


Abstract
RNA triphosphatase catalyzes the first step in mRNA cap formation which entails the cleavage of the β–γ phosphoanhydride bond of triphosphate-terminated RNA to yield a diphosphate end that is then capped with GMP by RNA guanylyltransferase. Here we characterize a 303 amino acid RNA triphosphatase (Pct1p) encoded by the fission yeast Schizosaccharomyces pombe. Pct1p hydrolyzes the γ phosphate of triphosphate-terminated poly(A) in the presence of magnesium. Pct1p also hydrolyzes ATP to ADP and Pi in the presence of manganese or cobalt (Km = 19 µM ATP; kcat = 67 s–1). Hydrolysis of 1 mM ATP is inhibited with increasing potency by inorganic phosphate (I0.5 = 1 mM), pyrophosphate (I0.5 = 0.4 mM) and tripolyphosphate (I0.5 = 30 µM). Velocity sedimentation indicates that Pct1p is a homodimer. Pct1p is biochemically and structurally similar to the catalytic domain of Saccharomyces cerevisiae RNA triphosphatase Cet1p. Mechanistic conservation between Pct1p and Cet1p is underscored by a mutational analysis of the putative metal-binding site of Pct1p. Pct1p is functional in vivo in S.cerevisiae in lieu of Cet1p, provided that it is coexpressed with the S.pombe guanylyltransferase. Pct1p and other yeast RNA triphosphatases are completely unrelated, mechanistically and structurally, to the metazoan RNA triphosphatases, suggesting an abrupt evolutionary divergence of the capping apparatus during the transition from fungal to metazoan species.
INTRODUCTION
The m7GpppN cap structure of eukaryotic mRNA is formed by three enzymatic reactions: (i) the 5′ triphosphate end of the nascent pre-mRNA is hydrolyzed to a diphosphate by RNA 5′ triphosphatase; (ii) the diphosphate end is capped with GMP by GTP:RNA guanylyltransferase; and (iii) the GpppN cap is methylated by AdoMet:RNA (guanine-N7) methyltransferase (1). The genetic and physical organization of the mRNA capping enzymes differs to a significant degree in higher versus lower eukaryotes. Mammals encode a two-component capping system consisting of a bifunctional triphosphatase–guanylyltransferase polypeptide and a separate methyltransferase polypeptide. The budding yeast Saccharomyces cerevisiae encodes a three-component system consisting of separate triphosphatase (Cet1p), guanylyltransferase (Ceg1p) and methyltransferase (Abd1p) gene products. The guanylyltransferase and methyltransferase components of the capping apparatus are structurally and mechanistically conserved between mammals and budding yeast. In contrast, the structures and catalytic mechanisms of the mammalian and yeast RNA triphosphatases are completely different (1).
The RNA triphosphatases of mammals and other metazoans belong to a superfamily of phosphatases that includes protein tyrosine phosphatases, dual specificity protein phosphatases and phosphoinositide phosphatases (2–5). Metazoan RNA triphosphatases catalyze γ phosphate cleavage via a two-step pathway. First, a cysteine thiolate nucleophile of the enzyme attacks the γ phosphorus to form a covalent protein-cysteinyl-S-phosphate intermediate and release diphosphate-terminated RNA. Then, the covalent intermediate is hydrolyzed to liberate inorganic phosphate. The metazoan RNA triphosphatases do not require a metal cofactor.
In contrast, the RNA triphosphatase of S.cerevisiae (Cet1p) is dependent on a divalent cation cofactor and there is no evidence of a covalent phosphoenzyme intermediate. Cet1p exemplifies a new family of metal-dependent phosphohydrolases that includes the RNA triphosphatases encoded by two groups of eukaryotic DNA viruses (poxviruses and baculoviruses) (6). The yeast/viral triphosphatase family is defined by two glutamate-rich peptide motifs (referred to as motifs A and C) that are essential for catalytic activity and comprise the metal binding site (6–9).
Genes encoding homologs of the three S.cerevisiae cap-forming enzymes have been identified in the pathogenic fungus Candida albicans (10–12). The C.albicans CaCET1 (triphosphatase), CGT1 (guanylyltransferase) and CCM1 (methyltransferase) genes can complement the growth of S.cerevisiae strains in which the endogenous triphosphatase, guanylyltransferase or methyltransferase genes are deleted. Indeed, the entire three-component capping system of S.cerevisiae can be replaced by the C.albicans capping system (13). The catalytic domain of the Candida RNA triphosphatase CaCet1p is similar in amino acid sequence to its S.cerevisiae counterpart Cet1p (Fig. 1). The active site of Cet1p is located within the hydrophilic core of a topologically closed eight-strand β barrel—the ‘triphosphate tunnel’ (14). The β strands comprising the tunnel are displayed over the Cet1p amino acid sequence in Figure 1. Most of the hydrophilic side chains of Cet1p that point into the tunnel are conserved in C.albicans CaCet1p. Extensive mutational analyses of the tunnel in both Cet1p and CaCet1p suggest that the active site structure and catalytic mechanism are virtually identical in the Saccharomyces and Candida triphosphatases (6,15,16).
Figure 1.

Schizosaccharomyces pombe RNA triphosphatase is structurally related to the RNA triphosphatases of S.cerevisiae and C.albicans. The complete amino acid sequence of S.pombe Pct1p is aligned to the sequences of the C-terminal catalytic domains of S.cerevisiae Cet1p (residues 241–539) and C.albicans CaCet1p (residues 202–520) and to S.cerevisiae Cth1p (residues 1–317). Gaps in the alignment are indicated by dashes. The β strands that comprise the triphosphate tunnel of Cet1p are denoted above the sequence. The peptide domain of Cet1p that mediates its interaction with the guanylyltransferase Ceg1p is conserved in CaCet1p and is shaded. This segment is not conserved in either Cth1p or Pct1p. Positions of side-chain identity or structural similarity in all four fungal RNA triphosphatases are denoted by dots. Conserved motifs A (β1) and C (β11) that define the metal-dependent RNA triphosphatase family are indicated below the sequence. Residues comprising the homodimer interface of Cet1p that are conserved in Pct1p are underlined below the Pct1p sequence. Residues Glu78, Glu80 and Glu260 of Pct1p that were targeted for alanine substitution in the present study are also shaded.
We consider the RNA triphosphatase to be an attractive target for antifungal drug development because: (i) the triphosphatase activity is essential for yeast cell growth; (ii) the enzyme is conserved among at least two species of fungi, including the bona fide human pathogen C.albicans and; (iii) the proteomes of several metazoan species (nematode, arthropod and mammal) include no identifiable homologs of the known yeast RNA triphosphatases. Yet it remains possible that other species of fungi employ either a metazoan-like RNA triphosphatase or a new class of RNA triphosphatase.
