

Abstract
Background
Chronic lymphocytic leukemia has a variable clinical course. Genomic aberrations identify prognostic subgroups, pointing towards distinct underlying biological mechanisms that are poorly understood. In particular it remains unclear whether the prognostic subgroups of chronic lymphocytic leukemia are characterized by different levels of leukemogenic proteins.
Design and Methods
Expression of 23 proteins involved in apoptosis, proliferation, DNA damage, and signaling or whose genes map to chromosomal regions known to be critical in chronic lymphocytic leukemia was quantified in 185 cytogenetically well characterized cases of chronic lymphocytic leukemia using immunoblotting. Cases were categorized hierarchically into deletion(17p), deletion(11q), trisomy 12, deletion(13q) as sole abnormality or normal karyotype. Statistical analysis was performed for expression differences between these subgroups. In addition, the expression levels of CDK4, P27 and P53 were quantified over the clinical course and compared to levels in immunopurified B cells from healthy individuals.
Results
In subgroups with a good prognosis, differential expression was mainly seen for proteins that regulate apoptosis. In contrast, in cytogenetic subgroups with a worse prognosis, differential expression was mostly detected for proteins that control DNA damage and proliferation. Expression levels of CDK4, P27 and P53 were higher compared to those in B cells from healthy individuals and significantly correlated with increasing hierarchical risk. In addition, no significant longitudinal changes of expression levels of CDK4, P27 and P53 could be detected in chronic lymphocytic leukemia patients.
Conclusions
Differences in expression levels of apoptosis- and proliferation-controlling proteins define distinct prognostic subgroups of chronic lymphocytic leukemia and uncover a correlation of levels of CDK4, P27 and P53 proteins with higher hierarchical risk.
Keywords: CLL, protein expression, genetics, prognosis
Introduction
Chronic lymphocytic leukemia (CLL), the most common leukemia in the Western world, has a highly variable clinical course with survival times ranging from months to decades.1,2 The clinical course correlates with inherent genetic features such as the mutational status of the immunoglobulin heavy chain locus (IGHV) and genomic aberrations.2 Genomic aberrations occur in about 80% of CLL. The most frequent ones are losses of chromosomes 13q14 [del(13q)], 11q22–q23 [del(11q)], 17p13 [del(17p)], and gains of chromosome 12 (trisomy 12). The association of genomic aberrations with clinical outcome resulted in the definition of a hierarchical model for prognostic prediction.1 In order to incorporate information from IGHV mutation status, a modified risk model was proposed that defines four subgroups of CLL patients based on 17p deletion, 11q deletion, and IGHV mutation status.3 In addition, usage of the V3–21 variable-region gene has been associated with an unfavorable clinical outcome similar to that of unmutated CLL cases, irrespective of IGHV mutation status.4 However, the underlying biological mechanisms leading to the different prognostic impact of these genetic markers are mostly unclear, with the exception of deletion of 17p.5,6
The most commonly found genomic aberration that occurs in about 55% of CLL patients is deletion of 13q14 which correlates with a favorable outcome.1 The biological role of this aberration is largely unresolved, but the candidate genes in the region are significantly down-regulated in CLL patients, pointing to an epigenetic tumor suppressor mechanism.7 In addition, two microRNA (miR-16-1 and miR-15a) were discovered in the critical region that could later be linked to CLL.7–9 MicroRNA exert their regulatory function at a post-transcriptional level. Effects derived from miR genes may not, therefore, be seen at the mRNA level but rather have to be evaluated by protein expression analysis.
Deletions of chromosome 17p affect the P53 tumor suppressor gene and are found in 5–10% of treatment-naive and 25–40% of treatment-refractory CLL cases.1,10 The responses and survival times after chemotherapy in the 17p-/P53 mutant CLL are poor.1,10 In CLL cells harboring P53 abnormalities, the miR genes miR-34a, miR-29 and miR-17-5p are down-regulated.6,11,12 The miR genes that are down-regulated by loss of function of P53 control target genes that are involved in cell cycle control, apoptosis, and DNA repair, such as CDK4, BCL2, MCL1 and TCL1.6,12–15 Similar to the effects of miR genes, post-transcriptional regulation also occurs through targeted degradation of proteins such as P27.5,16
Patients with del(11q) are also characterized by shorter treatment-free and overall survival times.1,10,17 The ATM gene is localized within the minimally deleted region on chromosome 11q. ATM mutations lead to P53 dysfunction in CLL after ionizing radiation.18
The presence of trisomy 12 was also associated with a more aggressive clinical course of CLL in several studies.10,17 However, in prospective trials by different large international groups, the previously described association with poor outcome has not been confirmed.1,17 Genes involved in the pathogenesis of trisomy 12 have not been identified so far, but RNA expression studies revealed an over-expression of genes located on chromosome 12 such as P27 and CDK4 in cases with trisomy 12.19,20
The different genomic aberrations result in distinct biological and clinical phenotypes in CLL patients. However, even though post-transcriptional regulation, e.g. by miRNA, plays a major role in tumorigenesis in general and in CLL in particular, the protein expression levels of candidate genes that are differentially expressed in the prognostic subgroups of CLL have not been quantified so far. The expression of 23 selected candidate gene products was, therefore, quantified in a large cohort of 185 cytogenetically well-characterized CLL cases. Proteins were chosen based on their genomic localization within a critical region affected in CLL (APAF1, ARF3, CCND2, CDK4, P27, Smac/DIABLO, STAT6 on chromosome 12; ATM, CIAP2 on chromosome 11q22; RB on chromosome 13q14; P53 on chromosome 17p13). In addition, we selected proteins prominently involved in hallmark pathways of tumorigenesis:21 apoptosis (AKT1, BAX, BCL2, CIAP2, MCL1, PI3K, Smac/DIABLO, SURVIVIN, XIAP), proliferation (CCND1/2/2, CDK4, PCNA, P21, P27), DNA damage (ATM, P53, RB) and signaling (STAT6, ZAP70) (Online Supplementary Table S1). The aim of this protein expression profiling was to provide information of potential use for prognostic prediction as well as for the development of new drugs for tailored use in particular patients or subgroups of patients with CLL.
