


Abstract
During macronuclear development in the ciliated protozoan Tetrahymena thermophila, extensive DNA deletions occur, eliminating thousands of internal eliminated sequences (IESs). Using an rDNA-based transformation assay we have analyzed the role during DNA deletion of DNA flanking mse2.9, an IES within the second intron of a gene encoding an as yet incompletely characterized protein. We establish that a cis-acting sequence for mse2.9 deletion acts at a distance to specify deletion boundaries. A complex sequence element necessary for efficient and accurate mse2.9 deletion is located in the region 47–81 bp from the right side of mse2.9. The ability of a variety of IES flanking sequences to rescue a processing deficient mse2.9 construct indicates that some cis-acting signal is shared among different IESs. In addition, the short intronic sequence that flanks mse2.9 is able to direct efficient and accurate processing. Despite no obvious sequence similarity between mse2.9 and other IESs, we suggest that a common mechanism is used to delete different families of IESs in Tetrahymena.
INTRODUCTION
Developmentally-programmed DNA rearrangements occur in a wide variety of organisms, from bacteria to humans (1). Functions such as altering gene dosage or directly regulating gene expression have been assigned to many, but not all, of these programmed DNA rearrangements. An important example of a programmed DNA rearrangement is V(D)J recombination, which leads to antibody diversity (2). Some parasites use programmed gene rearrangements to vary surface antigens to avoid host immune response (3). Extensive genome remodeling is required to assemble genes in some hypotrichous ciliated protozoa (4,5). The function of other programmed DNA rearrangements is not as clear. The extensive genome rearrangements that occur during nuclear development in the oligohymenophoran ciliate, Tetrahymena thermophila (6), provide an example of programmed DNA rearrangements with poorly understood function.
Like all ciliated protozoa, Tetrahymena displays a nuclear dimorphism with a mostly transcriptionally silent diploid germ-line nucleus (micronucleus) and a highly polyploid and transcriptionally active somatic nucleus (macronucleus) within the same cell. The macronucleus develops from the micronucleus during conjugation (7). When two cells of different mating types conjugate, the micronucleus in each divides meiotically and mitotically to generate a haploid gametic nucleus that is reciprocally exchanged and fuses with that of its partner to form a zygotic nucleus. This zygotic nucleus divides, and from one of the products develops a new macronucleus. The old macronucleus is concurrently degraded in an apoptotic-like manner (8). In Tetrahymena, macronuclear development involves extensive programmed DNA rearrangements including chromosome fragmentation at defined sites followed by telomere addition, DNA amplification and site-specific interstitial DNA deletion (reviewed in 6).
In Tetrahymena interstitial DNA deletion is responsible for the elimination of ~10–15% of the germ-line genome, representing more than 5000 single and multi-copy elements (9). The size of these internal eliminated sequences (IESs) in Tetrahymena ranges from 0.6 to over 13 kb. Different IESs are generally non-conserved in sequence, AT-rich, and most are flanked by short non-conserved direct repeats. Alternate forms of rearrangement may exist for up to 25% of IESs (10) and varying degrees of microheterogeneity are seen at macronuclear junctions (11–13). IESs have not yet been found in coding sequence in Tetrahymena, but two are located within introns (14,15).
Possible functions of IESs in the micronucleus and the reasons for their elimination remain unclear. An interesting possibility suggests they participate in functions unique to micronuclei such as mitosis, meiosis and associated chromosome condensation (6,16).
Several Tetrahymena IESs have been well characterized at the molecular level. The tightly linked M (17) and R elements (18) were the first IESs characterized in Tetrahymena. The M element uses an alternative left boundary resulting in either a 0.6 or 0.9 kb deletion. A 10 bp A5G5 tract is present ~45 bp outside of M on both sides in macronuclear sequence. It also appears at the same distance from the alternative left junction that results in the smaller deletion. The sequence has been shown to be necessary and sufficient for M element deletion, and to control deletion boundaries at a distance (19,20). This polypurine tract has not been found flanking any other IES to date. Other cis-acting sequences in flanking DNA have been shown to be necessary for programmed deletion of mse2.9, Tlr1 and the R element (12,21,22).
Like M, the controlling sequences for R deletion flank the IES on both sides. Although the exact nature of these sequences is unclear, they appear to act similarly to the polypurine tract of M to specify deletion boundaries at a distance, suggesting a common mechanism for the deletion of M and R (22).
The Tlr1 element is a large 13 kb transposon-like element (23). The boundaries of Tlr1 are characterized by an 825 bp inverted repeat. A cis-acting sequence in flanking DNA 51 bp or more from the right junction has recently been shown to be required for accurate deletion of Tlr1 (21). Other IESs in Tetrahymena include the IES found in DNA flanking the histone HI gene (24), and an IES found within an intron of the PGM gene (15).
A 2.9 kb IES, mse2.9 (14), occurs within the second intron of the ARP1 locus, a gene encoding a highly acidic protein of unknown function containing numerous internal repeats. Mse2.9 has 81% AT content and the termini are located within TTAT direct repeats. Extensive microheterogeneity is found at mse2.9 macronuclear junctions (12). More than 66 bp of flanking DNA at the left side of mse2.9 was shown to be required for processing (12). A candidate cis-acting sequence was proposed to be a 10 bp sequence that is present 70–90 bp from the left and right boundaries of mse2.9 (12). A similar sequence motif occurs 94 bp from an alternative junction on the left side of mse2.9 that is only used in the absence of left flanking sequences (12).
