 52 | 89 323 | 0.2671 0.0009 (0.34%) | 0.042 |
| 53 | 68 605 | 0.2680 0.0010 (0.38%) | 0.044 |
| 54 | 52 570 | 0.2686 0.0012 (0.44%) | 0.045 |
| 55 | 40 242 | 0.2690 0.0013 (0.50%) | 0.046 |
| 56 | 30 746 | 0.2690 0.0015 (0.57%) | 0.046 |
| 57 | 23 481 | 0.2688 0.0018 (0.65%) | 0.046 |
| 58 | 17 888 | 0.2678 0.0020 (0.75%) | 0.043 |
| 59 | 13 662 | 0.2673 0.0023 (0.86%) | 0.042 |
| 60 | 10 427 | 0.2664 0.0026 (0.98%) | 0.039 |
A total of 92 441 pairs of length 7000 random sequences were generated using a set of standard amino acid frequencies (26). Island scores were generated for each pair using an extension of the Smith–Waterman algorithm (5), modified for affine gap scores (28). Substitutions were scored using the BLOSUM-62 matrix (27), and gaps of length k were assessed the score –(11 + k). Scores were recorded only for islands anchored within the central 5000 × 5000 square of each pairwise comparison. Maximum-likelihood estimates for λ and K were obtained using equations 4–6.
Figure 3.

Estimates 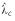 obtained via the island method with different
cutoffs c. Standard errors for the estimates are
shown with error bars. The plotted horizontal line indicates the
best estimate of the asymptotic λ. Details
of the simulation are given in the legend to Table 1.
obtained via the island method with different
cutoffs c. Standard errors for the estimates are
shown with error bars. The plotted horizontal line indicates the
best estimate of the asymptotic λ. Details
of the simulation are given in the legend to Table 1.
While the estimates ![]() of
Table 1 should be essentially free of
edge-effect bias, there is another systematic and easily understood
bias (12) evident for small values
of c. Optimal local alignments with low score are
unlikely to contain a gap, as will be discussed further below, and
for low thresholds
of
Table 1 should be essentially free of
edge-effect bias, there is another systematic and easily understood
bias (12) evident for small values
of c. Optimal local alignments with low score are
unlikely to contain a gap, as will be discussed further below, and
for low thresholds ![]() is therefore biased towards
the higher λ applicable to local alignments that exclude
gaps. In this example,
is therefore biased towards
the higher λ applicable to local alignments that exclude
gaps. In this example, ![]() falls monotonically for c ≥ 20, until it reaches the
value 0.2670 at c = 37; thereafter,
falls monotonically for c ≥ 20, until it reaches the
value 0.2670 at c = 37; thereafter, ![]() appears
to fluctuate randomly about this value. Of course a yet larger simulation,
yielding smaller stochastic errors, might detect systematic bias
even beyond c = 37.
appears
to fluctuate randomly about this value. Of course a yet larger simulation,
yielding smaller stochastic errors, might detect systematic bias
even beyond c = 37.
There is a tension between the bias of ![]() and
its precision, for the larger the value of c chosen,
the fewer the islands that attain score c, and
the larger the standard error of
and
its precision, for the larger the value of c chosen,
the fewer the islands that attain score c, and
the larger the standard error of ![]() . To illustrate the point,
consider a realistically sized random simulation, 10 000 times smaller
than that shown in Table 1, which would
require ~2 min on a modern workstation. The systematic bias in the
. To illustrate the point,
consider a realistically sized random simulation, 10 000 times smaller
than that shown in Table 1, which would
require ~2 min on a modern workstation. The systematic bias in the ![]() from
such a simulation should be the same as seen in Table 1, but the standard errors will be 100 times
larger. Table 2 shows the resulting tradeoff
between bias and precision. The best tradeoff probably occurs near c = 28, where the sum (~2.2%)
of the bias and the standard error are minimized. As the size of
the random simulation grows, the bias at a given cutoff remains
fixed, whereas the standard error decreases. Thus in general the
optimal tradeoff for larger simulations will tend to occur at higher
values of c.
from
such a simulation should be the same as seen in Table 1, but the standard errors will be 100 times
larger. Table 2 shows the resulting tradeoff
between bias and precision. The best tradeoff probably occurs near c = 28, where the sum (~2.2%)
of the bias and the standard error are minimized. As the size of
the random simulation grows, the bias at a given cutoff remains
fixed, whereas the standard error decreases. Thus in general the
optimal tradeoff for larger simulations will tend to occur at higher
values of c.
Table 2. Tradeoff between bias and precision in the estimation of λ.
|
c |
Bias (%) |
Standard error (%) |
| 22 | 3.1 | 0.6 |
| 23 | 2.6 | 0.7 |
| 24 | 2.1 | 0.8 |
| 25 | 1.7 | 0.9 |
| 26 | 1.4 | 1.0 |
| 27 | 1.1 | 1.2 |
| 28 | 0.9 | 1.3 |
| 29 | 0.7 | 1.5 |
| 30 | 0.6 | 1.8 |
| 31 | 0.4 | 2.0 |
| 32 | 0.3 | 2.3 |
| 33 | 0.2 | 2.6 |
| 34 | 0.1 | 3.0 |
| 35 | 0.1 | 3.5 |
| 36 | 0.0 | 4.0 |
| 37 | 0.0 | 4.5 |
The bias in the estimation of λ is calculated from Table 1, assuming λ’s true value is 0.2670. The standard error assumes an experiment generating 1/10 000 the number of island scores shown in Table 1.
For a given simulation one may estimate well the standard error
at any given c, but not the bias; if one could
estimate bias, one could correct for it. The analysis of a relatively
small simulation given in Table 2 is
possible only because a much larger simulation has in fact been
performed. In practice, one must choose the c at
which to estimate λ without knowing to any certainty how
much bias it entails. We have investigated automatic procedures
for choosing c, and found several reasonable methods,
but none for which an argument of optimality can be advanced. In
outline, ![]() decreases systematically for increasing c, until its increasing standard error obscures
any further change. It is at this point that the cutoff c should
be chosen.
decreases systematically for increasing c, until its increasing standard error obscures
any further change. It is at this point that the cutoff c should
be chosen.