Design and Methods
Patients
Tumor samples were obtained after informed consent in a procedure approved by the Ethics Committee of the University of Ulm (# 96/08). Mononuclear cells were isolated by Ficoll density gradient centrifugation from the peripheral blood of 185 CLL patients. Molecular cytogenetic analyses as well as IGHV sequencing were performed as described previously.1,3 CLL cases were categorized according to the hierarchical model of Döhner et al.1 into those with del(17p) (n=32), del(11q) (n=39), trisomy 12 (n=53), or del(13q) (n=35) as sole abnormality or a normal karyotype (n=25) (Table 1). Tumor cell content was defined by fluorescence in situ hybridization (FISH) or, in cases without cytogenetic aberrations, by high lymphocyte counts plus a high percentage of CD5+ and CD19+ cells. The median content of CLL cells in the cohort was above 85% (median, 86%; mean, 84%; range, 44%–98%; standard deviation, 9,22). IGHV mutation status was known for 179 of the 185 cases (Table 1). Patients’ samples used for detection of ZAP70 were purified to a tumor cell content of more than 96% by CD19+ selection using magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany). For further evaluation of the expression levels of the key proteins CDK4, P27 and P53, eight CD19-sorted CLL samples were analyzed in comparison to immunopurified B cells from eight healthy individuals (Online Supplementary Table S2). To detect potential changes of expression levels of the key proteins CDK4, P27 and P53 over time, samples harvested at different time points during the clinical course of the disease and before/after treatment were selected from four different CLL patients (Online Supplementary Tables S2 and S3).
Table 1.
Sample categorization according to the hierarchical model.1

Cell lines and western blot analyses
The cell lines and western blot analyses are described in the Online Supplementary Design and Methods section (see also Online Supplementary Figure S1).
Assessment of activity of ATM in patients with and without del(11q)
Viable frozen CLL cells were thawed and left in culture medium (RPMI medium 1640 with 10% fetal bovine serum and 1% glutamine) for several hours. Cells were then subjected to 5 Gray (Gy) ionizing radiation using a 37 Cs source and harvested after 1 h. The lysis buffer was supplemented with phosphatase inhibitors (10 mM NaF, 1 mM NaVO3, 1 mM Na2MoO4). Western blots were performed using an antibody detecting Phospho(Ser1981)-ATM (Cell Signaling Technology Inc., Beverly, USA). Anti-LAMIN-B (Santa Cruz Biotechnology, Heidelberg, Germany) was used to control for equal amounts of protein. Protein expression was quantified by densitometry using ImageJ 1.38x software (Online Supplementary Table S4).
P53 mutation analyses
Denaturing high-performance liquid chromatography “WAVE” analysis and bidirectional direct sequencing were used to identify mutations in the coding region of P53 (exons 2 – 11) as described elsewhere.22
Statistical analyses
Protein expression data were quantified for statistical analysis in 185 CLL patients. Differential protein expression among hierarchical subgroups of CLL and within IGHV mutated versus unmutated CLL was analyzed by Kruskal-Wallis tests across the five hierarchical subgroups (univariate analysis). In order to identify the cytogenetic subgroups that showed different protein levels, univariate logistic regressions were performed.
Protein-protein correlations were assessed by pair-wise correlations (univariate; no data imputation) as well as by partial correlations (multivariate, with data imputation). Continuation ratio models for correlation of protein expression with hierarchical risk (univariate analysis) were calculated based on the assumption of a hierarchical risk as a surrogate marker for survival.23,24 Deviations from this ordinal relationship were modeled by interaction terms where necessary.23,24 Kaplan-Meier curves for overall survival and treatment-free survival from diagnosis stratified by protein expression levels were calculated for the key proteins CDK4 (n=18), P27 (n=20), and P53 (n=25). All P values of univariate tests were adjusted for multiple testing using Holm’s method. Throughout the paper, except where explicitly stated otherwise, test results are called significant if the adjusted P value is less than 0.05, while test results with unadjusted P values less than 0.05 are interpreted as showing a trend. Further details on the statistical analysis are given in the Online Supplementary Appendix.
Results
We analyzed the expression of 23 proteins with potential functional relevance in CLL in a total of 185 cases of CLL (Table 1 and Online Supplementary Table S1). Expression of 19 of these proteins could be detected in CLL and was quantified by densitometry followed by statistical analyses.
Results of univariate analysis for differential protein expression among hierarchical subgroups of chronic lymphocytic leukemia
Significant differences in protein expression across all five hierarchical subgroups were found for ATM, BCL2, BAX, CCND3, CDK4, CIAP2, MCL1, P27, P53, RB and XIAP. Regarding AKT1, APAF1, ARF3, PCNA, STAT6 and ZAP70, statistical analysis did not reveal significant variations in expression across the hierarchical subgroups. No measurable expression was detected for CCND1/2, P21 and SUR-VIVIN. In order to identify differential protein expression, univariate logistic regressions were performed comparing the subgroup of interest with all other hierarchical subgroups (Online Supplementary Figure S2).
13q deletion as sole abnormality
The highest number of differentially expressed proteins was observed in cases with del(13q) as the sole abnormality (Figure 1 and Online Supplementary Figure S2). Levels of BCL2 and BAX were higher in this subgroup while levels of CDK4, P27 and XIAP were lower compared to those in other subgroups (adjusted P values: BCL2: <0.01 (n=134), BAX: 0.02 (n=116), CDK4: <0.01 (n=93), P27: <0.01 (n=99), XIAP: 0.05 (n=78); fold changes: BCL2: 1.7, BAX: 1.7, CDK4: 0.6, P27: 0.7, XIAP: 0.7) (Figure 1). The higher number of differentially expressed proteins involved in apoptosis regulation may indicate a stronger deregulation of apoptosis in the CLL subgroup with a single del(13q) compared to the other hierarchical subgroups. At the same time, levels of proliferation-associated proteins such as CDK4 and P27 were lower than those in all other subgroups (Figure 2).
Figure 1.