Here we have refined the cis-acting requirements for mse2.9 deletion. We have established that the cis-acting sequence for mse2.9 deletion acts at a distance to specify deletion boundaries. This establishes clear mechanistic links with the excision of the M and R elements. We have demonstrated that sequence important for mse2.9 elimination is located in flanking DNA, between 37 and 81 bp from the right deletion junction. A candidate cis-acting sequence was determined by deleting an A-rich sequence occurring between 48 and 59 bp that inhibits mse2.9 processing. We demonstrate that this sequence requires up to 20 bp of downstream sequence to specify efficient and accurate processing suggesting that the mse2.9 controlling sequence may contain distinct sequence elements. In addition we show that the intronic sequence flanking mse2.9 contains the controlling sequences for its accurate and efficient programmed deletion. We also show that a functional difference exists between flanking sequence at the left and right boundaries of mse2.9 as that on the left of mse2.9 can only partially substitute for that on the right. M and R flanking sequences are also able to partially substitute for mse2.9 flanking sequence. Flanking sequence from the H1 histone IES can almost fully substitute for mse2.9 flanking sequence, suggesting that the two IESs may be members of a class of elements that share similar cis-acting signals. Despite the lack of obvious sequence identity between the flanking sequences of mse2.9, and those of other IESs, we suggest that there are enough similarities in the excision process to argue that a common mechanism directs excision of different IES families in Tetrahymena.
MATERIALS AND METHODS
Cell strains
Tetrahymena thermophila strains Cu428 [Mpr/Mpr (VII, mp-s)] and B2086 [Mpr+/Mpr+ (II, mp-s)] of inbreeding line B were provided by Dr J. Gaertig (University of Georgia, Athens, GA). Cells were cultured axenically in 1× SPP (1% proteose peptone, 0.2% glucose, 0.1% yeast extract and 0.003% EDTA: ferric sodium salt) at 30°C as described (25).
Tetrahymena transformation
Tetrahymena strains B2086 and Cu428 were used throughout this study in transformation experiments. The procedure of Gaertig and Gorovsky (26) was used to electroporate conjugating Tetrahymena. Mid-log phase cells (100 ml) were washed with 10 mM Tris–HCl pH 7.4, resuspended at 3 × 105 cells/ml, and starved for 24 h. Conjugation was initiated by mixing 50 ml of each strain in a 2 l flask and incubating without shaking at 30°C. Ten hours after mixing, cells were harvested and washed once with 10 mM HEPES pH 7.5, resuspended at 3 × 107 cells/ml and 125 µl concentrated cells were mixed with 125 µl CsCl2-purified plasmid DNA (0.25 µg/ml) in 10 mM HEPES pH 7.5. This mixture was placed within a 0.4 cm electroporation cuvette and electroporated using a BRL Cell-Porator (settings: 330 µF, 400 V, high resistance). The electroporated cells were then placed in 150 ml 1× SPP from which 200 µl was added to each well of a 96-well microtitre plate. The electroporated cells in 150 ml of 1× SPP were also diluted 1:10 (in 1× SPP) and 200 µl/well were added to another 96-well microtitre plate. Paromomycin was added to a final concentration of 100 µg/ml 12–18 h after electroporation. Transformants were identified as saturated Tetrahymena cultures in microtitre wells after 2–3 days.
DNA purification and analysis
Extraction of whole-cell DNA from Tetrahymena was essentially according to Gaertig et al. (27). Vegetatively growing cells (10 ml) were concentrated by a brief centrifugation at 1125 g in 15 ml polypropylene tubes to 0.1 ml, and resuspended in 0.7 ml of lysis buffer (42% urea, 0.35 M NaCl, 10 mM Tris–HCl pH 7.4, 10 mM EDTA and 1% SDS). Contents were mixed by inversion until the solution was homogeneous. This was extracted twice with phenol/chloroform (1:1) and once with chloroform, and 0.6 ml was transferred to 0.2 ml 5 M NaCl and precipitated with 0.8 ml isopropanol. Whole-cell DNA was spooled on a glass rod and washed twice with 0.5 ml 70% ethanol. DNA was dried under vacuum before being resuspended in 200 µl double distilled H2O plus RNase at 10 µg/ml. The DNA was incubated at 37°C for 30 min, and subsequently stored at –20°C.
Restriction enzyme digestion, agarose gel electrophoresis, DNA ligation, Southern blotting and hybridization, and transformation of Escherichia coli were carried out using standard protocols as described by Sambrook et al. (28) or by following the manufacturer’s instructions. Probes for Southern analysis were labeled by random priming (28) with [α-32P]dATP (Amersham). Restriction enzymes, T4 DNA ligase, Klenow fragment of E.coli DNA polymerase I, mung bean nuclease and alkaline phosphatase were obtained from New England Biolabs. Southern blots were imaged with a Canberra Packard Instant Imager.
Oligonucleotides
Sequences of oligonucleotides used in PCR reactions and for direct sequencing of PCR products were: 5r, 5′-AATAAGATGCAAAGCAGC; heh1, 5′-CATGATATCATAATAATAACTTTAATTAGT; heh2, 5′-CATGATATCCAATATATAAACCAATTCAAT; heh3, 5′-ATGATATCCTTTATCATTAAATTAATTTC; heh4, 5′-CATGATATCGTTTTATTTTAAACGTGTTAA; heh5, 5′-CATGATATCAACGTGTTAATAAAATAAAAATT; heh6, 5′-CATGATATCTTAACACGTTTAAAATAAAAC; heh7, 5′-CATGATATCCCCAAGCTTATTTAAGAT; heh8, CATGATATCTAATTTTTATTTTATTAACAC; heh9, 5′-CATGATATCATTGAATTGGTTTATATA; H1flankF, 5′-CATGATATCTTTTAATAAATAATTGATATT; H1flankR, 5′-CATGATATCTCTTAAAATAAAATTAACTAAC; msef/r, 5′-CATGATATCATCTAGAAAATATGTATGT; intronF, 5′-CATGATATCGTAATTTGTTATTTTATTATT; intronR, 5′-CATGATATCCTAATTTTATAGTTAAGA; MflankF, 5′-CATGATATCTGGTTAAATTTTGCTTAC; MflankR, 5′-CATGATATCTATCTTCTTTTCTGCTAAT; RflankF, 5′-CATGATATCACAATTTGAATGAAAAAAT; RflankR, 5′-CATGATATCTTTAATATTCTAAGCAA; 3r, 5′-GCTTAAACACAACTATTC; LBa2, 5′-GATTCAGACGAAGATAACGGTGAT; RU4, 5′-CCATTTTCTAATTTTATAGTTAAGAAA.