We are therefore interested in tracking the stage in evolution at which the capping apparatus of lower and higher eukaryotes diverged with respect to triphosphatase structure and mechanism. Towards this end, we have initiated a molecular genetic and biochemical analysis of the cap-forming enzymes of the fission yeast Schizosaccharomyces pombe (12,17). A wide evolutionary distance separates fission yeast from budding yeast. Schizosaccharomyces is in many respects more closely related to metazoans in its genetic organization and pre-mRNA processing strategy than to Saccharomyces. For example: (i) fission yeast has a much higher proportion of intron-containing genes (∼45%) than budding yeast (∼5%); (ii) the components of the pre-mRNA splicing machinery in fission yeast are more similar to their mammalian counterparts than to the corresponding budding yeast proteins; and (iii) several fission yeast splicing factors have homologs in humans, but are missing from the proteome of budding yeast (18).
We have already identified the genes encoding the guanylyltransferase (PCE1) and methyltransferase (PCM1) components of the S.pombe capping apparatus (12,17). Neither gene contains an intron. Pce1p and Pcm1p are structurally similar to the S.cerevisiae guanylyltransferase (Ceg1p) and methyltransferase (Abd1p), respectively, and expression of the S.pombe guanylyltransferase or methyltransferase in S.cerevisiae is sufficient for the growth of cells lacking the endogenous guanylyltransferase or methyltransferase proteins. The S.pombe guanylyltransferase Pce1p displays extensive amino acid sequence similarity to the C-terminal guanylyltransferase domain of mammalian capping enzyme Mce1p (17), but lacks any counterpart of the N-terminal triphosphatase domain of Mce1p.
Here we show that the S.pombe RNA triphosphatase is encoded by a separate gene, which we named PCT1. PCT1 contains a 5′-proximal intron and encodes a 303 amino acid polypeptide with extensive structural similarity to the catalytic domains of Cet1p and CaCet1p. Purified recombinant Pct1 protein has intrinsic metal-dependent RNA triphosphatase and nucleoside triphosphatase activities. Mechanistic conservation between fission and budding yeast triphosphatases is suggested by mutational analysis of the putative metal-binding motifs of Pct1p. Our results are consistent with an abrupt divergence of the RNA triphosphatase component of the capping apparatus during the transition from fungal to metazoan species. We find that the S.pombe triphosphatase is functional in S.cerevisiae in lieu of Cet1p when it is coexpressed with the fission yeast guanylyltransferase Pce1p. To our knowledge, this is the first example of species-specific genetic interactions between fungal cap-forming enzymes.
MATERIALS AND METHODS
Yeast expression vectors for S.pombe RNA triphosphatase
The intron-containing gene SPAC644.04 which encodes a putative RNA triphosphatase (hereafter referred to as PCT1, for ‘Pombe Capping enzyme Triphosphatase’) was amplified from S.pombe genomic DNA using Pfu DNA polymerase (Stratagene) and oligonucleotide primers designed to introduce an NdeI restriction site at the translation start codon and a BamHI restriction site immediately downstream of the stop codon. The amplified gene was inserted into the pCR-Blunt II-TOPO vector (Invitrogen) to generate the plasmid pCR-PCT1(+intron). The complete nucleotide sequence of the PCT1 insert matched exactly the S.pombe sequence deposited in GenBank (accession no. AL355012). An intron-less PCT1 cDNA was synthesized by the two-stage overlap PCR method (19). The first-stage amplification of the exons was performed using primers designed to introduce in-frame overlaps at the exon boundaries. The second-stage PCR product was inserted into the pCR-Blunt II-TOPO vector; the entire insert was sequenced to confirm the continuity of the 303 amino acid open reading frame (ORF) and that no coding mutations were introduced during amplification. An NdeI–BamHI fragment containing the complete PCT1 cDNA was inserted into the yeast shuttle vector pYN132 (TRP1 CEN) so as to place PCT1 expression under the control of the constitutive TPI1 promoter; the resulting plasmid was named p132-PCT1. The cDNA was also inserted into the multicopy yeast shuttle vector pG1 (2µ TRP1) (20) so that PCT1 expression was driven by the constitutive GPD1 promoter; this plasmid was named pG1-PCT1.
Yeast vectors for coexpression of RNA triphosphatase and RNA guanylyltransferase
An NcoI–SmaI fragment containing the PCE1 gene encoding S.pombe RNA guanylyltransferase (17) was inserted into the yeast shuttle vector pYX132 (CEN TRP1) so that PCE1 expression was driven by the TPI1 promoter; this plasmid was named p132-PCE1. Plasmid p132-CEG1 (CEN TRP1) contains the CEG1 gene that encodes S.cerevisiae RNA guanylyltransferase. The dual triphosphatase/guanylyltransferase expression plasmids p132-PCT1/PCE1 (CEN TRP1) and p132-PCT1/CEG1 (CEN TRP1) were constructed by excising the PCT1 expression cassette from pG1-PCT1. This comprises a DNA fragment extending from 722 bp upstream of the PCT1 ORF (including the GPD1 promoter) to 444 bp downstream of the ORF. The cassette was then inserted into p132-PCE1 and p132-CEG1 with the triphosphatase and guanylyltransferase transcription units arrayed in a tail-to-tail configuration.
Chimeric S.pombe/mouse capping enzyme
A gene encoding Pct1p fused to the guanylyltransferase domain of the mouse capping enzyme [Mce1(211–597)p] was constructed as follows. The PCT1 coding sequence was PCR-amplified using an antisense primer that changed the PCT1 stop codon to His and introduced an NdeI restriction site at the C-terminus. The PCR product was digested with NdeI and then inserted into the NdeI site of pYX1-MCE1(211–597) (CEN TRP1) (4) to yield the fusion gene PCT1–MCE1(211–597). Expression of the chimeric gene is under the control of the TPI1 promoter.