Combined box plots/dot plots of differentially expressed proteins in CLL cases with del(13q), del(11q), trisomy 12q, del(17p) and a cytogenetically normal karyotype compared to all other hierarchical subgroups. Each dot represents the level of expression (y-axis) of a single CLL case. Negative values of expression are the result of log-transformation for statistical analysis.
Figure 2.

Overview of protein levels in hierarchical subgroups of CLL suggesting an imbalance of apoptosis and proliferation in CLL in benign versus aggressive subgroups. The figure illustrates an increasing number of differentially expressed proteins involved in proliferation/DNA damage and a decreasing number of differentially expressed proteins that control apoptosis with higher genetic risk category [two arrows: proteins with significant differential expression; single arrows: significant before adjustment for multiple testing (trend)].
Normal karyotype
In the subgroup with a normal karyotype, we detected higher levels of PI3K and SMAC (adjusted P values: PI3K: 0.01 (n=75), SMAC: 0.03 (n=79); fold changes: PI3K: 1.5, SMAC: 1.3) compared to the levels in all other genetic subgroups. This reflects a potential deregulation of apoptosis similar to that in the del(13q) subgroup.25 However, levels of proteins associated with proliferation were not lower in the CLL subgroup with a normal karyotype than in the other subgroups (Figures 1 and 2 and Online Supplementary Figure S2).
Trisomy 12
CLL cases with trisomy 12 showed significantly lower levels of P53 (adjusted P value: <0.01 (n=87); fold change: 0.2) than cases in the other genetic subgroups (Figure 1). A lower level of ATM in CLL with trisomy 12 has already been described at the mRNA level and is reflected at the protein level as a trend in this study [adjusted/unadjusted P values: 0.11/0.01 (n=87)].20 A gene-dosage dependent effect was previously reported for CDK4 at the RNA level in cases with trisomy 12.20 However, an increase in proteins whose genes map to chromosome 12 (APAF1, ARF3, CDK4, P27, SMAC/DIABLO, STAT6) was not detected in the current study. In contrast, P27 and SMAC were present at lower levels in cases with trisomy 12 (adjusted/unadjusted P values: P27: 0.11/<0.01 (n=99), SMAC: 0.14/<0.01 (n=79)) than in all other subgroups. PI3K, a kinase involved in anti-apoptotic signaling in CLL,25 was found to be significantly reduced (adjusted P value: 0.03 (n=75); fold change: 0.7) (Figure 1).
11q deletion
CLL samples from patients with del(11q) showed low levels of P53 protein (adjusted P value: 0.04 (n=118); fold change: 0.5) (Figure 1). A trend towards a higher expression of CDK4 was found in these cases compared to in the other subgroups [adjusted/unadjusted P value: 0.11/<0.01 (n=93)]. No significant differences in ATM protein levels were detected (Figure 1 and Online Supplementary Figure S2). In order to test whether activation of ATM, as assessed by its phosphorylation status, is dependent on the 11q deletion, we additionally quantified the phosphorylation levels of the ATM kinase in samples from 16 CLL patients with and without del(11q) in response to 5 Gy ionizing radiation. Interestingly, activation of ATM was independent of the presence of a del(11q) (Online Supplementary Figure S3 and Online Supplementary Table S4).
17p deletion
In the del(17p) subgroup, levels of P27 and P53 were significantly higher (adjusted P values: P27: <0.01 (n=99), P53: <0.001 (n=118); fold changes: P27: 1.6, P53: 3.7) (Figure 1). For CDK4 a trend to a stronger expression was observed [adjusted/unadjusted P value: 0.07/<0.01 (n=93)]. Strikingly, none of the apoptosis-regulating proteins investigated was found to be significantly differentially expressed in the del(11q) or the del(17p) subgroups (Figure 2). In order to test whether high levels of P53 protein are predictive for P53 gene mutation even in the absence of a deletion of 17p, we selected four cases without 17p deletion but strong expression of P53. A P53 gene mutation was detectable in three of these four cases (Online Supplementary Table S5).
IGHV mutation status
As expected, genomic aberrations associated with a good prognosis correlated significantly with mutated IGHV genes in our cohort [adjusted P values: del(13q): P=0.01 (n=35); normal karyotype: P=0.33 (n=25)]. In contrast, cytogenetic aberrations associated with a worse outcome, such as del(17p) and del(11q), are linked with unmutated IGHV genes2–4,26 [adjusted P values in this cohort: del(11q): P=0.04 (n=39); del(17p): P=0.02 (n=32)] and higher levels of ZAP702 (in this cohort: unadjusted P value: 0.03, adjusted P value: 0.54 (n=22); significance lost due to the small number of informative cases). The only correlation of IGHV status with protein expression detected in the present study was a lower level of BCL2 in IGHV unmutated cases [adjusted/unadjusted P values: 0.58/0.03 (n=132)]. Thus, as already found for genetic aberrations, BCL2 was expressed at lower levels in patients in higher risk categories.
Taken together, patients in hierarchical subgroups of CLL associated with a better prognosis showed differentially expressed levels of proteins associated with apoptosis. In contrast, worse prognosis subgroups were primarily characterized by different levels of proteins involved in proliferation and/or DNA-damage pathways. The observed protein expression patterns therefore suggest an imbalance of apoptosis and proliferation in CLL in benign versus aggressive subgroups, respectively.
Analysis of protein-protein correlations
To further test our hypothesis of an imbalance of apoptosis and proliferation in CLL, we investigated to what extent levels of proteins correlated among each other by performing univariate analyses as well as multivariate analysis.
In univariate analysis significant correlations were found between AKT1 and ARF3, BCL2 and CDK4, ATM and STAT6, CDK4 and P27, P53 and ARF3, CIAP2 and SMAC as well as P53 and RB (Table 2 and Online Supplementary Figure S4). The positive correlation of CDK4/P27, CIAP2/SMAC and P53/RB as well as the inverse correlation found for BCL2/CDK4 corroborate our hypothesis that there seems to be an imbalance between apoptosis and proliferation/DNA damage control in CLL when comparing genetically low risk with high risk cases (Figure 2).