PCR analysis
PCR was used to amplify junction sequences of processing constructs obtained by transformation. The primers 5r and RU4 were used to amplify processed mse2.9 junction sequences in transformed strains, except in the deletion clones where the primers Lba2 and 3r were used. These primer sets amplify junctions only in transformed strains, as 3r and 5r are complementary to rDNA sequence. PCR was performed in a 50 µl vol containing 0.5 µM each primer, 0.2 mM each dNTP, 10× tsg buffer, 1.5 mM MgCl2 and 2.5 U tsg (Biobasic, Toronto, Canada). PCR was performed in a Perkin-Elmer 9600 thermal cycler using the following cycling conditions: one cycle at 94°C for 3 min; 35 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s; one cycle at 72°C for 5 min. Long PCR was performed with the Expand Long Template PCR system (Boehringer Mannheim), using conditions as specified by the manufacturer.
DNA sequencing
Sequencing was performed by using automated cycle sequencing with dye-labeled di-deoxy terminators and a PE/ABI 373a or 377 sequencer at the Core Molecular Biology Facility (York University, Toronto, Ontario, Canada). PCR products of mse2.9 junctions were sequenced with the RU4 primer. Deletion clones were sequenced with the 3r primer.
Plasmid construction
The construction of J120 (heh2.2 in pHSS6) has been described (12). Derivatives of heh2.2 with small internal deletions at the right side of the micronuclear-limited sequence were constructed using inverse PCR with J120 as template. The constructs heh2.2Δ19, heh2.2Δ30, heh2.2Δ54 and heh2.2Δ97 were constructed using the primer sets heh1+heh4, heh1+heh5, heh1+heh2 and heh1+heh3, respectively. Each of these primers contains an EcoRV recognition site near its 5′-end. The PCR products from the respective PCR reactions were gel purified, digested with EcoRV, ligated using T4 DNA ligase under dilute conditions and transformed into E.coli DH5αF′. In this way the deleted sequence of these constructs was replaced with a 6 bp EcoRV site. Each pHSS6-based plasmid was digested with NotI and SphI and then ligated to NotI-digested pD5H8 (20) or pD5H8NI (22) (a gift from Dr D. Chalker and Dr M.-C. Yao, Fred Hutchinson Cancer Research Center, Seattle, WA) that had been treated with calf-intestinal alkaline phosphatase (CIP). The heh2.2Δ47-61 construct was constructed using inverse PCR of J120 with the primers heh2+heh6. The PCR product was digested with EcoRV and subcloned into pD5H8 as described above. To replace right flanking sequence of mse2.9 with its left flanking sequence, heh2.2Δ97:pHSS6 was digested with EcoRV, CIP-treated and ligated to the EcoRV-digested PCR product of intronf+msef/r with J120 as template. Flanking sequence from M and R elements was similarly inserted into EcoRV-digested heh2.2Δ97 (in pHSS6). M and R flanking sequence was amplified from pCA455-3 and pMY404-21, respectively (both gifts from Dr D. Chalker) using these primer sets: Mflankf+MflankR and Rflankf+RflankR. PCR products were digested with EcoRV and subcloned into pD5H8 as above. H1 IES flanking DNA was amplified from genomic DNA using the primer set H1flankF+H1flankR and cloned into heh2.2Δ95:pHSS6 and into pD5H8 as above.
The deletion clone heh2.2+38R was constructed as described by Li and Pearlman (12). The deletion clones heh2.2+60R and heh2.2+81R were constructed by cloning into SmaI-digested, CIP-treated pD5H8N1 the EcoRV-digested PCR products of J120 template with primer sets heh7+heh8 and heh7+heh9. The heh:intron construct was similarly constructed with J120 as a template and the primers intron F+intron R.
RESULTS
The cis-acting sequence of mse2.9 acts at a distance to specify deletion boundaries
We have further dissected the role of mse2.9 cis-acting sequences using the rDNA-based in vivo transformation assay (19,20) that was used previously (12). Derivatives of mse2.9 are cloned into the 3′ non-transcribed spacer region of the vector pD5H8 (20) [or pD5H8N1, which differs by the addition of a SmaI restriction site in the polylinker (22)] that contains the rDNA locus of Tetrahymena. When this plasmid is introduced into conjugating cells by electroporation, it is processed like the normal micronuclear rDNA locus: the locus is cleaved at the chromosome breakage sequence that flanks it on the 5′ and 3′ sides, telomeres are added to these breakage sites, the locus becomes palindromic, and this mini-chromosome is amplified to approximately 9000 copies per cell (6). The rDNA of pD5H8 outreplicates endogenous rDNA and confers resistance to the drug paromomycin (20).
Previous experiments examining mse2.9 deletion utilized a chimeric DNA, heh2.2, which is missing the internal 1.9 kb EcoRI fragment of mse2.9 (Fig. 1) (12). When heh2.2 was transformed into conjugating cells it was processed similarly to wild-type mse2.9, utilizing the same left and right junctions, and resulting in the same degree of microheterogeneity (12). External deletions of flanking sequences were used to establish the requirement for flanking DNA for mse2.9 excision (12). In this study we focus on DNA flanking the right side of heh2.2. Previously we have shown that an alternative deletion site exists in micronuclear sequence at the left side of heh2.2 (12) that would complicate the analysis described in this study.
Figure 1.

Organization of T.thermophila DNA at the mse2.9 locus and construction of heh2.2. MAC, macronuclear DNA; MIC, micronuclear DNA; ARP1, acidic repetitive protein. The area inside the dotted lines indicates micronuclear-limited DNA. Outside the lines macro- and micronuclear sequence are identical. Hatched and clear boxes denote ARP1 exons and introns, respectively. Construction of heh2.2 has been described previously (12). The solid line beneath unprocessed heh2.2 represents the probe used for Southern analysis. Intron boundaries that flank mse2.9 are indicated by thick vertical lines. H, HindIII; X, XbaI; E, EcoRI.