Expression and purification of recombinant S.pombe RNA triphosphatase
An NdeI–BamHI restriction fragment containing the PCT1 cDNA was inserted into the bacterial expression plasmid pET16b. The resulting plasmid, pET-PCT1, was introduced into Escherichia coli BL21(DE3). A 100 ml culture of BL21(DE3)/pET-PCT1 was grown at 37°C in Luria–Bertani medium containing 0.1 mg/ml ampicillin until the A600 reached 0.5. The culture was placed on ice for 30 min and then adjusted to 0.4 mM IPTG and 2% (v/v) ethanol. Incubation was continued for 20 h at 18°C with constant shaking. Cells were harvested by centrifugation and the pellets were stored at –80°C. All subsequent procedures were performed at 4°C. Thawed bacteria were resuspended in 5 ml of lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 10% sucrose). Lysozyme was added to a final concentration of 100 µg/ml and the suspension was incubated on ice for 15 min, adjusted to 0.1% Triton X-100 and then sonicated to reduce the viscosity of the lysate. Insoluble material was removed by centrifugation for 45 min at 17 000 r.p.m. in a Sorvall SS34 rotor. The soluble extract was applied to a 1 ml column of Ni2+-NTA agarose that had been equilibrated with lysis buffer containing 0.1% Triton X-100. The column was washed with the same buffer and then eluted step-wise with buffer B (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 10% glycerol, 0.05% Triton X-100) containing 50, 100, 200, 500 and 1000 mM imidazole. SDS–PAGE analysis showed that the recombinant Pct1p polypeptide was recovered predominantly in the 200 mM imidazole eluate fraction. The eluate was adjusted to 50 mM NaCl and 2 mM DTT by addition of an equal volume of dilution buffer (50 mM Tris–HCl pH 8.0, 4 mM DTT, 10% glycerol, 0.05% Triton X-100). This material was applied to a 1 ml column of phosphocellulose that had been equilibrated with buffer C (50 mM Tris–HCl pH 8.0, 2 mM DTT, 10% glycerol, 0.05% Triton X-100) containing 50 mM NaCl. The column was washed with the same buffer and eluted step-wise with buffer C containing 0.1, 0.2, 0.5 and 1.0 M NaCl. Pct1p was recovered in the 0.2 and 0.5 M NaCl eluate fractions. The 0.5 M NaCl fraction was pooled and the protein concentration was determined using the Bio-Rad dye-binding method with bovine serum albumin (BSA) as the standard. The yield of recombinant Pct1p was ∼0.4 mg.
Alanine-substitution mutations at positions Glu78, Glu80 and Glu260 were introduced into the PCT1 gene by the two-stage overlap extension method (21). The mutated genes were digested with NdeI and BamHI and then inserted into the bacterial expression vector pET16b. The inserts were sequenced completely to confirm the desired alanine mutation and exclude the acquisition of unwanted changes during amplification or cloning. The pET-PCT1-Ala plasmids were transformed into E.coli BL21(DE3) and the recombinant mutant proteins were expressed and purified as described above for wild-type Pct1p.
RESULTS
PCT1: a candidate S.pombe RNA triphosphatase gene
We identified, on phylogenetic grounds, a candidate RNA triphosphatase gene by searching the S.pombe Genome Sequencing Project Database (Sanger Center) for proteins related to the S.cerevisiae RNA triphosphatase Cet1p. The initial search revealed a gene fragment on S.pombe chromosome I encoding a polypeptide with significant similarity to the catalytic domain of Cet1p extending from motif A in strand β1 to motif C in strand β11. Subsequent annotation of this portion of chromosome I by the Sanger Center indicated that the S.pombe gene fragment corresponds to the 3′ exon of an intron-containing gene SPAC644.04 (GenBank accession no. AL355012). The predicted spliced mRNA product of this gene (which we have renamed PCT1) encodes a 303 amino acid polypeptide (Fig. 1). The 5′ exon of PCT1 encodes amino acids 1–78 and the 3′ exon encodes amino acids 79–303. Figure 1 shows a structure-based alignment of the sequence of Pct1p with the sequences of three known fungal RNA triphosphatases: S.cerevisiae Cet1p, C.albicans CaCet1p and S.cerevisiae Cth1p. [Cth1p is a non-essential S.cerevisiae enzyme of unknown function (15,22).] Positions of side-chain identity or conservation in all four proteins are denoted by dots. Reference to the crystal structure of Cet1p indicates that the identical and conserved residues are clustered in the β strands that comprise the walls of the triphosphate tunnel (Fig. 1) and include many of the hydrophilic amino acids that are essential for catalysis by Cet1p and CaCet1p. The 303 amino acid Pct1p protein is considerably smaller than Cet1p (549 amino acids) or CaCet1p (520 amino acids); Cet1p and CaCet1p contain non-essential N-terminal extensions that are missing from Pct1p. In the experiments presented below, we test biochemically and genetically whether Pct1p has the requisite activities of a cap-forming enzyme in vitro and in vivo.
Metal-dependent phosphohydrolase activity of Pct1p
A cDNA encoding the 303 amino acid S.pombe polypeptide was constructed by deleting the intron from a genomic DNA fragment containing the PCT1 gene. The cDNA was then cloned into a T7 RNA polymerase-based pET vector so as to place the ORF in-frame with an N-terminal leader encoding a 21 amino acid peptide with 10 tandem histidines. The expression plasmid was introduced into E.coli BL21(DE3), a strain that contains the T7 RNA polymerase gene under the control of the lac promoter. A new 38 kDa polypeptide corresponding to Pct1p was detectable by SDS–PAGE in extracts of IPTG-induced bacteria bearing the pET-PCT1 plasmid (not shown). Initial purification of the His-tagged fusion protein was achieved by adsorption to Ni-agarose and elution with 200 mM imidazole. Pct1p was purified further by adsorption to a column of phosphocellulose and step elution with NaCl. The phosphocellulose preparation was nearly homogenous with respect to the Pct1p polypeptide, as judged by SDS–PAGE (Fig. 2A). Further characterization of recombinant Pct1p was performed using the phosphocellulose fraction.
Figure 2.

RNA triphosphatase and ATPase activities of Pct1p. (A) Protein purification. Aliquots (4 µg) of the phosphocellulose fractions of wild-type Pct1p (WT) and mutants E78A, E80A and E260A were analyzed by electrophoresis in a 12% polyacrylamide gel containing 0.1% SDS. Polypeptides were visualized by staining with Coomassie blue dye. The positions and sizes (in kDa) of marker proteins are indicated to the left of the gel. (B) ATPase activity. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, 1 mM [γ-32P]ATP and either wild-type (WT) or mutant proteins as specified were incubated for 15 min at 30°C. The reactions were quenched by adding 2.5 µl of 5 M formic acid. Aliquots of the mixtures were applied to a polyethyleneimine–cellulose thin-layer chromatography (TLC) plate, which was developed with 1 M formic acid, 0.5 M LiCl. 32Pi release was quantitated by scanning the chromatogram with a FUJIX phosphorimager and was plotted as a function of input protein. (C) RNA triphosphatase activity. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 1 mM MgCl2, 20 pmol (of triphosphate termini) of [γ-32P]poly(A) and either WT or mutant proteins as specified were incubated for 15 min at 30°C. 32Pi release is plotted as a function of input protein.
We found that purified recombinant Pct1p is indeed an RNA triphosphatase. Activity was assayed by the liberation of 32Pi from 2 µM γ-32P-labeled triphosphate-terminated poly(A) in the presence of 1 mM magnesium chloride. The extent of γ phosphate hydrolysis during a 15 min incubation at 30°C was proportional to input protein (Fig. 2C). In the linear range of enzyme dependence, 180 fmol of 32Pi was released per fmol of Pct1p. This value corresponds to a turnover number of ∼0.2 s–1, which is lower than the values reported for the hydrolysis of [γ-32P]poly(A) by S.cerevisiae Cet1p (1 s–1), C.albicans CaCet1p (1.4 s–1), baculovirus LEF4 (1 s–1) and vaccinia D1 (0.8 s–1) (6,8,16,23).