EDGE EFFECTS AND THEIR CORRECTION
Independently of the type of bias in estimating λ described above, ![]() varies
substantially as a function of m and n when λ is
estimated from traditional borderless (i.e. b = 0) m × n sequence
comparisons (20). One may therefore
argue that one’s estimate of λ and K should
depend upon the lengths of the real sequences to which they will
be applied (22). We here take
the alternative view that the length-dependence of
varies
substantially as a function of m and n when λ is
estimated from traditional borderless (i.e. b = 0) m × n sequence
comparisons (20). One may therefore
argue that one’s estimate of λ and K should
depend upon the lengths of the real sequences to which they will
be applied (22). We here take
the alternative view that the length-dependence of ![]() is merely
an artifact of finite-length sequence comparison edge effects, and
that a correction for these effects is best applied to m and n in equations 1 and 2 rather
than to λ and K.
is merely
an artifact of finite-length sequence comparison edge effects, and
that a correction for these effects is best applied to m and n in equations 1 and 2 rather
than to λ and K.
The central idea of the ‘edge effect’ correction is that high-scoring local alignments from the comparison of two random sequences have an expected length l(x), dependent upon their score x, and therefore cannot begin arbitrarily close to the end of either sequence. Accordingly, in place of m and n in equations 1 and 2, the ‘effective’ lengths of the sequences should be taken to be m′ = m – l(x) and n′ = n – l(x) (20).
Empirically, the mean length l(x) of high-scoring random alignments with sufficiently large score x depends linearly on x
l(x) = αx + β. 7
We will discuss in a later section the interpretation of α and β, but note here that these parameters may be estimated by recording the lengths as well as the scores of optimal island alignments. The length of a gapped alignment is interpreted as the average length of the two segments it involves.
For the island method, the way that edge effects bias ![]() is easy
to understand. The decay in the observed number of alignments with
score at least x is steeper than would be estimated from
equation 1 because the effective lengths m′ and n′ shrink with
increasing x. Some simple calculus suggests the
apparent λ from the comparison of sequences of sufficient
lengths m and n should be given
approximately by
is easy
to understand. The decay in the observed number of alignments with
score at least x is steeper than would be estimated from
equation 1 because the effective lengths m′ and n′ shrink with
increasing x. Some simple calculus suggests the
apparent λ from the comparison of sequences of sufficient
lengths m and n should be given
approximately by
![]() . 8
. 8
For the specific scoring system studied in the massive random simulation above, we estimate α = 1.90 ± 0.02 (see discussion below). Therefore, we expect the apparent λ for n × n comparisons to follow the equation
![]() . 9
. 9
To test this theory, we used the island method to estimate λ for
the same scoring system studied in the simulation above. We generated
islands from many n × n random sequence comparisons, but with no border
for suppressing edge effects. Sufficient comparisons were performed
to yield over 106 islands with a score of at least 37
for each of the 12 lengths n studied; as described
above, using this threshold eliminates almost all cutoff-based bias.
The resulting maximum-likelihood estimates ![]() (n,n) have a standard error of
0.1%, and are shown as open circles in Figure 4. Given our small uncertainty in λ and α, for n > 400
(1/n < 0.0025 in Fig. 4) the data fit the theory of equation 9 to
within stochastic error (i.e. two standard deviations). Furthermore,
(n,n) have a standard error of
0.1%, and are shown as open circles in Figure 4. Given our small uncertainty in λ and α, for n > 400
(1/n < 0.0025 in Fig. 4) the data fit the theory of equation 9 to
within stochastic error (i.e. two standard deviations). Furthermore, ![]() (n,n) deviates from equation 9 by <0.5% for n > 218
(1/n < 0.0045), and by <1% throughout
the range studied. For each n, we calculated a χ2 goodness-of-fit test to the
geometric distribution; in all 12 cases, the data fit the model with
a P-value > 0.09.
(n,n) deviates from equation 9 by <0.5% for n > 218
(1/n < 0.0045), and by <1% throughout
the range studied. For each n, we calculated a χ2 goodness-of-fit test to the
geometric distribution; in all 12 cases, the data fit the model with
a P-value > 0.09.
Figure 4.

Estimates 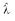 derived
from borderless n × n sequence comparisons by the island method as
a function of 1/n. Approximately 1 000
000 islands with a score of at least 37 were generated to produce
the estimates, which thus have a standard error of 0.1%;
the size of the symbols represents one standard error. The plotted
line represents the theory of equation 9 for the
apparent (n,n). The
scoring system and random sequence model are the same as those described
in the legend to Table 1.
derived
from borderless n × n sequence comparisons by the island method as
a function of 1/n. Approximately 1 000
000 islands with a score of at least 37 were generated to produce
the estimates, which thus have a standard error of 0.1%;
the size of the symbols represents one standard error. The plotted
line represents the theory of equation 9 for the
apparent (n,n). The
scoring system and random sequence model are the same as those described
in the legend to Table 1.
We emphasize that we do not argue that the line plotted in Figure 4 is more accurate in describing score distribution
tail behavior than the experimental ![]() (n,n) produced by the island method. Rather, the good
agreement implies the correction we recommend for finite lengths m and n should be sufficiently accurate
for comparing proteins of typical size. In evaluating the statistical
significance of actual sequence comparisons, one may apply edge-effect
corrections either to the sequence lengths, as we suggest, or to λ,
but one should not combine the two corrections. We emphasize further
that equation 8 does not permit one to estimate λ accurately
from a ‘finite-size’ simulation that estimates
(n,n) produced by the island method. Rather, the good
agreement implies the correction we recommend for finite lengths m and n should be sufficiently accurate
for comparing proteins of typical size. In evaluating the statistical
significance of actual sequence comparisons, one may apply edge-effect
corrections either to the sequence lengths, as we suggest, or to λ,
but one should not combine the two corrections. We emphasize further
that equation 8 does not permit one to estimate λ accurately
from a ‘finite-size’ simulation that estimates ![]() (m,n) because such a simulation
will not yield an estimate of the asymptotic value of α.