We have used inverse PCR to make a series of internal deletions of macronuclear-retained flanking DNA at the right boundary of micronuclear-limited sequence of heh2.2, and examined the ability of these constructs to be accurately processed. The first two constructs, heh2.2Δ19 and heh2.2Δ30 (Fig. 2A), have deletions of 25 and 36 bp, respectively, replaced with a 6 bp EcoRV site to give net shorternings of 19 and 30 bp. Deleted in both constructs is an unusual palindromic sequence that immediately flanks the right TTAT direct repeat (14). These constructs were cloned into pD5H8 and introduced by electroporation into conjugating cells. Genomic DNA was purified from paromomycin-resistant transformants, digested with HindIII, separated by agarose gel electrophoresis, blotted and probed with an [α-32P]dATP-labeled fragment of heh2.2 (Fig. 1). Figure 2B (lanes 1–4) shows that heh2.2Δ19 rearranges as efficiently as a heh2.2 control. This indicates that the 25 bp of deleted sequence, including the unusual palindrome, have no essential role in mse2.9 rearrangement. Figure 2B (lanes 5–9) shows that heh2.2Δ30 processed quite efficiently as well. To see if processing was accurate as well as efficient in these transformants, we amplified and directly sequenced rearranged junctions from several transformed lines. Microheterogeneity was not seen within the individual transformants analyzed but was seen when different transformants were compared. Figure 2B shows that for the two heh2.2Δ19 transformants sequenced, while the proper left boundary was used, the right boundary was shifted 16 or 20 bp into micronuclear sequence, approximately the same amount of sequence that was deleted. Similarly the heh2.2Δ30 transformant analyzed (Fig. 2B) showed that its right deletion boundary was shifted into micronuclear DNA by 28 bp, again approximately the same distance as the amount of sequence deleted. The results from the processing of these two constructs suggest that the regulatory sequence flanking mse2.9 specifies the deletion boundary at a fixed distance.
Figure 2.


Analysis of a series of small deletions at the right flank of heh2.2. (A) The thick horizontal line represents the micronuclear-limited sequence of heh2.2. The triangle (Δ) indicates the site of the deletions. (B) Southern blot analysis of total genomic DNA from cells transformed with heh2.2, heh2.2Δ19 (lanes 1–4) and heh2.2Δ30 (lanes 5–9) constructs. The amount of DNA (∼0.5 µg) digested in each experiment in this study ensures that only heh2.2 constructs cloned onto the high-copy rDNA-based vector are seen. DNA was digested with HindIII and probed with the HindIII–XbaI macronuclear-retained fragment of heh2.2 (Fig. 1). Sequence of several transformant deletion junctions is shown below the Southern blot. Macronuclear-destined sequence is in capital letters and deleted sequence is in lowercase. The TTAT direct repeats that flank mse2.9/heh2.2 are in bold text. Arrows emphasize shifted right (and one left) boundaries. The numbering corresponds to the distance from the underlined nucleotide within the respective direct repeat TTAT (left side of heh2.2) and TTAT (right side of heh2.2). Restriction sites are italicized. (C) Southern blot and sequence analysis of heh2.2Δ54 transformants (as in B). (D) Southern blot and sequence analysis of heh2.2Δ97 transformants (as in B). H, HindIII; X, XbaI; E, EcoRI; R, EcoRV.
In another construct, heh2.2Δ54 (Fig. 2A), 60 bp of Tetrahymena DNA at the right side of heh2.2 was replaced with an EcoRV site to give a net shorterning of 54 bp. This was again cloned into pD5H8 and introduced into conjugating cells. Southern analysis of DNA purified from transformants is shown in Figure 2C. Although the putative cis-acting sequence identified previously (12) is present in this construct, heh2.2Δ54 rearrangement is not efficient indicating that the putative cis-acting sequence is not required for mse2.9 processing. Only one transformant out of the eight analyzed shows fully processed DNA (as one of two observed products: Fig. 2C, lane 6). This suggests that the deleted DNA contains sequence necessary for mse2.9 processing. To determine if proper boundaries were utilized in the transformants that exhibit rearrangement, we amplified and directly sequenced any transformant junctions that were the size expected for accurate deletion. Direct sequencing of the PCR product from the transformant in Figure 2C (lane 6) that is near the size expected for a proper rearrangement of this construct, indicates it used the proper left boundary with the right boundary shifted 57 bp into micronuclear sequence (Fig. 2C). Combined with the previous observation that the mse2.9 controlling sequence specifies rearrangement boundaries at a distance, we suggest that this transformant represents accurately rearranged DNA. Thus although rearrangement is not efficient in the heh2.2Δ54 transformants, a small percentage of accurate rearrangement may occur with the rightward boundary shifted by approximately the same amount as the amount of sequence that was deleted. This result, combined with that from the previous two constructs, suggests that flanking DNA in the region between 37 and 60 bp from the right side of mse2.9 is required for efficient rearrangement. However, sequence to the right of the deleted region can still specify accurate rearrangement in rare cases.
Another construct, heh2.2Δ97 (Fig. 2A), is missing 103 bp of sequence to the right of heh2.2. This was introduced as a pD5H8 construct into conjugating cells and genomic DNA was purified from transformants. Figure 2D shows the Southern analysis of these transformants. Like heh2.2Δ54, rearrangement of heh2.2Δ97 is inefficient. All transformants show at least some unrearranged DNA. However, a few transformants do show some ability to aberrantly rearrange DNA (for example Fig. 2D, lanes 9 and 10). We amplified and directly sequenced transformant junctions from any PCR fragments that were near the expected size for a normal rearrangement of heh2.2Δ97. Amplification of the transformants did not yield a PCR product with the expected size for accurate deletion of heh2.2Δ97 (data not shown). Sequencing of one of the aberrantly-sized fragments that did amplify (from Fig. 2D, lane 8) indicated utilization of alternate processing sites at the left and right boundaries of heh2.2 (Fig. 2D). Another transformant (Fig. 2D, lane 9) utilized an incorrect right boundary but a correct left boundary. Thus like heh2.2Δ54, efficient rearrangement was compromised in heh2.2Δ97 transformants. However, unlike heh2.2Δ54, transformants of heh2.2Δ97 were unable to accurately rearrange the right boundary and this inaccuracy may be seen at the left boundary as well. This suggests that sequences in the flanking DNA between 61 and 103 bp from the right side of micronuclear-limited sequence may contain additional information necessary for accurate deletion of mse2.9.