The signature feature of the fungal/viral triphosphatase family members is their ability to hydrolyze nucleoside triphosphates to nucleoside diphosphates and inorganic phosphate in the presence of manganese or cobalt (6,8,9,16). The divalent cation specificity of the NTPase is distinct from the RNA triphosphatase function, which is optimal in magnesium. We found that recombinant Pct1p catalyzed the release of 32Pi from [γ-32P]ATP in the presence of manganese and that the extent of ATP hydrolysis increased as a function of input enzyme (Fig. 2B). There was no detectable ATP hydrolysis in the absence of a divalent cation (Fig. 3A). Hydrolysis of 1 mM ATP was optimal at 1–2 mM MnCl2 and declined slightly at 3–5 mM MnCl2 (Fig. 3A). ATP hydrolysis with cobalt as the cofactor was optimal at 1.5–6 mM CoCl2 (Fig. 3A). The titration curves were sigmoidal at manganese and cobalt concentrations below the level of input ATP. ATPase activity was tested with a battery of divalent actions added at 2 mM concentration: cobalt was ~70% as effective as manganese, whereas magnesium, calcium, copper and zinc were inactive (Fig. 3B). The manganese-dependent ATPase activity of Pct1p in 50 mM Tris–HCl buffer was optimal between pH 7.5 and 9.0; the activity at pH 6.0 was 33% of the activity at pH 7.5 (data not shown).
Figure 3.

Divalent cation requirement for ATP hydrolysis. (A) Dependence of ATPase activity on manganese and cobalt cation concentration. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 1 mM [γ-32P]ATP, 20 ng of Pct1p and MnCl2 or CoCl2 as specified were incubated for 15 min at 30°C. 32Pi release is plotted as a function of divalent cation concentration. (B) Divalent cation specificity. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 1 mM [γ-32P]ATP, 20 ng of Pct1p and 2 mM divalent cation as specified were incubated for 15 min at 30°C. Mg, Mn, Ca and Co were added as chloride salts; Cu and Zn were added as sulfates.
Kinetic analysis of ATP hydrolysis
The rate of release of 32Pi from [γ-32P]ATP was nearly identical to the rate of conversion of [α-32P]ATP to [α-32P]ADP in a parallel reaction mixture containing the same concentration of Pct1p (Fig. 4A). We detected no formation of [α-32P]AMP during the reaction. Hence, we conclude that Pct1p catalyzes the hydrolysis of ATP to ADP and Pi. Pct1p catalyzed the conversion [α-32P]GTP to [α-32P]GDP at the same rate as it hydrolyzed ATP to ADP (data not shown). Pct1p also hydrolyzed [α-32P]dATP to [α-32P]dADP at approximately half the rate of ATP hydrolysis (not shown). Other nucleotides were not tested.
Figure 4.

Kinetic analysis of ATP hydrolysis. (A) Reaction mixtures (150 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, 1 mM [α-32P]ATP or [γ-32P]ATP and 1.5 µg of Pct1p were incubated at 30°C. Aliquots (10 µl) were withdrawn at the times indicated and quenched immediately with formic acid. The reaction products were analyzed by polyethyleneimine–cellulose TLC. The extent of 32Pi or [α-32P]ADP formation (from 10 nmol of input ATP per sample) is plotted as a function of time. (B) Dependence of ATP hydrolysis on ATP concentration. Reaction mixtures (20 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, 40 pg of Pct1p and [γ-32P]ATP as specified were incubated for 15 min at 30°C. The extent of 32Pi release is plotted as a function of ATP concentration. The insert shows a double-reciprocal plot of the rate of 32Pi formation (s–1 = pmol 32Pi formed/900) versus [ATP].
Kinetic parameters for Pct1p were determined by measuring the extent of 32Pi formation during a 15 min reaction as a function of input [γ-32P]ATP concentration in the range 2.5–60 µM. A double-reciprocal plot of the data fits well to a linear function (Fig. 4B). We calculated a Km of 19 µM ATP and a kcat of 67 s–1. The turnover number of Pct1p in ATP hydrolysis is higher than the values reported for Cet1p (25 s–1), CaCet1p (17 s–1), Cth1p (2 s–1), D1 (10 s–1) and LEF4 (30 s–1) (6,8,15,16,24). The Km of Pct1p for ATP (19 µM) falls in the middle of the range reported for other family members: Cet1p (3 µM), CaCet1p (9 µM), LEF4 (43 µM), Cth1p (75 µM) and D1 (800 µM).
Pct1p activity is abolished by replacement of motif A and C glutamates with alanine
Pct1p residues Glu78 and Glu80 in motif A and Glu260 in motif C (Fig. 1) were replaced individually by alanine. The E78A, E80A and E260A proteins were expressed as His-tagged fusions and purified from soluble bacterial lysates by Ni-agarose and phosphocellulose column chromatography. The purity of the recombinant E78A, E80A and E260A proteins was comparable to that of wild-type Pct1p (Fig. 2A). The E78A, E80A and E260A mutants were unable to hydrolyze γ-32P-labeled poly(A) or ATP even when the levels of input enzyme were far in excess of the amount sufficient for maximal release of 32Pi by wild-type Pct1p (Fig. 2B and C). Using these data, we calculated that the specific RNA triphosphatase and ATPase activities of E78A, E80A and E260A were <0.1% of the activity of wild-type enzyme. We conclude that both glutamates of motif A and the central glutamate of motif C are essential for the phosphohydrolase activity of Pct1p.
This trio of acidic amino acids is broadly conserved in the RNA triphosphatases encoded by S.cerevisiae, C.albicans, poxviruses, African swine fever virus and baculoviruses. The equivalent three glutamates in motif A and motif C directly coordinate manganese at the metal binding site of Cet1p and are essential for catalysis by Cet1p in vitro and for Cet1p function in vivo (6,14). The motif A and C glutamates are also essential for catalysis by CaCet1p, Cth1p, vaccinia D1 and baculovirus LEF4 (7,9,15,16,25). Motifs A and C are located within strands β1 and β11 of Cet1p, which are widely separated in the primary structure, but closely approximated in the tertiary structure along the ‘floor’ of the tunnel (14). In motifs A and C of the budding yeast RNA triphosphatases, alternating charged side chains are interdigitated with alternating aliphatic/aromatic side chains (Fig. 1). This sequence pattern is reprised in motifs A and C of the S.pombe RNA triphosphatase, suggesting that the metal-binding residues of the fission yeast enzyme are also located within a pair of β strands.