Table 2.
Results of univariate analysis (pair-wise marginal protein-protein correlations). The positive correlation of CDK4/P27, CIAP2/SMAC and P53/RB as well as the inverse correlation found for BCL2/CDK4 corroborate our hypothesis that there seems to be an imbalance of apoptosis and proliferation/DNA damage control in CLL cells, when comparing genetically low-risk with high-risk cases.

For multivariate analysis, missing data were statistically imputed. For each pair of proteins we controlled effects of all remaining proteins and of cytogenetic aberrations. Proteins and patients with the largest proportion of missing values were excluded from the analysis (Online Supplementary Design and Methods). The analyzed dataset with imputed missing values comprised 15 proteins and 139 patients. The strongest correlations were found for ARF3 and STAT6, ARF3 and AKT1 as well as for BCL2 and MCL1 (correlation coefficients: 0.22, −0.21, and 0.20, respectively) (Figure 3). The indirect negative correlation between AKT1 and STAT6 suggests that either STAT6-signaling or AKT1-signaling is affected in CLL (Online Supplementary Figure S5). Only a low number of significant correlations between proteins were revealed in multivariate analysis. This could be partly due to the missing values that had to be imputed. However, it also shows an influence of genetic aberrations on the protein expression pattern of CLL cells.
Figure 3.
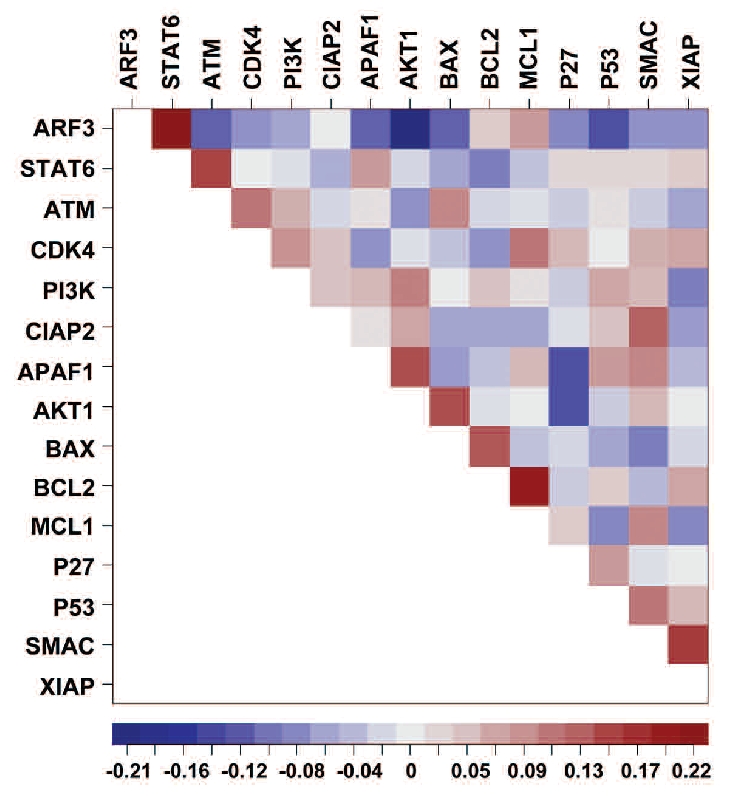
Heat map showing multivariate analysis of protein-protein correlations taking cytogenetics into account (imputed data set) (N=139). The order of the proteins is determined by a seriation method which sorts the pairs of proteins with the strongest correlations close to the diagonal. The strongest correlations were found for ARF3 and STAT6, ARF3 and AKT1 as well as for BCL2 and MCL1 (correlation coefficients: 0.22, −0.21, and 0.20, respectively).
Expression of CDK4, P27 and P53 correlates with hierarchical risk
To test whether any proteins predict for prognosis of CLL patients we correlated protein levels to ordinal hierarchical risk as a surrogate marker for survival. Patients were stratified into four categories representing increasing risk [IGHV mutated < IGHV unmutated/V3–21 < del(11q) < del(17p)]. These categories have been proposed as a model for prognostic prediction.3,26 Continuation ratio models for the correlation of protein expression with hierarchical risk were calculated. Interestingly, a significant univariate effect on the hierarchical risk group was detected for CDK4, P27 and P53 (adjusted P values: CDK4: P<0.001 (N=90); P27: P<0.001 (N=98); P53: P<0.01 (N=117)). For all three proteins, higher levels were associated with an increase in the risk category (Figure 4A).
Figure 4.

(A) Diagrams of ordinality risk for CDK4, P27 and P53. Protein expression was correlated to ordinal hierarchical risk as a surrogate marker for survival. Patients were stratified into four categories representing increasing risk [IGHV mutated < IGHV unmutated/V3–21 < del(11q) < del(17p)].(3, 26) Continuation ratio (CR) models for correlation of protein expression with hierarchical risk (univariate analysis) were calculated based on the data detected in this study and the assumption of a hierarchical risk as a surrogate marker for survival. Solid lines connect the observed mean log expression values per risk category, while dashed lines show the estimated lines according to the CR model without interaction effects. Diagrams show an increase of the risk category with increasing expression of CDK4, P27 and P53 [Wald-tests for overall effects at a significance level of 0.05; adjusted P values: CDK4: P<0.001 (n=90); P27 P<0.001 (n=98); P53: P<0.01 (n=117)]. (B) Kaplan-Meier curves for overall survival from diagnosis stratified by protein expression levels (high: log(expression) > median, low: log(expression) ≤ median). The given P values correspond to the log-rank tests between these groups (CDK4 (n=18), P27 (n=20), and P53 (n=25)). As the relation between death hazard and P53 protein expression level is highly non-linear three strata (with cut-offs at the 33%-quantile and 67%-quantile) were used for P53. The figure shows that survival rates are better for the intermediate group. This fits nicely with the observed non-linear effect of P53 on the hierarchical risk (Figure 4a), where the two high-risk groups del(11q) and del(17p) correspond to either very low P53 expression (11q) or very high P53 expression (17p), while the lower-risk groups showed intermediate P53 expression.