Deletion of sequence between 47 and 61 bp to the right of mse2.9 inhibits rearrangement
On inspection of flanking sequence in the region between 37 and 61 bp to the right of mse2.9, we noticed an A-rich sequence between 47 and 61 bp (5′-TAAAATAAAAATTA-3′). This A-rich sequence is reminiscent of the polypurine-rich tract that flanks the M element on both sides at about the same distance from the left and right boundaries. To test whether this sequence element is important for mse2.9 processing we deleted it from heh2.2, replacing the sequence with an EcoRV site (Fig. 3A).
Figure 3.

Deletion of a 14 bp A-rich sequence that is situated in DNA flanking the right side of micronuclear sequence inhibits heh2.2 processing. (A) An EcoRV site replaces the 14 bp at a distance of 47–60 bp from the right boundary of mse2.9. (B) Southern blot analysis of whole-cell DNA purified from heh2.2Δ47–60 bp transformants. DNA was digested and probed as in Figure 2. H, HindIII; X, XbaI; E, EcoRI; R, EcoRV.
This construct yielded no accurate rearrangement (Fig. 3B), indicating that a cis-acting sequence for programmed mse2.9 deletion is at least in part located in this region.
External deletions of flanking sequence at the right side of mse2.9 suggest that sequence between 61 and 81 bp to the right of mse2.9 is also important for rearrangement
To test if all sequence necessary to specify accurate and efficient programmed mse2.9 deletion is contained within the 61 bp of flanking sequence to the right of mse2.9, we made several external deletions of heh2.2 and tested them for rearrangement. A heh2.2 deletion clone that contained only 38 bp of flanking sequence on the right side of mse2.9 (Fig. 4A, heh2.2+38R) failed to rearrange (Fig. 4B, lanes 1–5). This is not unexpected when considering that the clone does not contain the cis-acting sequence identified above. A deletion clone that contained only 60 bp of flanking sequence to the right of heh2.2 (Fig. 4A, heh2.2+60R) also failed to process (Fig. 4B, lanes 5–9). There was, however, some aberrant processing in this clone (Fig. 4C). Unlike heh2.2+38R, this clone did contain the cis-acting sequence identified above. This result suggests that although important for processing, this sequence is not by itself able to specify mse2.9 rearrangement. The next deletion clone we tested contained 81 bp of flanking sequence to the right of heh2.2 (Fig. 4A, heh2.2+81R). Rearrangement of this clone as assayed by Southern analysis was efficient (Fig. 4B, lanes 10–13). The sequencing of one of the transformant deletion junctions indicated that processing was accurate as well (Fig. 4C) in that it matched the major macronuclear mse2.9 junction (12). The differential ability of this series of external deletion clones to be processed suggests that there is additional sequence important for programmed mse2.9 deletion situated 61 to 81 bp to the right of the IES.
Figure 4.

Deletion analysis reveals that mse2.9 requires 81 bp of sequence on the right side for efficient and accurate processing. (A) The thick horizontal line represents micronuclear-limited sequence of heh2.2. (B) Southern blot analysis of genomic DNA purified from transformants of heh2.2+38R (lanes 1–5), heh2.2+60R (lanes 6–9) and heh2.2+81R (lanes 10–13) clones. Whole-cell DNA was digested and probed as in Figure 2. Longer exposure for some lanes was performed to increase visualization of DNA fragments in low abundance. (C) Sequence analysis of several deletion clone transformants as in Figure 2.
Sequences necessary for mse2.9 deletion are contained within intronic sequence
To determine if controlling sequences for mse2.9 deletion are contained within the intronic flanking sequence of mse2.9, we made a construct of heh2.2 containing the full micronuclear sequence, but containing only flanking intronic sequence (Fig. 5A, 110 bp on the left side, 140 bp on the right). We electroporated this construct as a pD5H8 clone into conjugating cells and purified the genomic DNA of transformants. PCR analysis was used to assay for efficient processing. Figure 5B indicates that processing appears to be efficient, in that only one PCR fragment is seen. Figure 5C shows that the junction of transformant in Figure 5B (lane 1) was the same as the major macronuclear junction previously identified (12). This indicates that the cis-acting requirements for efficient and accurate mse2.9 deletion are fully contained within intronic sequence.
Figure 5.
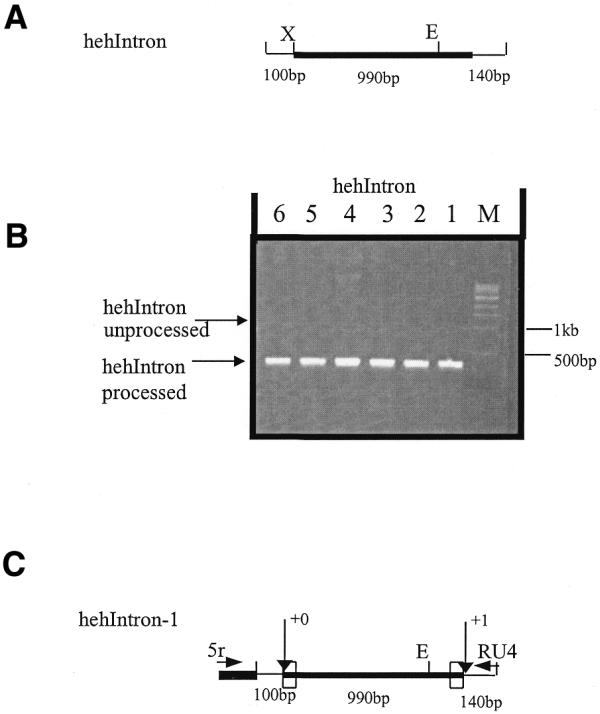
Intronic flanking sequences are sufficient for processing of heh2.2. (A) The thick horizontal line represents micronuclear-limited sequence of heh2.2. (B) Agarose gel analysis (1.2%) of PCR amplification of whole-cell DNA from hehIntron transformants. Primers 5r and RU4 were used to ensure only transformant junctions were amplified. Correct processing yields a product of ∼400 bp. (C) Arrows designate deletion junctions indicating that processing is accurate. The boxes represent the TTAT direct repeats that border mse2.9. The numbering corresponds to the distance from the underlined nucleotide within the respective direct repeat TTAT (left side of heh2.2) and TTAT (right side of heh2.2). Primers 5r (complementary to rDNA sequence) and RU4 (complementary to mse2.9 flanking sequence) are shown. X, XbaI; E, EcoRI.