(m,n) because such a simulation
will not yield an estimate of the asymptotic value of α.
The island method with borders allows one to estimate the ‘infinite-length’ or asymptotic parameters λ and K directly, and simultaneously to estimate, as described below, the edge-effect correction parameters α and β. A single simulation that estimates these four parameters thus permits the statistical evaluation of comparisons of sequences of arbitrary length.
COMPARISON OF THE DIRECT AND ISLAND METHODS
For estimating the asymptotic parameters λ, K, α and β,
the island method has a distinct speed advantage over the direct method,
as we will discuss below. However, it is easiest first to compare
the two methods on the problem of estimating ![]() (n,n) studied in the previous section.
In this ‘finite size’ case, the methods have contrasting
advantages. To achieve a standard error σ in
(n,n) studied in the previous section.
In this ‘finite size’ case, the methods have contrasting
advantages. To achieve a standard error σ in ![]() (n,n)/
(n,n)/![]() (n,n), the island method must generate approximately
1/σ2 data points
(see equation 5), while the direct method need
generate only about 0.61/σ2 points
(23). Furthermore, the algorithm
for generating island scores requires more computation than that
for generating maximal local alignment scores because it must keep
track to which island the score of each path graph cell belongs.
Our implementation and timing experiments show the direct method
uses only ~70% of the time per cell that the island method
does. These two factors combined yield a speed advantage of ~240% for
the direct method. On the other hand, the island method may generate multiple
data points from each n × n sequence comparison. The expected number of such
points depends both upon the length of the sequences being compared
and upon the threshold score c as given by equation 1. The total speed advantage of the island over
the direct method is then Kn2e–λc/2.4.
In our case study we have been employing c = 37
and very many data points to obtain extremely accurate parameter
estimates, but as stated above c = 28
would be appropriate for a comparison of more typical accuracy.
At this threshold, the comparison of two sequences of length 340
yields about 2.4 islands on average, counterbalancing the direct
method’s speed advantages. For comparisons larger than
this, the island method will be faster than the direct method, and
slower for smaller comparisons.
(n,n), the island method must generate approximately
1/σ2 data points
(see equation 5), while the direct method need
generate only about 0.61/σ2 points
(23). Furthermore, the algorithm
for generating island scores requires more computation than that
for generating maximal local alignment scores because it must keep
track to which island the score of each path graph cell belongs.
Our implementation and timing experiments show the direct method
uses only ~70% of the time per cell that the island method
does. These two factors combined yield a speed advantage of ~240% for
the direct method. On the other hand, the island method may generate multiple
data points from each n × n sequence comparison. The expected number of such
points depends both upon the length of the sequences being compared
and upon the threshold score c as given by equation 1. The total speed advantage of the island over
the direct method is then Kn2e–λc/2.4.
In our case study we have been employing c = 37
and very many data points to obtain extremely accurate parameter
estimates, but as stated above c = 28
would be appropriate for a comparison of more typical accuracy.
At this threshold, the comparison of two sequences of length 340
yields about 2.4 islands on average, counterbalancing the direct
method’s speed advantages. For comparisons larger than
this, the island method will be faster than the direct method, and
slower for smaller comparisons.
This analysis, however, tells only part of the story, because the
biases of the direct and island methods in estimating ![]() (n,n) vary with n.
To study the extent of this bias, for each length n considered
in the previous section we generated sufficient data points for
both the direct method and the island method with c = 28
to produce estimates
(n,n) vary with n.
To study the extent of this bias, for each length n considered
in the previous section we generated sufficient data points for
both the direct method and the island method with c = 28
to produce estimates ![]() (n,n) with
a standard error of 0.1%. We then compared these estimates
to the independent and effectively unbiased estimates (also with
standard error 0.1%) shown by the points plotted in Figure 4; the resulting estimates of bias are given
in Table 3. For sequence lengths n ≤ 343 the direct method tends to overestimate
(n,n) with
a standard error of 0.1%. We then compared these estimates
to the independent and effectively unbiased estimates (also with
standard error 0.1%) shown by the points plotted in Figure 4; the resulting estimates of bias are given
in Table 3. For sequence lengths n ≤ 343 the direct method tends to overestimate ![]() (n,n) by >1%.
Some reflection reveals why this should be the case. For the scoring system
under study, ~81% of all optimal alignments from 343 × 343 comparisons have a score
less than 37, and >7% have a score less than 28.
As we learned from our analysis of the island method, including
low scoring, largely ungapped, alignments introduces noticeable
bias into estimates of λ. The problem is amplified for
the direct method because, due to the extremely fast decay of the
left-hand tail of the extreme-value distribution, the data points
upon which the maximum-likelihood estimate most strongly depends
are those with lowest score.
(n,n) by >1%.
Some reflection reveals why this should be the case. For the scoring system
under study, ~81% of all optimal alignments from 343 × 343 comparisons have a score
less than 37, and >7% have a score less than 28.
As we learned from our analysis of the island method, including
low scoring, largely ungapped, alignments introduces noticeable
bias into estimates of λ. The problem is amplified for
the direct method because, due to the extremely fast decay of the
left-hand tail of the extreme-value distribution, the data points
upon which the maximum-likelihood estimate most strongly depends
are those with lowest score.