Sedimentation analysis
The native size of recombinant Pct1p was analyzed by sedimentation of the protein through a 15–30% glycerol gradient containing 100 mM NaCl. Marker proteins catalase, BSA and cytochrome c were included as internal standards in the same glycerol gradient. After centrifugation, the polypeptide compositions of the gradient fractions were analyzed by SDS–PAGE (Fig. 5, top). A plot of the S values of the three standards versus fraction number yielded a straight line (not shown). Pct1p (a 38 kDa polypeptide) was resolved into two discrete peaks: a predominant 4.7 S ‘light’ component sedimenting just ahead of BSA (a 68 kDa monomeric polypeptide) and a minor 12 S ‘heavy’ component sedimenting just ahead of catalase (a 248 kDa tetramer of a 62 kDa subunit). We conclude that the predominant form of Pct1p is a homodimer and the minor species is at least a hexamer, if not an octamer. The gradient fractions were then assayed for manganese-dependent ATPase. The bimodal ATPase activity profile coincided with the distribution of Pct1p polypeptide (Fig. 5, bottom). The same bimodal distribution of Pct1p protein and ATPase activity was observed when the enzyme was sedimented in the absence of internal standards (data not shown).
Figure 5.
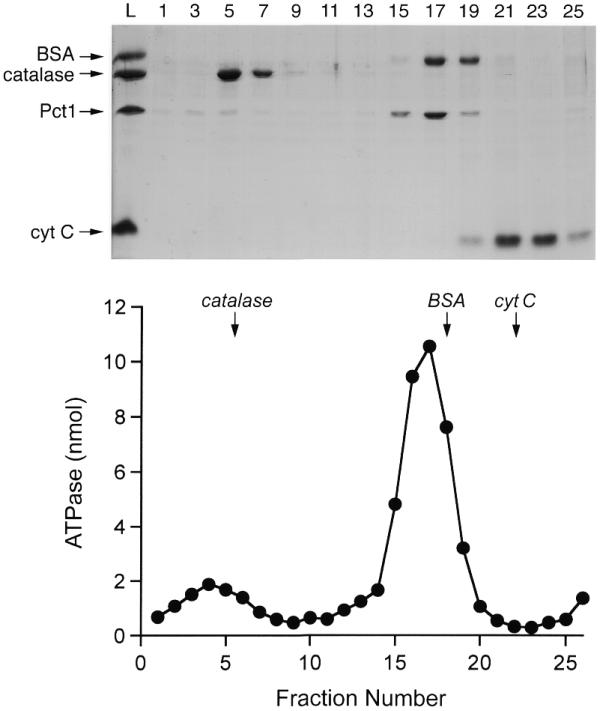
Glycerol gradient sedimentation. Pct1p (40 µg) was mixed with catalase (40 µg), BSA (40 µg) and cytochrome c (40 µg) in 0.2 ml of buffer G (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 2 mM DTT, 0.05% Triton X-100). The mixture was layered onto a 4.8 ml 15–30% glycerol gradient containing buffer G. The gradient was centrifuged in a Beckman SW50 rotor at 50 000 r.p.m. for 15 h at 4°C. Fractions (∼0.2 ml) were collected from the bottom of the tube. (Top) Aliquots (18 µl) of odd numbered fractions were analyzed by SDS–PAGE along with samples of the input protein mixture (L). Polypeptides were visualized by staining with Coomassie blue dye. The identities of the polypeptides are indicated at the left of the gel. (Bottom) ATPase activity profile. Reaction mixtures (20 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, 1 mM [γ-32P]ATP and 2 µl of the indicated glycerol gradient fractions were incubated for 15 min at 30°C. The peaks of the marker proteins are indicated by arrows.
The homodimeric quaternary structure of S.pombe Pct1p is a feature that is shared with the RNA triphosphatase of S.cerevisiae (14,26). Indeed, many of the amino acids that comprise the mostly hydrophobic dimerization surface in the crystal structure of S.cerevisiae Cet1p (14) are conserved in the S.pombe protein (Fig. 1).
Inhibition of Pct1p ATPase by phosphate, pyrophosphate and tripolyphosphate
We tested the effects of various phosphate derivatives on the ability of 30 nM Pct1p to hydrolyze 1 mM ATP in the presence of 2 mM manganese (Fig. 6). Inorganic phosphate, which is a product of the phosphohydrolase reaction, inhibited activity in a dose-dependent manner. ATP hydrolysis was inhibited by 50% of the control value at 1 mM Pi (Fig. 6). Inorganic pyrophosphate was a better inhibitor than phosphate (50% inhibition at 0.4 mM PPi) while tripolyphosphate was >10-fold more potent than pyrophosphate (50% inhibition at 30 µM tripolyphosphate) (Fig. 6). The finding that tripolyphosphate elicited 50% inhibition when present at a concentration 33-fold less than input ATP and 67-fold less than input manganese argues against the possibility that tripolyphosphate inhibits by acting as a chelator to compete with ATP (or enzyme) for the metal cofactor. A simple explanation for the potent inhibition by tripolyphosphate is that it binds more avidly than ATP to the triphosphate-binding pocket within the active site tunnel.
Figure 6.

Inhibition of ATP hydrolysis by phosphate, pyrophosphate and tripolyphosphate. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, 1 mM [γ-32P]ATP, 11 ng of Pct1p and either sodium phosphate, sodium pyrophosphate or sodium tripolyphosphate as specified were incubated for 15 min at 30°C. The reactions were initiated by the addition of Pct1p. The extents of ATP hydrolysis were normalized to those of control reactions from which inhibitory phosphates were omitted (values of 2.5–3.5 nmol 32P released). The normalized activities (control value = 1.0) are plotted as a function of inhibitor concentration.
Pct1p by itself cannot replace Cet1p in budding yeast
The PCT1 cDNA was cloned into a yeast CEN TRP1 expression plasmid under the control of the strong constitutive S.cerevisiae TPI1 promoter and also into a multicopy 2µ TRP1 expression plasmid under the control of the strong constitutive GPD1 promoter. The PCT1 expression plasmids were then transformed into S.cerevisiae cet1Δ strain YBS20, in which the chromosomal CET1 locus was deleted and replaced by LEU2. Growth of YBS20 is dependent on maintenance of a wild-type CET1 allele on a CEN URA3 plasmid. Therefore, YBS20 is unable to grow on agar medium containing 5-fluoroorotic acid (5-FOA), which selects against the URA3 plasmid, unless it is first transformed with either another CET1 allele or a biologically active RNA triphosphatase gene from a heterologous source (26). Introduction of the CEN PCT1 or 2µ PCT1 plasmid into YBS20 did not allow for growth of the transformants on 5-FOA, whereas cells transformed with a CEN CET1 plasmid readily yielded FOA-resistant colonies (Fig. 7). We surmise that Pct1p by itself was unable to substitute for Cet1p in vivo, even when expressed at a high gene dosage.
Figure 7.