To investigate whether this association also leads to a correlation with treatment-free survival or overall survival, Kaplan-Meier curves were calculated for protein levels of CDK4, P27 and P53. Clinical data were only available together with protein expression data for a small number of patients, and associations should, therefore, be viewed with caution. However, in this small group of patients, higher levels of CDK4, P27 and P53 were associated with shorter treatment-free survival and overall survival (Figure 4A, B, Online Supplementary Figure S6). Combination of expression levels of the three key proteins does not increase the predictive value (Online Supplementary Table S6). In order to test whether expression levels of CDK4, P27 and P53 were stable over the clinical course, their levels of expression were analyzed in samples from two CLL patients over the course of 2 years without treatment and in samples from two patients before and after treatment (Online Supplementary Tables S2 and S3). No significant longitudinal changes in expression levels were be detected (Online Supplementary Figure S7). Suggesting an important role in CLL, expression of P27 and CDK4 was substantially higher in CLL cell samples than in CD19-sorted non-malignant B-cell samples from healthy donors (Online Supplementary Table S2, Online Supplementary Figure S7). Expression levels of P53 were very low compared to levels in EHEB cells.
Discussion
Despite extensive research, the underlying pathomechanism of CLL is still unclear.2,5 Recurrent genomic aberrations are clearly defined in CLL and likely reflect different biological and prognostic subgroups of patients.1,2 Recent advances in uncovering post-transcriptional regulation, especially by the identification of miR genes, strongly encourage the investigation of the proteome to understand CLL pathophenotypes.7 Due to substantial post-transcriptional regulation, protein levels should reflect cellular phenotype and pathogenic differences better than levels of mRNA alone. In order to assess the contribution of proteins that control apoptosis, proliferation, DNA damage and signaling in CLL, we analyzed a set of proteins from molecular pathways that are central in carcinogenesis21 in a large and well-characterized cohort of CLL patients. Cytogenetic aberrations were not defined in previous protein expression studies on CLL. These studies were, therefore, not able to test whether hierarchical genetic and prognostic CLL subgroups differ with regard to protein expression.27,28
High expression of BCL2 was observed in patients with del(13q). The role of BCL2 as a target of miR-16-1 and miR-15a has been disputed.8,9,29 However, the observed significantly higher levels of BCL2 in patients with del(13q) suggest a link to miR-16-1 and miR-15a. Levels of proteins such as CDK4 and P27, which are involved in cell cycle control, were significantly lower than in the rest of the cohort of patients, possibly as a result of stronger expression of miR-34a which targets translation of these proteins. However, it should be remembered that CDK4 has also been reported to be a target of miR-16-1 and miR-15a,30 which would lead to higher expression upon their loss. In CLL patients with del(11q) or del(17p), in whom expression of miR-34a is low,6,11,12 levels of P27 and CDK4 were significantly higher pointing to a stronger potential of the cells to proliferate. Even if peripheral CLL cells are arrested in the G1/G0 phase of the cell cycle, high levels of CDK4 could be a marker for recent proliferation in microenvironmental niches that support proliferation (i.e. lymph nodes).31
Cases with trisomy 12 rarely show P53 mutations or acquire these over time.32 In addition, CLL with trisomy 12 seems to be more sensitive to chemotherapy than other genetic subgroups of CLL.17 Intriguingly, in this study we found that CLL with trisomy 12 was characterized by low levels of P53. As cells that harbor mutations of P53 show strong induction of the aberrant P53 tumor suppressor protein,22,32,33 our finding of low levels of P53 protein in CLL cases with trisomy 12 fits with an absence of P53 mutations in this subgroup. The levels of other proteins involved in response to DNA damage, such as ATM and RB, were also different in CLL cases with trisomy 12, although only as a trend (Figure 2). Interestingly, this trend towards inactivation of ATM has also been found in mantle cell lymphoma with trisomy 12.34 Together with our findings for PI3K, BCL2 (trend) and CCND3 (trend), this may point to a higher potential of CLL cells with trisomy 12 to undergo apoptosis than CLL cells of the del(13q) subgroup, and a lower disposition of these cells to proliferate when compared to the high risk subgroups. Overall, regarding protein expression, CLL patients with trisomy 12 seem to represent an intermediate functional entity in line with their intermediate prognosis (Figure 2).1
In CLL with del(11q), mRNA levels of P53 are not significantly different to those in other subgroups.19,20 Nevertheless, we found low levels of P53 protein in this subgroup. Differences in P53 protein levels are, therefore, likely caused by post-transcriptional regulation, possibly due to reduced inhibition of MDM2 by loss-of-function of ATM in the del(11q) subgroup, leading to enhanced P53 degradation.35 In contrast to a reduction of ATM transcript levels in cases with del(11q), probably as a result of lower gene dosage, no significant differences in levels of ATM protein were detectable between the cohort with del(11q) and all other patients.19,20 This may be because ATM protein expression is affected only in a subset of the del(11q) cases. Assessment of the phosphorylation status of ATM kinase in response to ionizing radiation did not show differences in cases with or without del(11q). These findings could indicate a possible involvement of genes or microRNA other than ATM in the pathogenesis of CLL cases with del(11q).36 As in the del(17p) subgroup, a trend towards a higher expression of CDK4 was found in CLL with del(11q). Both deletions are associated with an unfavorable clinical course, and, interestingly, the deletion 11q23 harbors miR-34-b and –c, which target CDK4.1,6,11,12,14 We were, however, unable to detect expression of miR-34b/c in malignant and non-malignant lymphocytes (data not shown) and could not test whether increased levels of CDK4 are the result of reduced miR-34b/c levels. Nevertheless, our findings indicate that CLL cells with del(11q) tend towards enhanced proliferation (Figure 2).
The significantly higher expression of P53 in the del(17p) group confirms its role as the affected gene on 17p, with mutations that cause accumulation of aberrant/non-functional P53 protein.22 The high levels of P27 and CDK4 detected in the del(17p) subgroup could potentially be a result of down-regulated miR-34a with consequently reduced translational inhibition of the proteins.6 High levels of P27 protein and their association with a poor prognosis in CLL have been demonstrated before.37,38 As in the del(11q) subgroup, the higher expression of CDK4 and P27 suggests more pronounced proliferative signals in patients with del(17p).