Differential rescue of the heh2.2Δ97 processing defect by flanking sequence from a variety of IESs
We attempted to rescue the processing defect of heh2.2Δ97 by inserting into it flanking sequences from the other side of mse2.9. We amplified the 110 bp of intronic sequence flanking the left side of mse2.9 and inserted it into the EcoRV site in heh2.2Δ97. The sequence was inserted in the same orientation relative to its position at the left side so that the flanking sequences in the new clone, heh2.2Δ97+mseflanks, are in an inverted repeat orientation (Fig. 6A). The construct was cloned into pD5H8 and electroporated into conjugating Tetrahymena as above. Southern blot analysis of genomic DNA from transformants indicated an improvement in processing efficiency [compare Fig. 6B, lanes 1–3 (heh2.2Δ97) with lanes 4–10 (heh2.2Δ97+mseflanks)]. A wide variety of aberrant products were seen, as well as a product the size expected for accurate deletion. This suggests that the sequence was not sufficient to restore full efficiency or accuracy of processing. We amplified and attempted to directly sequence products that had the expected size for accurately processed heh2.2. We were unable to determine sequence from those PCR products, presumably due to the inverted repeat structure expected from an accurately processed PCR product. However from their size on the Southern blot (Fig. 6B), and the size of their PCR products (data not shown), we suggest that among the aberrant products are those that represent accurate deletion of heh2.2. The failure of this construct to completely rescue mse2.9 processing suggests that there are at least some differences between the controlling sequences flanking the left and right sides of mse2.9.
Figure 6.


Partial rescue of the heh2.2Δ97 processing defect. (A) Insertion of flanking sequence from the left flank of mse2.9, and right flanking sequence from the M and R element, and the H1 IES (thick horizontal rule). The arrows at the left and right flanks of construct heh2.2Δ97+mseflanks emphasize that they are in an inverted repeat configuration. (B) Southern blot analysis of whole-cell DNA from heh2.2Δ97+mseflanks transformants, as well as heh2.2Δ97 transformants (from a separate experiment than Fig. 2C), and a heh2.2 control for fully processed DNA. DNA was digested and probed as in Figure 2. (C) Southern blot analysis of heh2.2Δ97+Rflank (as in B). Sequence from a transformant deletion junction is shown under the Southern blot, as described in Figure 2. (D) Southern blot and sequence analysis of heh2.2Δ97+Mflank (as in C). (E) Southern blot and sequence analysis of heh2.2Δ97+H1IESflank (as in C). H, HindIII; X, XbaI; E, EcoRI; R, EcoRV.
To determine if flanking sequence from other IESs could functionally replace mse2.9-flanking sequence, we made chimeric heh2.2Δ97 constructs that contain flanking sequence from the right side of the R (170 bp) and M elements (135 bp), as well as the H1 IES (126 bp) (Fig. 6A). These constructs were transformed into conjugating Tetrahymena and genomic DNA of transformants was analyzed by Southern analysis. Figure 6C shows that there is a mild stimulation of the ability of heh2.2Δ97+Rflank transformants to process compared to heh2.2Δ97. Several of the transformants feature products the correct size for accurately processed heh2.2 (Fig. 6C, lanes 2, 3 and 6), which we confirmed by directly sequencing an amplified transformant junction (Fig. 6C). Figure 6D shows that similar to the previous experiment there is a mild stimulation of the ability of heh2.2Δ97+Mflank to process compared to heh2.2Δ97. A variety of aberrantly processed products are seen along with several transformants that feature fragments representing accurate heh2.2 deletion (Fig. 6D, lanes 1, 3 and 5). Again this was confirmed by directly sequencing an amplified transformant junction (Fig. 6D). Figure 6E shows that the heh2.2Δ97+H1IESflank transformants exhibited a greater stimulation of accurate deletion, with several transformants featuring completely rearranged DNA (Fig. 6E, lanes 1, 3 and 7). The accuracy of processing was confirmed by directly sequencing an amplified transformant junction (Fig. 6E). This result strongly suggests that the histone H1 IES also contains cis-acting determinants for its own developmentally-programmed excision in flanking DNA. Thus flanking DNA from the right side of the M and R elements, and the H1 IES, may substitute with differing efficiencies for that at the right side of mse2.9.
DISCUSSION
Our data suggest that the cis-acting regulatory sequence controlling mse2.9 deletion is more complex than previously suggested. We propose that the regulatory sequence at the right side of mse2.9 spans the area from 47 to 81 bp from the right boundary of mse2.9, and that it may contain distinct sequence elements that enhance rearrangement and specify proper deletion boundaries.
Regulatory cis-acting sequences have been found in DNA flanking several IESs in Tetrahymena. The polypurine tract located ∼45 bp outside of each end of the M element was the first cis-acting regulatory element reported in Tetrahymena (19). This cis-acting sequence is important in determining proper deletion boundaries of the M element, although further sequence context is likely required for fully efficient processing (20). This A5G5 tract is not present in an analogous location in other IESs in Tetrahymena suggesting initially that alternate deletion mechanisms could exist (20). However, the subsequent discovery of regulatory cis-acting sequences in DNA flanking mse2.9 (12), Trl1 (21), the R element (22) and the H1 IES (this study), despite no obvious primary sequence similarity, implies that similarities exist in the mechanism that is used by Tetrahymena to delete IESs.