Genetic interaction between S.pombe RNA triphosphatase and S.pombe RNA guanylyltransferase. Yeast strain YBS20 (cet1Δ) was transformed with CEN TRP1 plasmids containing either CET1, PCT1, PCT1 + PCE1 or PCT1 + CEG1 and with a 2µ plasmid containing PCT1. Trp+ isolates were streaked on an agar plate containing 0.75 mg/ml 5-FOA. The plate was photographed after incubation for 3 days at 30°C.
Fusion of S.pombe Pct1p to the guanylyltransferase domain of mouse capping enzyme confers the ability to complement S.cerevisiae Cet1p function in vivo
Saccharomyces cerevisiae RNA triphosphatase Cet1p and RNA guanylyltransferase Ceg1p interact in vivo and in vitro to form a heteromeric bifunctional capping enzyme complex (26). Formation of a Cet1p–Ceg1p complex in trans allows the yeast guanylyltransferase to target the yeast triphosphatase to the nascent pre-mRNA. This is achieved via the binding of the guanylyltransferase component to the phosphorylated C-terminal domain (CTD) of the RNA polymerase II transcription elongation complex (27–30). The guanylyltransferase-binding domain of S.cerevisiae Cet1p is localized to a 21 amino acid segment from residues 239–259 that flanks the triphosphatase catalytic domain (Fig. 1) (31). The guanylyltransferase-binding peptide is located on the surface of Cet1p and is conserved in the C.albicans RNA triphosphatase CaCet1p (11,13,14,31). Yet S.pombe Pct1p does not contain a recognizable counterpart of the guanylyltransferase-binding sites of Cet1p and CaCet1p (Fig. 1).
A plausible scenario to explain why Pct1p is unable to complement Cet1p function is that Pct1p does not interact with the budding yeast guanylyltransferase and therefore has no chaperone to direct it to nascent pre-mRNAs. Lehman et al. (26) showed that the in vivo requirement for a Ceg1p binding site on RNA triphosphatase can be bypassed by linking the Cet1p triphosphatase catalytic domain (minus the Ceg1p binding domain) in cis to the C-terminal guanylyltransferase domain of the mammalian capping enzyme, Mce1(211–597)p. Mce1(211–597)p binds avidly to the phosphorylated CTD of RNA polymerase II (32). The mammalian guanylyltransferase can also act as a chaperone for the S.cerevisiae Cth1p RNA triphosphatase when the two are fused, thereby allowing Cth1p to complement the cet1Δ mutation (15). It can even confer in vivo activity in yeast on vaccinia virus RNA triphosphatase when the viral and mammalian proteins are linked (25). Therefore, we tested whether fusion of S.pombe Pct1p to the mouse guanylyltransferase might correctly target Pct1p and thereby result in a gain-of-function in vivo.
A chimeric gene PCT1–MCE1(211–597) containing the PCT1 cDNA fused in-frame to the ORF for the mouse guanylyltransferase domain was cloned into a CEN TRP1 vector so that its expression was under the control of the yeast TPI1 promoter. The PCT1–MCE1(211–597) plasmid was then transformed into the S.cerevisiae cet1Δ ceg1Δ double-deletion strain YBS50. Transformation of YBS50 with CEN TRP1 plasmids bearing MCE1 or MCE1(211–597) provided positive and negative controls, respectively. Growth of YBS50 depends on maintenance of a CEN URA3 CET1 CEG1 plasmid (26). Transformation of YBS50 with MCE1, which encodes the bifunctional mammalian triphosphatase–guanylyltransferase, allowed growth of YBS50 on medium containing 5-FOA (Fig. 8). Transformants bearing MCE1(211–597), which encodes only the guanylyltransferase domain of mouse capping enzyme, failed to give rise to FOA-resistant colonies. This was expected because a functional triphosphatase on the CEN TRP1 plasmid is needed to complement the cet1Δ ceg1Δ double deletion. The instructive finding was that YBS50 cells bearing the PCT1–MCE1(211–597) plasmid did grow on 5-FOA (Fig. 8). Moreover, FOA-selected PCT1–MCE1(211–597) cells grew as well as MCE1 cells on rich medium (YPD agar) at 30 and 37°C, as gauged by colony size (data not shown). We conclude from this experiment that S.pombe Pct1p can function in vivo in mRNA capping when it is appropriately targeted to the transcription complex.
Figure 8.
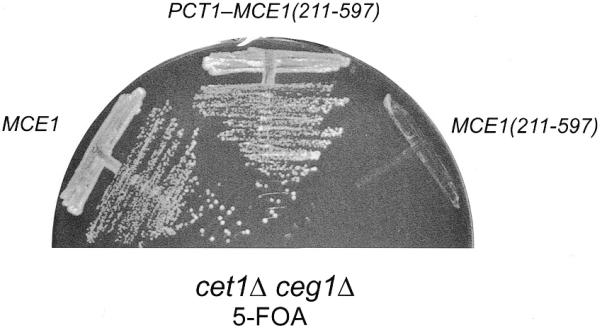
Pct1p functions in vivo in S.cerevisiae when fused to the guanylyltransferase domain of mammalian capping enzyme. Yeast strain YBS50 (cet1Δ ceg1Δ) was transformed with CEN TRP1 plasmids containing either MCE1, MCE1(211–597), or the PCT1–MCE1(211–597) chimera. Trp+ isolates were streaked on an agar plate containing 0.75 mg/ml 5-FOA. The plate was photographed after incubation for 5 days at 30°C.
Specific genetic interaction between S.pombe triphosphatase and guanylyltransferase
The fact that S.pombe guanylyltransferase Pce1p by itself can replace Ceg1p in vivo in S.cerevisiae (17) implies that S.pombe guanylyltransferase can interact with the S.cerevisiae triphosphatase. However, S.pombe triphosphatase by itself is not functional in budding yeast—possibly because it does not interact with Ceg1p. If Pct1p is a bona fide capping enzyme in S.pombe, then we postulate that it is targeted to the transcription complex via a chaperone specific to S.pombe. Given that an association of the triphosphatase and guanylyltransferase components of the capping apparatus is conserved in evolution (with the interaction being in trans in S.cerevisiae and in cis in metazoans, poxviruses and baculoviruses), the simplest model would be that S.pombe triphosphatase interacts with S.pombe guanylyltransferase Pce1p. To test this hypothesis, we asked whether the in vivo function of Pct1p in budding yeast could be restored by coexpressing Pce1p.
Yeast CEN TRP1 expression plasmids were constructed that contained a PCT1 cDNA driven by the yeast GDP1 promoter plus either the S.pombe PCE1 gene or the S.cerevisiae CEG1 gene driven by the TPI1 promoter. These dual expression vectors were transformed into the yeast cet1Δ strain and RNA triphosphatase activity of the plasmid-borne PCT1 allele was tested by plasmid shuffle (Fig. 7). The salient and instructive findings were that coexpression of Pct1p with S.pombe Pce1p completely revived the in vivo activity of Pct1p (as measured by growth on 5-FOA), whereas coexpression with the budding yeast guanylyltransferase Ceg1p had no salutary effect on Pct1p (Fig. 7). (Control experiments verified that the plasmid containing PCT1 plus CEG1 did complement the S.cerevisiae ceg1Δ mutant.) We subsequently found that budding yeast cells containing the triphosphatase and guanylyltransferase components of the fission yeast capping apparatus grew as well on rich medium (YPD agar) at 30 and 37°C as cells containing the wild-type Saccharomyces capping enzymes (not shown).