Until recently, CLL has been regarded as a disease resulting from an inherent apoptotic defect involving the entire mass of leukemic cells.2 Especially when compared to more aggressive but genetically similar lymphomas such as mantle cell lymphoma, deregulation of primarily apoptosis-associated genes has been detected in CLL.39,40 However, CLL cells also proliferate, even if at lower rates than normal B cells.2,41,42 Here, this concept is strengthened by stratifying CLL subgroups according to their protein expression profiles into three functional categories: regulation of apoptosis, proliferation and DNA damage (Figure 2). It seems that in patients with low-risk genetic lesions such as del(13q) or a normal karyotype, a higher number of apoptosis-regulating proteins are differentially expressed, whereas proteins responsible for cell cycle or DNA damage control are present at the same or lower levels. In contrast, in CLL subgroups with a high risk of rapid disease progression, a more pronounced deregulation of proteins involved in DNA damage and proliferation can be observed. Based on these results we hypothesize an imbalance of proliferation and apoptosis in CLL, depending on the genetic subgroup affiliation (Figure 2).
Moreover, our study identifies CDK4, P27 and P53 as proteins whose levels correlate significantly with increasing hierarchical risk subgroups.3,26 P53 has already been described as a marker of disease progression and poor prognosis in CLL.18,22,33 High protein levels of P53 are consistently associated with a mutation in the P53 gene that render the protein functionally defective.18,22,23 In the series of patients analyzed here, four CLL cases were identified with high levels of P53 protein but without a del(17p). In three of the four cases a loss-of function point mutation was found in the P53 gene. This finding reinforces the model that loss-of-function of P53 by point mutations leads to elevated levels of P53 protein.18,22,23 In addition, we reproducibly found different levels of P53 protein in CLL patients with del(17p) (high P53) and del(11q) (low P53).18,22,23 Thus, elevated levels of P53 protein seem to be helpful for identifying patients without del(17p) but with point mutations in the P53 gene and an unfavorable prognosis.18,22,23 Intriguingly, the cell cycle regulatory protein P27 is also over-represented in patients with loss of 17p and correlates with hierarchical subgroups.43 P27 regulates cell proliferation, cell motility and apoptosis.16 P27 protein can exert both positive and negative effects on these processes, and up-regulation of P27 can participate in the resistance of tumor cells to anticancer treatments.16 This may explain previous associations of high levels of P27 with rapid disease progression and poor outcome in CLL, whereas in solid tumors an inverse correlation has been shown.16,37,38 P27 is an example of the importance of quantifying protein levels, as its function is tightly regulated by protein degradation.16
Apart from P53 and P27, CDK4 is associated with hierarchical risk. CDK members are components of the cell cycle machinery that governs the transition between phases during cell cycle progression, and deregulated CDK activity is a hallmark of malignancy.44 This knowledge provides a rationale for regarding CDK as potential targets for novel therapeutic compounds.44 Interestingly, inhibitors of CDK such as roscovitine or flavopiridol seem to have the ability to induce apoptosis also in CLL with loss-of-function of the P53/ATM pathway.45,46 The strong expression of CDK4 in high risk CLL and its association with hierarchical risk may partly explain this phenomenon. Only a few proteins were found at different levels independently of genetic lesions. This supports the concept of cytogenetic lesions being causative in defining distinct biological subgroups of CLL. A novel finding was a significant inverse partial correlation between STAT6 and AKT1, which was independent of genetic aberrations suggesting that either the STAT-signaling pathway or the AKT-signaling pathway is deregulated in CLL. Both signaling pathways ultimately result in anti-apoptotic signaling.47,48 We observed higher levels of AKT1 in cases with del(13q) as a sole abnormality, concordant with previous observations regarding AKT1 mRNA.20 This strengthens the notion that drugs that interfere with the AKT/PI3K pathway are a highly attractive new approach for the treatment of CLL and other malignancies.49,50 In fact, a novel inhibitor of AKT has been shown to exert anti-proliferative activity on CLL cell lines and to induce cell death of CLL cells in vitro with typical features of apoptosis, such as activation of caspase-3 and release of cytochrome c.49,50
Earlier studies found an association of high levels of BCL2 and MCL1 proteins with poor prognostic factors, shortened survival and failure to achieve remission in response to established CLL therapies.27,28,51,52 As reported previously,51 levels of MCL1 were independent of cytogenetics in the present study. Concerning BCL2, higher levels were associated with lower risk categories. Although this finding is in line with an earlier study showing that low levels of BCL2 mRNA were associated with the poor-risk subgroup del(17p),20 the place of BCL2 protein in prognostic prediction of CLL remains ambiguous.27,28
In summary, the present study shows that. (i) cytogenetic aberrations in CLL influence the biological phenotype of the malignant cells, resulting in aberrant protein levels; (ii) differences in expression levels of proteins controlling apoptosis and proliferation seem to define distinct prognostic subgroups of CLL; and (iii) levels of P27, CDK4 and P53 proteins correlate with and are potential surrogate markers for hierarchical risk.
The promising findings of this present study, which investigated 23 proteins in a cohort of 185 patients, suggest that more comprehensive proteome analyses with more advanced technologies will be rewarding in CLL. Studies of the CLL proteome will likely be of help to expand the spectrum of patient/subgroup-tailored therapies for CLL.
Acknowledgments
the A-T cell lines AO and ER were kindly provided by Drs. AMR Taylor and T Stankovic. We thank Christina Galler and Natalie Kinzer for excellent technical assistance. We would also like to acknowledge Nupur Bhattacharya and Susanne Stöhr for helpful discussions and critical reading of the manuscript.