Table 3. Bias in the estimation of λ(n,n) of the island, direct and censored direct methods, and their relative speeds.
| n | Bias (%) | Speed ratio | |||
| |
Island |
Direct |
Censored |
Island:direct |
Island:censored |
| 2400 | +0.9 | –0.2 | –0.2 | 60 | 60 |
| 1200 | +0.7 | 0.0 | 0.0 | 14 | 14 |
| 800 | +0.8 | +0.4 | +0.4 | 6 | 6 |
| 600 | +0.6 | +0.5 | +0.5 | 3 | 3 |
| 480 | +0.5 | +0.8 | +0.6 | 2 | 2 |
| 400 | +0.4 | +0.8 | +0.3 | 1.4 | 1.5 |
| 343 | +0.4 | +1.2 | +0.4 | 1.0 | 1.2 |
| 300 | +0.3 | +1.4 | +0.5 | 0.8 | 1.1 |
| 267 | +0.1 | +1.4 | +0.3 | 0.6 | 1.0 |
| 240 | 0.0 | +1.6 | +0.1 | 0.5 | 0.9 |
| 218 | –0.1 | +1.4 | –0.1 | 0.4 | 0.8 |
| 200 | 0.0 | +1.6 | –0.1 | 0.3 | 0.8 |
Maximum-likelihood estimates λ were derived using the island, direct and censored methods from n × n sequence comparisons. For the island method with cutoff score c = 28, approximately 106 scores were generated, yielding a standard error of 0.1%. For the direct method, approximately 6.1 × 105 scores were generated, yielding a standard error of 0.1%. For the censored method, sufficient scores above the threshold c = 28 were generated to yield a standard error of 0.1% (23); this number ranged from 6.1 × 105 for n = 2400 to 8.4 × 105 for n = 200. Biases are calculated assuming the correct values for λ(n,n) are those of the points plotted in Figure 4. Speed ratios are the computation times required, respectively, by the direct and censored methods divided by the time required by the island method; high speed ratios favor the island method.
Borrowing from our analysis of the island method, it is possible to greatly reduce the bias of the direct method by basing its maximum-likelihood estimate only on those scores that reach a minimum threshold c (23) (see Appendix). This refinement is achieved at a cost in speed, however, because not every n × n comparison will yield a data point, and because such ‘censoring’ increases the number of data points required to achieve a given standard error (23). For example, only 56% of 200 × 200 comparisons have a maximal alignment score of at least 28, and with this degree of censoring the number of data points required for a given error increases by 40% (23). For this size comparison, the censored direct method is thus 2.5 times slower than the unmodified method, while still 20% faster than the island method. A fuller analysis gives the speed advantage to the island over the censored method only for comparisons larger than about 280 × 280. Of course as the size of the comparisons grows, so does the island method’s relative speed advantage (Table 3), reaching a factor greater than 3 for comparisons of size 600 × 600.
The island method has a major speed advantage for comparisons of size ≥ 800 × 800, but it appears to have a corresponding disadvantage with respect to bias (Table 3). This arises because over 99.8% of the optimal alignment scores from 800 × 800 comparisons are at least 31, and at least 34 from 1200 × 1200 comparisons. Effectively, the score ‘threshold’ for the direct method increases with comparison size, while we have used a fixed score threshold of 28 for all the island comparisons in Table 3. Were this threshold raised for large comparisons, it would be possible to achieve equivalent bias to the direct method, while retaining a >3-fold speed advantage. However, for large comparisons, one has the option with the island method to choose greater speed over smaller bias.
If all the island method had to offer were a 2–3-fold speed advantage for comparisons in the size range 500 × 500 to 800 × 800, it would hardly constitute a significant advance. However, our main point is that in lieu of estimating statistical parameters for various finite-size comparisons, a single estimate of the asymptotic λ and K along with the edge-effect correction parameters α and β will suffice. In this context, the island method has major advantages to the direct method. Most simply, the island method can accurately estimate the asymptotic λ by increasing the dimensions of its comparisons to such an extent that finite-size effects become negligible. Because the number of data points the island method generates grows in proportion to the area of its comparisons, there is no loss in speed. In contrast, as we have seen, the direct method pays a heavy penalty in speed as the size of its comparisons grow.
To avoid unduly increasing the comparison size, one might consider adding borders to direct method comparisons, as described above for the island method (Fig. 2). This, however, imposes substantial computational overheads. First, one must record where local alignments are ‘rooted’, to avoid counting local alignments rooted outside the central square. The extra computation per cell is similar to keeping track of which island a cell belongs to and increases run time by a factor greater than 1.4. Second, borders can greatly increase the computational area of medium-sized comparisons. For example, a border of moderate length 200 (see the next section) increases the area of a 600 × 600 comparison by a factor of 2.8. The two effects combined would slow such a comparison down by a factor close to 4. In contrast, for asymptotic parameter estimation, borders may be added to the island method comparisons at essentially no computational cost: first because the island method must record the roots of local alignments in any case; second because the comparisons’ underlying dimensions may be enlarged arbitrarily, rendering inconsequential the additional area entailed by the inclusion of borders.
In conclusion, for finite-size parameter estimation, the island method begins to have a speed advantage only for the comparison of sequences of moderate length. However, for the asymptotic parameter estimation we recommend, the island method has a speed advantage to the direct method approaching an order of magnitude.
THE ESTIMATION OF α AND β
For optimal local alignments of a given score x,
the standard deviation in the distribution of alignment lengths
is large: about the same as the mean length. Nevertheless, the mean length
can be seen to grow approximately linearly with x,
as illustrated by data from the massive simulation above, plotted in
Figure 5. The slope of this dependence does
not approach its asymptotic value until x is sufficiently
large. Therefore, as with estimates of λ, estimates of
the parameters α and β in
equation 7 are best calculated by confining attention
to alignments with a score greater than or equal to a threshold
value c. In Table 4 we
give, for various thresholds, estimates of α and β obtained by linear regression on the
lengths of the optimal island alignments. Once again, choosing a
threshold that balances bias and stochastic error is to some degree
arbitrary. We show in Figure 5 the line
implied by the estimates ![]() = 1.90 and
= 1.90 and ![]() = –30, yielded by the threshold c = 47. These estimates agree within stochastic
error to those for all c ≥ 44.
= –30, yielded by the threshold c = 47. These estimates agree within stochastic
error to those for all c ≥ 44.
Figure 5.

The mean length l(x) of optimal island alignments, as a function of the alignment score x. Error bars, representing one standard error, grow with score primarily because the number of alignments on which the mean length estimates are based decreases. The plotted line represents a linear regression on the data for scores ≥47. Details of the simulation are given in the legend to Table 1.