DISCUSSION
Our biochemical and genetic studies of the S.pombe RNA triphosphatase Pct1p provide new insights into the evolution and divergence of the mRNA capping apparatus in lower and higher eukaryotes. Given the sequence similarities between the S.pombe RNA triphosphatase and the catalytic domains of the S.cerevisiae and C.albicans RNA triphosphatases, the similar catalytic repertoires of these enzymes in metal-dependent hydrolysis of triphosphate-terminated RNA and free nucleoside triphosphates and the concordance of mutational effects in the metal-binding motifs A and C of all three proteins, we conclude that the active site architectures of fission yeast and budding yeast RNA triphosphatases are highly conserved. A reasonable extrapolation from the analysis of three fungal species is that all fungi are likely to encode the same basic capping machinery. This machinery is composed of separate triphosphatase, guanylyltransferase and methyltransferase gene products, with triphosphatase components from the metal-dependent family of Cet1p-like phosphohydrolases rather than the metal-independent cysteine-phosphatase family found in metazoans and plants. Thus, RNA triphosphatase retains its potential for being a target for mechanism-based antifungal drug discovery, insofar as an inhibitor that either occupies or occludes the active site of C.albicans RNA triphosphatase will probably be active against RNA triphosphatases from other pathogenic fungi. Tripolyphosphate is a relatively potent inhibitor of the Pct1p phosphohydrolase activity and a worthwhile platform on which to build and test new derivatives.
The guanylyltransferase and triphosphatase activities are linked in cis within a single polypeptide in all metazoan organisms examined to date [including the nematode Caenorhabditis elegans (2), the amphibian Xenopus laevis (33) and the arthropods Drosophila melanogaster and Artemia salina (34), as well as mice and humans] and in the plant Arabidopsis thaliana. The metazoan and plant RNA triphosphatases, members of the cysteine-phosphatase superfamily, are structurally and mechanistically unrelated to the fungal RNA triphosphatases. In all fungi examined to date, the triphosphatase and guanylyltransferase activities reside in separate polypeptides that interact in trans. The guanylyltransferase components are structurally and mechanistically similar in all eukaryotes. The mammalian guanylyltransferase domain can support yeast cell growth (albeit slower growth relative to wild-type yeast) with the yeast triphosphatase Cet1p present in trans (4). Moreover, the mouse guanylyltransferase displays a vestigial low-affinity interaction in vitro with the Ceg1p-binding peptide domain of yeast Cet1p (31). These findings suggest that the guanylyltransferases of higher eukaryotes evolved from an ancestral enzyme that did interact in trans with a triphosphatase, but that selection for such an interaction was relaxed during the emergence of metazoa.
The enzymes that catalyze the basic nucleic acid transactions (DNA replication, DNA repair, RNA synthesis and RNA processing) are generally very well conserved in lower and higher eukaryotes. Yet, in the case of RNA triphosphatase, we see a complete divergence of structure and mechanism either at the time of metazoan evolution or prior to the branching of the metazoan phyla listed above. How might this have occurred? We envisage a gene rearrangement event early in metazoan evolution that transferred a cysteine-phosphatase domain into the same transcription unit as the guanylyltransferase transcription unit, leading to creation of the triphosphatase–guanylyltransferase fusion protein that we see today in higher eukaryotic species. Note that lower and higher eukaryotes encode a plethora of cysteine-phosphatase family members with diverse substrate specificity and one can easily imagine the evolution of an RNA substrate preference prior to or not long after the proposed gene fusion. Note also that eukaryotes and eukaryotic viruses encode additional RNA 5′ phosphatases of the cysteine-phosphatase family. These proteins are not linked in cis to a guanylyltransferase and are of unknown function (35–37). RNA triphosphatase-like cysteine-phosphatases are not evident in the yeast proteome. Also, we cannot discern a homolog of fungal RNA triphosphatases in the metazoan proteomes. We suspect that the fusion of metazoan guanylyltransferase to a cysteine-type triphosphatase allowed for the loss of a Cet1p-like enzyme from the early metazoan genome, or else permitted the divergence of such a protein to a point where it now serves a new function in metazoan species and is no longer discernable as Cet1p-like. The only yeast-like RNA triphosphatases that exist in metazoans are those encoded by large DNA viruses such as poxviruses and baculoviruses.
Although the RNA triphosphatase components of the fungal capping systems have the same catalytic domain, the present study illustrates that the capping components are not always functionally interchangeable in vivo. There is an apparent hierarchy of cross-species complementation, whereby all of the known fungal guanylyltransferases can function in S.cerevisiae with the endogenous Cet1p triphosphatase (10,17), but the ability of heterologous triphosphatases to function with the S.cerevisiae guanylyltransferase is variable and correlates with the presence or absence of a conserved guanylyltransferase-binding domain on the surface on the heterologous RNA triphosphatase. Schizosaccharomyces pombe triphosphatase Pct1p, which lacks the surface domain and does not function on its own in S.cerevisiae, can support cell growth when S.pombe guanylyltransferase Pce1p is provided in trans. Thus, the fission yeast capping components display a species-specific genetic interaction in vivo. Insights into the physical basis of the Pct1p–Pce1p interaction will be dependent on crystallization of the S.pombe enzymes and fine mapping of their functional domains by mutagenesis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Susanne Schneider for technical assistance. This research was supported by NIH grant GM52470.