Footnotes
Funding: this research was supported by grants from the “Deutsche José Carreras Leukämie-Stiftung” (DJCLS R06/13), the “Max-Eder Program of the Deutsche Krebshilfe” (#107239), the Helmholtz SBCancer Alliance for Systems Biology, the “Else Kröner Fresenius Stiftung” (P20/07//A11/07), the Landesstiftung Baden-Württemberg (PLS-Prot15), and the Global CLL Research Foundation.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 2.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–15. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 3.Krober A, Seiler T, Benner A, Bullinger L, Bruckle E, Lichter P, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood. 2002;100(4):1410–6. [PubMed] [Google Scholar]
- 4.Thorselius M, Krober A, Murray F, Thunberg U, Tobin G, Buhler A, et al. Strikingly homologous immunoglobulin gene rearrangements and poor outcome in VH3-21-using chronic lymphocytic leukemia patients independent of geographic origin and mutational status. Blood. 2006;107(7):2889–94. doi: 10.1182/blood-2005-06-2227. [DOI] [PubMed] [Google Scholar]
- 5.Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10(1):37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 6.Zenz T, Mohr J, Eldering E, Kater AP, Buhler A, Kienle D, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801–8. doi: 10.1182/blood-2008-08-172254. [DOI] [PubMed] [Google Scholar]
- 7.Mertens D, Philippen A, Ruppel M, Allegra D, Bhattacharya N, Tschuch C, et al. Chronic lymphocytic leukemia and 13q14: miRs and more. Leuk Lymphoma. 2009;50(3):502–5. doi: 10.1080/10428190902763509. [DOI] [PubMed] [Google Scholar]
- 8.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99(24):15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 10.Winkler D, Dohner H, Stilgenbauer S. Genetics, gene expression, and targeted therapies in chronic lymphocytic leukemia. Curr Drug Targets. 2006;7(10):1313–27. doi: 10.2174/138945006778559184. [DOI] [PubMed] [Google Scholar]
- 11.Dijkstra MK, van Lom K, Tielemans D, Elstrodt F, Langerak AW, van ‘t Veer MB, et al. 17p13/TP53 deletion in B-CLL patients is associated with microRNA-34a downregulation. Leukemia. 2009;23(3):625–7. doi: 10.1038/leu.2008.264. [DOI] [PubMed] [Google Scholar]
- 12.Mraz M, Pospisilova S, Malinova K, Slapak I, Mayer J. MicroRNAs in chronic lymphocytic leukemia pathogenesis and disease subtypes. Leuk Lymphoma. 2009;50(3):506–9. doi: 10.1080/10428190902763517. [DOI] [PubMed] [Google Scholar]
- 13.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA. 2002;99(10):6955–60. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X, He L, Hannon GJ. The guardian‘s little helper: microRNAs in the p53 tumor suppressor network. Cancer Res. 2007;67(23):11099–101. doi: 10.1158/0008-5472.CAN-07-2672. [DOI] [PubMed] [Google Scholar]
- 15.Petlickovski A, Laurenti L, Li X, Marietti S, Chiusolo P, Sica S, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood. 2005;105(12):4820–7. doi: 10.1182/blood-2004-07-2669. [DOI] [PubMed] [Google Scholar]
- 16.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253–67. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 17.Stilgenbauer S, Eichhorst BF, Busch R, Zenz T, Winkler D, Buhler A, et al. Biologic and clinical markers for outcome after fludarabine (F) or F plus cyclophosphamide (FC) -comprehensive analysis of the CLL4 Trial of the GCLLSG. ASH Annual Meeting Abstracts 2008. 2008;112(11):2089. [Google Scholar]
- 18.Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood. 2001;98(3):814–22. doi: 10.1182/blood.v98.3.814. [DOI] [PubMed] [Google Scholar]
- 19.Haslinger C, Schweifer N, Stilgenbauer S, Dohner H, Lichter P, Kraut N, et al. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. J Clin Oncol. 2004;22 (19):3937–49. doi: 10.1200/JCO.2004.12.133. [DOI] [PubMed] [Google Scholar]
- 20.Kienle DL, Korz C, Hosch B, Benner A, Mertens D, Habermann A, et al. Evidence for distinct pathomechanisms in genetic subgroups of chronic lymphocytic leukemia revealed by quantitative expression analysis of cell cycle, activation, and apoptosis-associated genes. J Clin Oncol. 2005;23(16):3780–92. doi: 10.1200/JCO.2005.02.568. [DOI] [PubMed] [Google Scholar]
- 21.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 22.Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112(8):3322–9. doi: 10.1182/blood-2008-04-154070. [DOI] [PubMed] [Google Scholar]
- 23.Bender R, Benner A. Calculating ordinal regression models in SAS and S-Plus. Biometrical J. 2000;42(6):677–99. [Google Scholar]
- 24.Harrell F. Regression Modeling Strategies. Springer-Verlag; New York: 2001. [Google Scholar]
- 25.Cuni S, Perez-Aciego P, Perez-Chacon G, Vargas JA, Sanchez A, Martin-Saavedra FM, et al. A sustained activation of PI3K/NF-kappaB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia. 2004;18(8):1391–400. doi: 10.1038/sj.leu.2403398. [DOI] [PubMed] [Google Scholar]
- 26.Krober A, Bloehdorn J, Hafner S, Buhler A, Seiler T, Kienle D, et al. Additional genetic high-risk features such as 11q deletion, 17p deletion, and V3-21 usage characterize discordance of ZAP-70 and VH mutation status in chronic lymphocytic leukemia. J Clin Oncol. 2006;24(6):969–75. doi: 10.1200/JCO.2005.03.7184. [DOI] [PubMed] [Google Scholar]
- 27.Faderl S, Keating MJ, Do KA, Liang SY, Kantarjian HM, O’Brien S, et al. Expression profile of 11 proteins and their prognostic significance in patients with chronic lymphocytic leukemia (CLL) Leukemia. 2002;16(6):1045–52. doi: 10.1038/sj.leu.2402540. [DOI] [PubMed] [Google Scholar]
- 28.Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood. 1998;91(9):3379–89. [PubMed] [Google Scholar]