Table 4. The estimation of α and β.
|
c |
αc |
βc |
| 33 | 1.840 0.002 | –26.9 0.1 |
| 34 | 1.847 0.002 | –27.2 0.1 |
| 35 | 1.852 0.002 | –27.4 0.1 |
| 36 | 1.858 0.003 | –27.7 0.1 |
| 37 | 1.864 0.003 | –27.9 0.1 |
| 38 | 1.869 0.004 | –28.2 0.2 |
| 39 | 1.873 0.005 | –28.4 0.2 |
| 40 | 1.877 0.005 | –28.5 0.2 |
| 41 | 1.874 0.006 | –28.4 0.3 |
| 42 | 1.877 0.007 | –28.5 0.3 |
| 43 | 1.88 0.01 | –28.8 0.4 |
| 44 | 1.89 0.01 | –29.0 0.5 |
| 45 | 1.89 0.01 | –29.3 0.6 |
| 46 | 1.91 0.01 | –30.2 0.7 |
| 47 | 1.90 0.02 | –30 1 |
| 48 | 1.89 0.02 | –29 1 |
| 49 | 1.88 0.02 | –29 1 |
| 50 | 1.91 0.03 | –30 1 |
| 51 |
1.89 0.03 |
–29 2 |
| 52 | 1.90 0.03 | –30 2 |
| 53 | 1.94 0.04 | –32 2 |
| 54 | 1.96 0.05 | –33 3 |
Estimates for α and β were obtained by linear regression of alignment length versus score for islands with score at least c. Details of the simulation are given in the legend to Table 1.
While the standard error for ![]() is 1% at c = 47,
one is forced to settle for much larger errors in simulations of
more realistic size. However, α and β are used only to correct the lengths
of the sequences being compared, and the significance of alignment scores
depends only linearly upon these lengths. Therefore it is generally
quite acceptable to estimate α to within
10 or even 20%. The data generated to provide reasonably accurate
estimates of the far more important parameter λ easily
suffice for this purpose.
is 1% at c = 47,
one is forced to settle for much larger errors in simulations of
more realistic size. However, α and β are used only to correct the lengths
of the sequences being compared, and the significance of alignment scores
depends only linearly upon these lengths. Therefore it is generally
quite acceptable to estimate α to within
10 or even 20%. The data generated to provide reasonably accurate
estimates of the far more important parameter λ easily
suffice for this purpose.
At a score of 95, the highest score achieved in this simulation, the predicted mean length is less than 150. Therefore, even though the standard deviation of the alignment length is approximately equal to the mean length, the border of length 1000 used in our simulation should be much more than sufficient for estimating the asymptotic values of the parameters λ, K, α and β, corresponding to ‘infinite length’ comparisons. For comparisons performed without borders, or with borders of insufficient length, estimates of α and β deviate from the asymptotic values, just as estimates of λ were shown to deviate above.
The expected length of gapped alignments with a high score clearly places limits on the applicability of equations 1 and 2 to the comparison of short sequences, even after edge effects have been corrected for. Specifically, if the expected length of an optimal alignment is longer than the shorter of the two sequences being compared, then one has effectively entered the realm of global sequence comparison, to which our theory no longer applies. This is perhaps best seen as an indication that the combination of substitution and gap costs being employed are tailored for too ‘distant’ similarities, and that a scoring system with a greater relative entropy should be used instead (32).
RELATIVE ENTROPY AND THE RELATION OF α TO β
It has recently been established under certain simplifying assumptions that in the no-gap case, the edge-effect correction outlined above is the proper first-order correction to equations 1 and 2 for finite-length sequences (J.L.Spouge, personal communication). For high-scoring local alignments without gaps, it can be shown (33) that the average length of alignments with score x is well approximated by
![]() , 10
, 10
where Hu is the relative entropy of the scoring system in nats (32), and the subscript u indicates we are speaking of ungapped alignments. It is therefore reasonable to define, and estimate, the relative entropy per amino acid pair for gapped alignments by the formula
Hg = λg/αg, 11
where the subscript g indicates the gapped case.
Given this definition, we estimate Hg for the scoring system studied above to be 0.141 ± 2% nats. Note that for the identical scoring system, Altschul and Gish (20) obtained the much greater estimate of 0.25 nats for Hg, due primarily to their assumption that β is 0 in equation 7. This assumption yields a good estimate of Hg only in the limit of very large scores x, a limit not nearly approached in simulations of practical size.
Given that for ungapped alignments βu is near zero, as seen experimentally (see Table 5 for some examples), one may ask why βg should be distinctly negative. An understanding is to realize that for a scoring system in which a gap of length 1 has score –G, at each end of an optimal alignment there must be a section with score +G that does not include gaps. The average lengths of these sections will be described better by the ungapped than by the gapped α. This is a much stronger effect than the fact that an optimal alignment may not begin or end with a negatively scoring aligned pair of letters, which causes βu to be slightly negative. Together, these two effects lead to the prediction that the parameter βg can be approximated by the formula
Table 5. The estimation of β using α.
| Matrix |
BLOSUM-45 |
BLOSUM-62 |
BLOSUM-80 |
PAM-70 |
PAM-30 |
| αu | 0.9113 | 0.7916 | 0.5222 | 0.3250 | 0.1938 |
| βu | –5.7 | –3.2 | –1.6 | –0.7 | –0.3 |
| Gap existence | 14 | 11 | 10 | 10 | 9 |
| Gap extension | 2 | 1 | 1 | 1 | 1 |
| αg | 1.92 0.03 | 1.90 0.02 | 1.07 0.02 | 0.70 0.01 | 0.48 0.01 |
| βg | –37.2 1.6 | –29.7 1.0 | –12.5 0.8 | –8.1 0.5 | –5.9 0.3 |
| 2G(αu – αg) + βu | –38.0 1.0 | –29.8 0.5 | –13.7 0.4 | –9.0 0.3 | –6.0 0.2 |
Estimates for α and β were obtained by linear regression of alignment length versus score, for scores attaining at least a cutoff value. Sequences were generated using a set of standard amino acid frequencies (26). Substitution scores were from either the BLOSUM (27) or PAM (37,38) series. Affine gap scores charged an existence penalty for each gap, and an extension penalty for each residue within a gap. Cutoff scores were chosen sufficiently high to avoid detectable bias in estimating α. Sufficient data points were generated to estimate α with a standard error of <2%. For each scoring system studied, this required over 500 pairs of random sequences, recording islands anchored within the central 5000 × 5000 square of each pairwise comparison. Borders of length 1000 were used for the BLOSUM-45 and BLOSUM-62 scoring systems, and of length 500 for all others.