Our results strongly support the idea that a common mechanism is used to delete different IESs in Tetrahymena. We have shown that the flanking regulatory sequences of mse2.9, like those of M and R, act at a distance to specify deletion boundaries. We have identified an important sequence determinant (the A-rich sequence) flanking mse2.9 that is important for processing. It appears at a distance (47–60 bp) from the right boundary of mse2.9. This is analogous to the M element A5G5 tract [45–55 bp from each boundary (19)], as well as a possible boundary determinant for the R element [within 60 bp (22)]. In addition we have shown that flanking sequences from a variety of IESs can substitute with varying efficiencies for the right flanking sequence of mse2.9.
Chalker et al. (22) have proposed that the flanking regulatory sequences for the M and R elements could be bipartite, containing sequences that primarily determine accuracy and others that enhance efficiency of processing. There is evidence that the flanking regulatory sequence of mse2.9 contains distinct sequence elements, in that the A-rich tract requires at least 20 bp of downstream sequence to specify accurate and efficient deletion. Processing of the heh2.2Δ54 construct supports this theory as well, although there is a formal possibility that it reflects coincidental aberrant rearrangement. Combined with the fact that Li and Pearlman (12) showed that >66 bp of flanking DNA is required on the left side of heh2.2 for accurate rearrangement, our data suggest that both sequences must be present for efficient processing to occur. Further studies are necessary to examine this possibility.
Chalker et al. (22) have also suggested that a pairing of flanking regulatory sequences, similar to that which occurs during V(D)J recombination (29), may be required for M and R element deletion in Tetrahymena. The fact that the M and R flanking sequence did not efficiently rescue the processing defect of heh2.2Δ97 is consistent with this idea.
The similarities of the action of the cis-acting sequence contained in flanking sequences of mse2.9, M and R argues that a common mechanism is utilized to delete IESs in Tetrahymena. There are, however, at least some differences between the deletion of mse2.9 and that of M and R. The microheterogeneity at mse2.9 junctions is more extensive than that found at junctions of M and R (12). The fact that the left flanking sequence of mse2.9 cannot fully substitute for its right flanking sequence is also an important difference. The use of identical flanking sequences completely rescued R processing (22) but only partially rescued that of heh2.2. Despite the seeming importance of the A-rich tract in sequence flanking the right side of mse2.9, a similar tract is not apparent in flanking sequence upstream of mse2.9, or upstream of the alternative processing site identified previously (12). The interaction between the left and right flanking sequences required for mse2.9 deletion is likely different from that required for R element processing.
It has been proposed that different IES families may exist in Tetrahymena that are deleted by a common mechanism but differ in their regulatory signals (20). We suggest that mse2.9 is likely to be a member of a family of IESs that uses the same mechanism for deletion as that of M and R, but that utilizes different regulatory signals in its flanking sequences. We suggest from the heh2.2Δ97 rescue experiment that another candidate member of the mse2.9 family of IESs is the H1 IES. The H1 IES flanking sequence rescued heh2.2Δ97 processing quite well implying they share some common cis-acting elements. There does not seem to be strong sequence similarity between the right flanking sequence of mse2.9 and the H1 IES other than a general AT-richness (data not shown). The lack of strong primary sequence similarity between the sequences that flank IESs in Tetrahymena, and the results of the heh2.2Δ97 rescue experiment, suggests that these regulatory signals may be related in ways other than primary sequence, perhaps possessing similar structural elements.
Saveliev and Cox (30,31) have used anchored PCR to examine reaction intermediates of M and R element excision. Their data suggest that IES excision is initiated by a 4 bp double strand break at one end of the IES. The chromosomal junction is created on one strand by a direct trans-esterification reaction of the 3′-OH of an adenosine on the opposite side of the IES. The mapping of developmentally-specific mse2.9 breakpoints will allow a test of our prediction of a common mechanism for IES excision in Tetrahymena.
The microheterogeneity found at macronuclear junctions suggests that an imprecise mechansism is used for IES excision in Tetrahymena. No IES has yet been found within a coding region in Tetrahymena. This imprecise reaction mechanism likely places constraints on the location of IESs within the genome. Microheterogeneity in processing in sequence destined to be macronuclear coding region could result in deleterious missense or nonsense mutations. Although mse2.9 is located within an intron, it is possible that cis-acting regulatory sequences could be located in coding sequence. We have shown that the regulatory sequences important for accurate and efficient excision of mse2.9 are located completely within intronic sequence. This might be expected if IESs in Tetrahymena are found to have a transpositional origin (5) as has been suggested by the mechanistic studies of Saveliev and Cox (30,31).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Anita Samardzic for expert technical assistance. We thank also Nora Tsao and Emina David for helpful discussions throughout the course of this work, and Jason Bielas and Dr Scott Beeser for comments. DNA Sequencing was done by Lee Wong (Core Molecular Biology Facility, York University). This work was supported by a grant from the Medical Research Council (MRC) of Canada to R.E.P. J.S.F. is supported by an MRC studentship.