- 29.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102(39):13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, Hasan L, et al. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009;69(13):5553–9. doi: 10.1158/0008-5472.CAN-08-4277. [DOI] [PubMed] [Google Scholar]
- 31.Schmid CIPG. Proliferation centres in B-cell malignant lymphoma, lymphocytic (B-CLL): an immunophenotypic study. Histopathology. 1994;24(5):445–51. doi: 10.1111/j.1365-2559.1994.tb00553.x. [DOI] [PubMed] [Google Scholar]
- 32.Zenz T, Smadova J, Vollmer D, Trbusek M, Benner A, Habe S, et al. TP53 Abnormalities in chronic lymphocytic leukemia exhibit a disease specific profile: meta-analysis of 270 Mutations. ASH Annual Meeting Abstracts 2008. 2008;112(11):2077. [Google Scholar]
- 33.Cordone I, Masi S, Mauro FR, Soddu S, Morsilli O, Valentini T, et al. p53 expression in B-cell chronic lymphocytic leukemia: a marker of disease progression and poor prognosis. Blood. 1998;91(11):4342–9. [PubMed] [Google Scholar]
- 34.Camacho E, Hernandez L, Hernandez S, Tort F, Bellosillo B, Bea S, et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood. 2002;99(1):238–44. doi: 10.1182/blood.v99.1.238. [DOI] [PubMed] [Google Scholar]
- 35.Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci USA. 1999;96(26):14973–7. doi: 10.1073/pnas.96.26.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalla C, Scheuermann MO, Kube I, Schlotter M, Mertens D, Dohner H, et al. Analysis of 11q22–q23 deletion target genes in B-cell chronic lymphocytic leukaemia: evidence for a pathogenic role of NPAT, CUL5, and PPP2R1B. Eur J Cancer. 2007;43(8):1328–35. doi: 10.1016/j.ejca.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Vrhovac R, Delmer A, Tang R, Marie JP, Zittoun R, Ajchenbaum-Cymbalista F. Prognostic significance of the cell cycle inhibitor p27Kip1 in chronic B-cell lymphocytic leukemia. Blood. 1998;91(12):4694–700. [PubMed] [Google Scholar]
- 38.Wolowiec D, Wojtowicz M, Ciszak L, Kosmaczewska A, Frydecka I, Potoczek S, et al. High intracellular content of cyclin-dependent kinase inhibitor p27(Kip1) in early- and intermediate stage B-cell chronic lymphocytic leukemia lymphocytes predicts rapid progression of the disease. Eur J Haematol. 2009;82(4):260–6. doi: 10.1111/j.1600-0609.2008.01196.x. [DOI] [PubMed] [Google Scholar]
- 39.Bentz M, Plesch A, Bullinger L, Stilgenbauer S, Ott G, Muller-Hermelink HK, et al. t(11;14)-positive mantle cell lymphomas exhibit complex karyotypes and share similarities with B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer. 2000;27(3):285–94. [PubMed] [Google Scholar]
- 40.Korz C, Pscherer A, Benner A, Mertens D, Schaffner C, Leupolt E, et al. Evidence for distinct pathomechanisms in B-cell chronic lymphocytic leukemia and mantle cell lymphoma by quantitative expression analysis of cell cycle and apoptosis-associated genes. Blood. 2002;99(12):4554–61. doi: 10.1182/blood.v99.12.4554. [DOI] [PubMed] [Google Scholar]
- 41.Defoiche J, Debacq C, Asquith B, Zhang Y, Burny A, Bron D, et al. Reduction of B cell turnover in chronic lymphocytic leukaemia. Br J Haematol. 2008;143(2):240–7. doi: 10.1111/j.1365-2141.2008.07348.x. [DOI] [PubMed] [Google Scholar]
- 42.Macallan DC, Wallace DL, Zhang Y, Ghattas H, Asquith B, de Lara C, et al. B-cell kinetics in humans: rapid turnover of peripheral blood memory cells. Blood. 2005;105(9):3633–40. doi: 10.1182/blood-2004-09-3740. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101(4):1220–35. doi: 10.1182/blood-2002-07-2009. [DOI] [PubMed] [Google Scholar]
- 44.Diaz-Padilla I, Siu LL, Duran I. Cyclin-dependent kinase inhibitors as potential targeted anticancer agents. Invest New Drugs. 2009 Mar 5; doi: 10.1007/s10637-009-9236-6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 45.Alvi AJ, Austen B, Weston VJ, Fegan C, MacCallum D, Gianella-Borradori A, et al. A novel CDK inhibitor, CYC202 (R-roscovitine), overcomes the defect in p53-dependent apoptosis in B-CLL by down-regulation of genes involved in transcription regulation and survival. Blood. 2005;105(11):4484–91. doi: 10.1182/blood-2004-07-2713. [DOI] [PubMed] [Google Scholar]
- 46.Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, Fischer B, et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood. 2007;109(2):399–404. doi: 10.1182/blood-2006-05-020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruns HA, Kaplan MH. The role of constitutively active Stat6 in leukemia and lymphoma. Crit Rev Oncol Hematol. 2006;57(3):245–53. doi: 10.1016/j.critrevonc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 48.Pogue SL, Kurosaki T, Bolen J, Herbst R. B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J Immunol. 2000;165(3):1300–6. doi: 10.4049/jimmunol.165.3.1300. [DOI] [PubMed] [Google Scholar]
- 49.Levy DS, Kahana JA, Kumar R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood. 2009;113(8):1723–9. doi: 10.1182/blood-2008-02-137737. [DOI] [PubMed] [Google Scholar]
- 50.Winkler D, Zenz T, Mertens D, Habermann A, Dohner H, Stilgenbauer S. In-vitro treatment with the AKT-inhibitor GSK 690693 induces cell death in lymphoma cell lines and in primary CLL cells and is followed by caspase-3 activation and cytochrome C release. ASH Annual Meeting Abstracts 2008. 2008;112(11):1589. [Google Scholar]
- 51.Awan FT, Kay NE, Davis ME, Wu W, Geyer SM, Leung N, et al. Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood. 2009;113(3):535–7. doi: 10.1182/blood-2008-08-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112(9):3807–17. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]