βg ≈ 2G(αu – αg) + βu. 12
For the particular scoring system and random letter frequencies we have been studying, G = 12, αu = 0.79 and βu = –3.2. In conjunction with our estimate of 1.90 ± 0.02 for αg, this yields an estimate of –29.8 ± 0.5 for βg, which coincides with the experimental value of –30 ± 1 within the precision of measurement. Similar agreement is found for other gap costs and scoring systems that are not too close to the log-linear transition (see Table 5).
Equation 12 suggests that, with a knowledge of the easily accessible αu and βu, the estimation of αg alone is sufficient for the edge-effect correction. In practice, however, estimating βg requires no more work than estimating αg, so one might as well use the experimental value.
DISCUSSION AND CONCLUSION
It was originally claimed that the primary advantage of the island over the direct method for estimating statistical parameters lay in speed (12). We have shown here that this is substantially true only for asymptotic parameter estimation. However, we also have argued that edge-effect parameters allow a single estimation of asymptotic parameters to replace all finite-size parameter estimates. Furthermore, the island method permits simple maximum-likelihood estimation of λ that accounts for discrete score data, and it allows for simultaneous parameter estimation using various score thresholds c, and thus the controlled tradeoff of systematic bias and stochastic error.
The parameter λ depends not only upon the scoring system employed, but also upon the letter frequencies of the sequences being compared. In practice, λ may sometimes vary by >10% from one pair of sequences to another, due merely to variations in sequence composition. Yet, in the context of a database search, it is simply too time consuming to re-estimate λ for each pairwise comparison of potential interest: one moderately accurate estimate of λ requires as much time as searching a typical current database using standard heuristic methods (4). Thus, while one may precompute highly accurate estimates of λ for a fixed ‘standard’ composition, isn’t this accuracy vitiated by varying compositions?
Two solutions to the problem of varying background frequencies
have been proposed, both of which can make use of accurate parameter
estimation procedures. Altschul et al. (4) have suggested that for non-standard
letter frequencies, the substitution scores be rescaled so as to
set the calculable (13) parameter λu equal
to that for the original substitution scores used with standard
frequencies. The conjecture is that the precalculated λg will then apply to gapped alignments
using the rescaled substitution scores in the context of the non-standard frequencies.
This procedure has been implemented with good results (34). Alternatively, Mott (22)
has used random simulations for a very large number of different
scoring systems, gap costs, sequence compositions and sequence lengths
to derive an empirical formula for λ,
dependent upon variables calculable from the scoring system, letter
frequencies and sequence lengths. Because the values of ![]() used
in deriving this formula were calculated by the direct method, frequently
with short sequences, some improvement in Mott’s formula
may be obtainable using the methods described here. To be more conservative,
statistical parameters may be based upon residue compositions within
sequence regions containing the aligned segments of interest. In
general, by improving the precision with which statistical parameters
are estimated for local sequence alignment, more accurate judgments
can be rendered concerning the biological relevance of protein and
DNA sequence similarities.
used
in deriving this formula were calculated by the direct method, frequently
with short sequences, some improvement in Mott’s formula
may be obtainable using the methods described here. To be more conservative,
statistical parameters may be based upon residue compositions within
sequence regions containing the aligned segments of interest. In
general, by improving the precision with which statistical parameters
are estimated for local sequence alignment, more accurate judgments
can be rendered concerning the biological relevance of protein and
DNA sequence similarities.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr John Spouge for helpful conversations. This research is supported by the National Science Foundation through grants nos DMR-9971456 and DBI-9970199. R.B. and T.H. are grateful to the hospitality of the N.C.B.I. through its Scientific Visitors Program. In addition, R.B. acknowledges a Hochschulsonderprogramm III fellowship of the DAAD, R.O. acknowledges an LJIS fellowship by the Wellcome-Burroughs Fund and T.H. a Beckman Young Investigator Award.
APPENDIX
Maximum-likelihood fitting
In this appendix we explain the maximum-likelihood fitting technique in the presence of discrete scores. In the case of the extreme-value distribution this extends the more commonly used maximum-likelihood fitting for continuous scores as it is, e.g. presented by Lawless (23). By analogy to some analytical results on discrete extreme-value distributions (35), for small lattice spacing we expect only small deviations in the estimated parameters due to the discreteness of the scores as long as we perform uncensored fits. However, for alignment scores it is often necessary to estimate the parameters only for a subset of the observed scores. For such a censored fit, the discreteness of the scores must be taken into account, as discussed here, to obtain correct maximum-likelihood estimates of the parameters of the underlying distribution.