References
- 1.Borst P. and Greaves,D.R. (1987) Programmed gene rearrangements altering gene expression. Science, 235, 658–667. [DOI] [PubMed] [Google Scholar]
- 2.Lewis S.M. (1994) The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv. Immunol., 56, 27–150. [DOI] [PubMed] [Google Scholar]
- 3.Rudenko G., Cross,M. and Borst,P. (1998) Changing the end: antigenic variation orchestrated at the telomeres of African trypanosomes. Trends Microbiol., 3, 113–117. [DOI] [PubMed] [Google Scholar]
- 4.Prescott D.M. (1994) The DNA of ciliated protozoa. Microbiol. Rev., 58, 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klobutcher L.A. and Herrick,G. (1997) Developmental genome reorganization in ciliated protozoa: the transposon link. Prog. Nucleic Acids Res. Mol. Biol., 56, 1–62. [DOI] [PubMed] [Google Scholar]
- 6.Coyne R.S., Chalker,D.L. and Yao,M.-C. (1996) Genome downsizing during ciliate development: nuclear division of labor through chromosome restructuring. Annu. Rev. Genet., 30, 557–578. [DOI] [PubMed] [Google Scholar]
- 7.Martindale D.W., Allis,C.D. and Bruns,P.J. (1982) Conjugation in Tetrahymena thermophila (a temporal analysis of cytological stages). Exp. Cell Res., 140, 227–236. [DOI] [PubMed] [Google Scholar]
- 8.Davis M.C., Ward,J.G., Herrick,G. and Allis,C.D. (1992) Programmed nuclear death: apoptotic-like degradation of specific nuclei in conjugating Tetrahymena. Dev. Biol., 154, 419–432. [DOI] [PubMed] [Google Scholar]
- 9.Yao M.-C. (1996) Programmed DNA deletions in Tetrahymena: mechanisms and implications. Trends Genet., 12, 26–30. [DOI] [PubMed] [Google Scholar]
- 10.Chau M.-F. and Orias,E. (1996) Developmentally programmed DNA rearrangement in Tetrahymena thermophila: isolation and sequence characterization of three new alternative deletion systems. Biol. Cell., 86, 111–120. [DOI] [PubMed] [Google Scholar]
- 11.Austerberry C.F., Snyder,R.O. and Yao,M.-C. (1989) Sequence microheterogeneity is generated at junctions of programmed DNA deletions in Tetrahymena thermophila. Nucleic Acids Res., 17, 7263–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J. and Pearlman,R.E. (1996) Programmed DNA rearrangement from an intron during nuclear development in Tetrahymena thermophila: molecular analysis and identification of potential cis-acting sequences. Nucleic Acids Res., 24, 1943–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patil N.S., Hempen,P.M., Udani,R.A. and Karrer,K.M. (1997) Alternate junctions and microheterogeneity of Tlr1, a developmentally regulated DNA rearrangement in Tetrahymena thermophila. J. Euk. Microbiol., 44, 518–522. [DOI] [PubMed] [Google Scholar]
- 14.Heinonen T.Y. and Pearlman,R.E. (1994) A germ line-specific sequence element in an intron in Tetrahymena thermophila. J. Biol. Chem., 269, 17428–17433. [PubMed] [Google Scholar]
- 15.Chilcoat N.D. and Turkewitz,A.P. (1997) In vivo analysis of the major exocytosis-sensitive phosphoprotein in Tetrahymena. J. Cell Biol., 139, 1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horsfall W.H. and Pearlman,R.E. (1988) Micronuclear DNA sequence from Tetrahymena do not confer mitotic stability on ARS plasmids in Saccharomyces. Genome, 30, 690–696. [Google Scholar]
- 17.Austerberry C.F. and Yao,M.-C. (1988) Sequence structures of two developmentally regulated, alternative DNA deletion junctions in Tetrahymena thermophila. Mol. Cell. Biol., 8, 3947–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Austerberry C.F. and Yao,M.-C. (1987) Nucleotide sequence structure and consistency of a developmentally regulated DNA deletion in Tetrahymena thermophila. Mol. Cell. Biol., 7, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godiska R. and Yao,M.-C. (1990) A programmed site-specific DNA rearrangement in Tetrahymena thermophila requires flanking polypurine tracts. Cell, 61, 1237–1246. [DOI] [PubMed] [Google Scholar]
- 20.Godiska R., James,C. and Yao,M.-C. (1993) A distant 10-bp sequence specifies the boundaries of a programmed DNA deletion in Tetrahymena. Genes Dev., 7, 2357–2365. [DOI] [PubMed] [Google Scholar]
- 21.Patil N.S. and Karrer,K.M. (2000) A developmentally regulated deletion element with long terminal repeats has cis-acting sequences in the flanking DNA. Nucleic Acids Res., 28, 1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chalker D.L., La Terza,A., Wilson,A., Kroenke,C.D. and Yao,M.-C. (1999) Flanking regulatory sequences of the Tetrahymena R deletion element determine the boundaries of DNA rearrangement. Mol. Cell. Biol., 19, 5631–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wells J.M., Ellingston,J.L., Catt,D.M., Berger,P.J. and Karrer,K.M. (1994) A small family of elements with long inverted repeats is located near sites of developmentally regulated DNA rearrangement in Tetrahymena thermophila. Mol. Cell. Biol., 14, 5939–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huvos P.E., Wu,M. and Gorovsky,M.A. (1998) A developmentally eliminated sequence in the flanking region of the histone H1 gene in Tetrahymena thermophila contains short repeats. J. Euk. Microbiol., 45, 189–197. [DOI] [PubMed] [Google Scholar]
- 25.Orias E., Hamilton,E.P. and Orias,J.D. (2000) Tetrahymena as a laboratory organism: useful strains, cell culture, and cell line maintenance. Methods Cell Biol., 62, 189–211. [DOI] [PubMed] [Google Scholar]
- 26.Gaertig J. and Gorovsky,M.A. (1992) Efficient mass transformation of Tetrahymena thermophila by electroporation of conjugants. Proc. Natl Acad. Sci. USA, 89, 9196–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaertig J., Thatcher,T.H., Gu,L. and Gorovsky,M.A. (1994) Electroporation-mediated replacement of a positively and negatively selectable beta-tubulin gene in Tetrahymena thermophila. Proc. Natl Acad. Sci. USA, 91, 4549–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J., Fritsch,E.T. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Hiom K. and Gellert,M. (1998) Assembly of a 12/23 paired signal complex: a critical control point in V(D)J recombination. Mol. Cell, 1, 1011–1019. [DOI] [PubMed] [Google Scholar]
- 30.Saveliev S.V. and Cox,M.M. (1995) Transient DNA breaks associated with programmed genomic deletion events in conjugating cells of Tetrahymena thermophila. Genes Dev., 9, 248–255. [DOI] [PubMed] [Google Scholar]
- 31.Saveliev S.V. and Cox,M.M. (1996) Developmentally programmed DNA deletion in Tetrahymena thermophila by a transposition-like reaction pathway. EMBO J., 15, 2858–2869. [PMC free article] [PubMed] [Google Scholar]