Throughout the appendix we will assume that sufficiently large island scores S follow a geometric distribution
Prob(S = x) = Dpx, 13
where we write p = exp(–λ) in order to emphasize the discrete character of the scores S. In the simplest case, the distribution is of this geometric form for all x ≥ 0 which fixes the prefactor D through normalization to D = 1 – p. Let us assume that we observed n islands with the scores x1, …, xN and we wish to find the value of p (i.e. λ) which best describes these observed scores. Since the probability of observing these scores is just the product of their individual probabilities the logarithm of the total probability (i.e. the log-likelihood) is
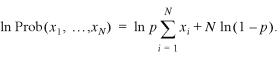 14
14
The best value ![]() of p is the one which maximizes
this expression. We can obtain it by equating the first derivative
of this expression to zero. This yields after some simple algebra
of p is the one which maximizes
this expression. We can obtain it by equating the first derivative
of this expression to zero. This yields after some simple algebra
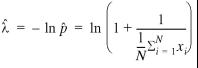 . 15
. 15
In our application, the distribution of island scores is not of the geometric form (equation 13) for all scores x. It follows this form only asymptotically for large scores x. In this case, the prefactor D is no longer fixed by normalization. Rather, it depends on the shape of the distribution for small x. In order to get a good estimate for p we have to perform a censored fit, i.e. we keep only those island scores x with value at least c. The integer cutoff c is chosen so that the geometric form (equation 13) is a reasonable description of the data. This is commonly called Type I censoring (23). We expect the censored scores to be distributed according to the restricted probabilities
![]() 16
16
which is independent of the unknown normalization factor D. If, out of n total scores, the scores x1, …, xM attain a value of c or larger, the logarithm of the probability is
![]()
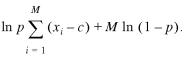 17
17
This log-likelihood function is identical to the one without censoring
presented in equation 14 except for the shift of
all scores by the cutoff c. Thus, the optimal value ![]() is
given by
is
given by
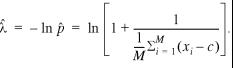 18
18
From this expression it becomes obvious why it is important to take
the discreteness of the scores into account for censored fits. The
maximum-likelihood estimate ![]() depends explicitly on
the cutoff, as c appears in equation 18,
but the set of scores x1, …, xM remains unchanged
as the cutoff c is varied between two adjacent
integers. Therefore, it is important to demand that c be
integral, taking the discreteness of the scores into account. In
order to get an estimate
depends explicitly on
the cutoff, as c appears in equation 18,
but the set of scores x1, …, xM remains unchanged
as the cutoff c is varied between two adjacent
integers. Therefore, it is important to demand that c be
integral, taking the discreteness of the scores into account. In
order to get an estimate ![]() for the other distribution
parameter K, we have to employ the expected number E(x) of islands with score at
least x given by equation 1. If
we observe Rc islands
with score at least c in B pairwise
comparisons we get Rc ≈ BE(c) ≈ B
for the other distribution
parameter K, we have to employ the expected number E(x) of islands with score at
least x given by equation 1. If
we observe Rc islands
with score at least c in B pairwise
comparisons we get Rc ≈ BE(c) ≈ B![]() mn
mn![]() c which can be rearranged
into equation 6 for the maximum-likelihood estimate
c which can be rearranged
into equation 6 for the maximum-likelihood estimate ![]() . If we choose the direct rather than the island
method to estimate λ, we are interested
in the distribution
. If we choose the direct rather than the island
method to estimate λ, we are interested
in the distribution
Prob(S′ = x′) = Prob(S′ ≤ x′) – Prob(S′ ≤ x′ – 1) 19
of the optimal local alignment scores. The optimal local alignment score S′ is the maximum of all the approximately ρmn island scores of the two sequences compared, where ρ describes the typical island density. Thus, the two probabilities on the right-hand side of equation 19 can be expressed by the distribution of island scores S, and S′ is distributed according to
Prob(S′ = x′) = Prob(S ≤ x′)ρmn – Prob(S ≤ x′ – 1)ρmn. 20
Since each island score follows the geometric distribution (equation 13) we get
![]()
![]() 21
21
with K = ρD/(1 – p). The last approximation is justified since Dpx′/(1 – p) is a small number for all the scores x′ that we are interested in. Equation 21 suggests that the optimal alignment scores are indeed extreme-value distributed. The only influence of the discreteness of the scores is that the probability Prob(S′ = x′) is given by finite differences of the extreme-value distribution instead of being a proper probability density, i.e. the derivative of the extreme-value distribution. Let us now assume that we performed B comparisons. We again choose a cutoff c, and keep only the m optimal local alignment scores x′1, …, x′M that are greater than or equal to c. (If we are interested in an uncensored fit, we can always choose c = 0 since all local alignment scores are non-negative.) These scores are expected to follow the distribution
![]()
![]() 22
22
The logarithm L(p,K) = ln Prob(x′1, …, x′M | x′1 ≥ c, …, x′M ≥ c) of the probability of observing the censored scores x′1, …, x′M then becomes
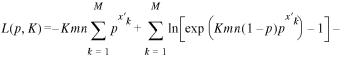
![]() 23
23
As before, the best estimates ![]() = exp(–
= exp(–![]() ) and
) and ![]() of the two parameters p and K, given the observed data x′1, …, x′M,
are the ones which maximize the function L(p,K). We could try to find this maximum by taking
the derivatives of L(p,K)
with respect to p and K and equating
them to zero. However, this leads to a pair of equations that can
only be solved numerically. Therefore, it is better to directly
use a numerical minimization algorithm applied to the function –L(p,K). We used
the downhill simplex method in two dimensions (36).
In order to improve convergence, it can be conveniently started
at the values of
of the two parameters p and K, given the observed data x′1, …, x′M,
are the ones which maximize the function L(p,K). We could try to find this maximum by taking
the derivatives of L(p,K)
with respect to p and K and equating
them to zero. However, this leads to a pair of equations that can
only be solved numerically. Therefore, it is better to directly
use a numerical minimization algorithm applied to the function –L(p,K). We used
the downhill simplex method in two dimensions (36).
In order to improve convergence, it can be conveniently started
at the values of ![]() and
and ![]() which are
obtained by the relatively simple uncensored, continuous extreme
value fit to the data. Then, it converges rapidly towards the global
minimum (
which are
obtained by the relatively simple uncensored, continuous extreme
value fit to the data. Then, it converges rapidly towards the global
minimum (![]() ,
,![]() ) of the function –L(p,K).
) of the function –L(p,K).
References
- 1.Pearson W.R. and Lipman,D.J. (1988) Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 3.Gish W. and States,D.J. (1993) Identification of protein coding regions by database similarity search. Nature Genet., 3, 266–272. [DOI] [PubMed] [Google Scholar]
- 4.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith T.F. and Waterman,M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197. [DOI] [PubMed] [Google Scholar]
- 6.Sellers P.H. (1984) Pattern recognition in genetic sequences by mismatch density. Bull. Math. Biol., 46, 501–514. [Google Scholar]
- 7.Waterman M.S., Gordon,L. and Arratia,R. (1987) Phase transitions in sequence matches and nucleic acid structure. Proc. Natl Acad. Sci. USA, 87, 1239–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arratia R. and Waterman,M.S. (1994) A phase transition for the score in matching random sequences allowing deletions. Ann. Appl. Prob., 4, 200–225. [Google Scholar]
- 9.Dembo A., Karlin,S. and Zeitouni,O. (1994) Critical phenomena for sequence matching with scoring. Ann. Prob., 22, 1993–2021. [Google Scholar]
- 10.Vingron M. and Waterman,M.S. (1994) Sequence alignment and penalty choice. Review of concepts, case studies and implications. J. Mol. Biol., 235, 1–12. [DOI] [PubMed] [Google Scholar]
- 11.Gumbel E.J. (1958) Statistics of Extremes. Columbia University Press, New York, NY.
- 12.Olsen R., Bundschuh,R. and Hwa,T. (1999) Rapid assessment of extremal statistics for gapped local alignment. In Lengauer,T., Schneider,R., Bork,P., Brutlag,D., Glasgow,J., Mewes,H.-W. and Zimmer,R. (eds), Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Menlo Park, CA, pp. 211–222. [PubMed]
- 13.Karlin S. and Altschul,S.F. (1990) Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl Acad. Sci. USA, 87, 2264–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dembo A., Karlin,S. and Zeitouni,O. (1994) Limit distribution of maximal non-aligned two-sequence segmental score. Ann. Prob., 22, 2022–2039. [Google Scholar]
- 15.Smith T.F., Waterman,M.S. and Burks,C. (1985) The statistical distribution of nucleic acid similarities. Nucleic Acids Res., 13, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins J.F., Coulson,A.F.W. and Lyall,A. (1988) The significance of protein sequence similarities. Comput. Appl. Biosci., 4, 67–71. [DOI] [PubMed] [Google Scholar]
- 17.Mott R. (1992) Maximum-likelihood estimation of the statistical distribution of Smith-Waterman local sequence similarity scores. Bull. Math. Biol., 54, 59–75. [DOI] [PubMed] [Google Scholar]
- 18.Waterman M.S. and Vingron,M. (1994) Rapid and accurate estimates of statistical significance for sequence database searches. Proc. Natl Acad. Sci. USA, 91, 4625–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waterman M.S. and Vingron,M. (1994) Sequence comparison significance and Poisson approximation. Stat. Sci., 9, 367–381. [Google Scholar]
- 20.Altschul S.F. and Gish,W. (1996) Local alignment statistics. Methods Enzymol., 266, 460–480. [DOI] [PubMed] [Google Scholar]
- 21.Pearson W.R. (1998) Empirical statistical estimates for sequence similarity searches. J. Mol. Biol., 276, 71–84. [DOI] [PubMed] [Google Scholar]
- 22.Mott R. (2000) Accurate formula for P-values of gapped local sequence and profile alignments. J. Mol. Biol., 300, 649–659. [DOI] [PubMed] [Google Scholar]
- 23.Lawless J.F. (1982) Statistical Models and Methods for Lifetime Data. Wiley, New York, NY, pp. 141–202.
- 24.Altschul S.F. and Erickson,B.W. (1986) Locally optimal subalignments using nonlinear similarity functions. Bull. Math. Biol., 48, 633–660. [DOI] [PubMed] [Google Scholar]
- 25.Waterman M.S. and Eggert,M. (1987) A new algorithm for best subsequence alignments with applications to tRNA-rRNA comparisons. J. Mol. Biol., 197, 723–728. [DOI] [PubMed] [Google Scholar]
- 26.Robinson A.B. and Robinson,L.R. (1991) Distribution of glutamine and asparagine residues and their near neighbors in peptides and proteins. Proc. Natl Acad. Sci. USA, 88, 8880–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henikoff S. and Henikoff,J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gotoh O. (1982) An improved algorithm for matching biological sequences. J. Mol. Biol., 162, 705–708. [DOI] [PubMed] [Google Scholar]
- 29.Fitch W.M. and Smith,T.F. (1983) Optimal sequence alignments. Proc. Natl Acad. Sci. USA, 80, 1382–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul S.F. and Erickson,B.W. (1986) Optimal sequence alignment using affine gap costs. Bull. Math. Biol., 48, 603–616. [DOI] [PubMed] [Google Scholar]
- 31.Myers E.W. and Miller,W. (1988) Optimal alignments in linear space. Comput. Appl. Biosci., 4, 11–17. [DOI] [PubMed] [Google Scholar]
- 32.Altschul S.F. (1991) Amino acid substitution matrices from an information theoretic perspective. J. Mol. Biol., 219, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dembo A. and Karlin,S. (1991) Strong limit theorems of empirical functionals for large exceedances of partial sums of i.i.d. variables. Ann. Prob., 19, 1737–1755. [Google Scholar]
- 34.Schäffer A.A., Wolf,Y.I., Ponting,C.P., Koonin,E.V., Aravind,L. and Altschul,S.F. (1999) IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics, 15, 1000–1011. [DOI] [PubMed] [Google Scholar]
- 35.Arratia R., Gordon,L. and Waterman,M.S. (1986) An extreme value theory for sequence matching. Ann. Stat., 14, 971–993. [Google Scholar]
- 36.Press W.H., Teukolsky,S.A., Vetterling,W.T. and Flannery,B.P. (1992) Numerical Recipes in C. The Art of Scientific Computing, Second Edition. Cambridge University Press, New York, NY, pp. 408–412.
- 37.Dayhoff M.O., Schwartz,R.M. and Orcutt,B.C. (1978) A model of evolutionary change in proteins. In Dayhoff,M.O. (ed.), Atlas of Protein Sequence and Structure. National Biomedical Research Foundation, Washington, DC, Vol. 5, Suppl. 3, pp. 345–352.
- 38.Schwartz R.M. and Dayhoff,M.O. (1978) Matrices for detecting distant relationships. In Dayhoff,M.O. (ed.), Atlas of Protein Sequence and Structure. National Biomedical Research Foundation, Washington, DC, Vol. 5, Suppl. 3, pp. 353–358.