References
- 1.Shuman S. (2000) Structure, mechanism and evolution of the mRNA capping apparatus. Prog. Nucleic Acid Res. Mol. Biol., 66, 1–40. [DOI] [PubMed] [Google Scholar]
- 2.Takagi T., Moore,C.R., Diehn,F. and Buratowski,S. (1997) An RNA 5′-triphosphatase related to the protein tyrosine phosphatases. Cell, 89, 867–873. [DOI] [PubMed] [Google Scholar]
- 3.Wen Y., Yue,Z. and Shatkin,A.J. (1998) Mammalian capping enzyme binds RNA and uses protein tyrosine phosphatase mechanism. Proc. Natl Acad. Sci. USA, 95, 12226–12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho C.K., Sriskanda,V., McCracken,S., Bentley,D., Schwer,B. and Shuman,S. (1998) The guanylyltransferase domain of mammalian mRNA capping enzyme binds to the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem., 273, 9577–9585. [DOI] [PubMed] [Google Scholar]
- 5.Martins A. and Shuman,S. (2000) Mechanism of phosphoanhydride cleavage by baculovirus phosphatase. J. Biol. Chem., 275, 35070–35076. [DOI] [PubMed] [Google Scholar]
- 6.Ho C.K., Pei,Y. and Shuman,S. (1998) Yeast and viral RNA 5′ triphosphatases comprise a new nucleoside triphosphatase family. J. Biol. Chem., 273, 34151–34156. [DOI] [PubMed] [Google Scholar]
- 7.Yu L., Martins,A., Deng,L. and Shuman,S. (1997) Structure-function analysis of the triphosphatase component of vaccinia virus mRNA capping enzyme. J. Virol., 71, 9837–9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross C.H. and Shuman,S. (1998) RNA 5′-triphosphatase, nucleoside triphosphatase and guanylyltransferase activities of baculovirus LEF-4 protein. J. Virol., 72, 10020–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin J., Dong,W. and Guarino,L.A. (1998) The LEF-4 subunit of baculovirus RNA polymerase has RNA 5′-triphosphatase and ATPase activities. J. Virol., 72, 10011–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada-Okabe T., Shimmi,O., Doi,R., Mizumoto,K., Arisawa,M. and Yamada-Okabe,H. (1996) Isolation of the mRNA-capping enzyme and ferric-reductase-related genes from Candida albicans. Microbiology, 142, 2515–2523. [DOI] [PubMed] [Google Scholar]
- 11.Yamada-Okabe T., Mio,T., Matsui,M., Kashima,Y., Arisawa,M. and Yamada-Okabe,H. (1998) Isolation and characterization of the Candida albicans gene for mRNA 5′ triphosphatase: association of mRNA 5′ triphosphatase and mRNA 5′ guanylyltransferase activities is essential for the function of mRNA 5′ capping enzyme in vivo. FEBS Lett., 435, 49–54. [DOI] [PubMed] [Google Scholar]
- 12.Saha N., Schwer,B. and Shuman,S. (1999) Characterization of human, Schizosaccharomyces pombe and Candida albicans mRNA cap methyltransferases and complete replacement of the yeast capping apparatus by mammalian enzymes. J. Biol. Chem., 274, 16553–16562. [DOI] [PubMed] [Google Scholar]
- 13.Schwer B., Lehman,K., Saha,N. and Shuman,S. (2001) Characterization of the mRNA capping apparatus of Candida albicans. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 14.Lima C., Wang,L.K. and Shuman,S. (1999) Structure and mechanism of yeast RNA triphosphatase: an essential component of the mRNA capping apparatus. Cell, 99, 533–543. [DOI] [PubMed] [Google Scholar]
- 15.Pei Y., Ho,C.K., Schwer,B. and Shuman,S. (1999) Mutational analyses of yeast RNA triphosphatases highlight a common mechanism of metal-dependent NTP hydrolysis and a means of targeting enzymes to pre-mRNAs in vivo by fusion to the guanylyltransferase component of the capping apparatus. J. Biol. Chem., 274, 28865–28874. [DOI] [PubMed] [Google Scholar]
- 16.Pei Y., Lehman,K., Tian,L. and Shuman,S. (2000) Characterization of Candida albicans RNA triphosphatase and mutational analysis of its active site. Nucleic Acids Res., 28, 1885–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shuman S., Liu,Y. and Schwer,B. (1994) Covalent catalysis in nucleotidyl transfer reactions: essential motifs in Saccharomyces cerevisiae RNA capping enzyme are conserved in Schizosaccharomyces pombe and vaccinia capping enzymes and among DNA ligases. Proc. Natl Acad. Sci. USA, 91, 12046–12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Käufer N.F. and Potashkin,J. (2000) Analysis of the splicing machinery in fission yeast: a comparison with budding yeast and mammals. Nucleic Acids Res., 28, 3003–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horton R.M., Hunt,H.D., Ho,S.N., Pullen,J.K. and Pease,L.R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene, 77, 61–68. [DOI] [PubMed] [Google Scholar]
- 20.Schena M., Picard,D. and Yamamoto,K. (1991) Vectors for constitutive and inducible gene expression in yeast. Methods Enzymol., 194, 389–398. [DOI] [PubMed] [Google Scholar]
- 21.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez C.R., Takagi,T., Cho,E. and Buratowski,S. (1999). A Saccharomyces cerevisiae RNA 5′-triphosphatase related to mRNA capping enzyme. Nucleic Acids Res., 27, 2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myette J. and Niles,E.G. (1996) Domain structure of the vaccinia virus mRNA capping enzyme: expression in Escherichia coli of a subdomain possessing the RNA 5′-triphosphatase and guanylyltransferase activities and a kinetic comparison to the full-size enzyme. J. Biol. Chem., 271, 11936–11944. [DOI] [PubMed] [Google Scholar]
- 24.Myette J. and Niles,E.G. (1996) Characterization of the vaccinia virus RNA 5′-triphosphatase and nucleoside triphosphate phosphohydrolase activities: demonstration that both activities are carried out at the same active site. J. Biol. Chem., 271, 11945–11952. [DOI] [PubMed] [Google Scholar]
- 25.Ho C.K., Martins,A. and Shuman,S. (2000) A yeast-based genetic system for functional analysis of viral mRNA capping enzymes. J. Virol., 74, 5486–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehman K., Schwer,B., Ho,C.K., Rouzankina,I. and Shuman,S. (1999) A conserved domain of yeast RNA triphosphatase flanking the catalytic core regulates self-association and interaction with the guanylyltransferase component of the mRNA capping apparatus. J. Biol. Chem., 274, 22668–22678. [DOI] [PubMed] [Google Scholar]
- 27.McCracken S., Fong,N., Rosonina,E., Yankulov,K., Brothers,G., Siderovski,D., Hessel,A., Foster,S., Shuman,S. and Bentley,D.L. (1997) 5′ Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated C-terminal domain of RNA polymerase II. Genes Dev., 11, 3306–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho E., Takagi,T., Moore,C.R. and Buratowski,S. (1997) mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxyl-terminal domain. Genes Dev., 11, 3319–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroeder S.C., Schwer,B., Shuman,S. and Bentley,D. (2000) Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev., 14, 2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komarnitsky P., Cho,E. and Buratowski,S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev., 14, 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho C.K., Lehman,K. and Shuman,S. (1999) An essential surface motif (WAQKW) of yeast RNA triphosphatase mediates formation of the mRNA capping enzyme complex with RNA guanylyltransferase. Nucleic Acids Res., 27, 4671–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho C.K. and Shuman,S. (1999) Distinct roles for CTD Ser2 and Ser5 phosphorylation in the recruitment and allosteric activation of mammalian capping enzyme. Mol. Cell, 3, 405–411. [DOI] [PubMed] [Google Scholar]
- 33.Yokoska J., Tsukamoto,T., Miura,K., Shiokawa,K. and Mizumoto,K. (2000) Cloning and characterization of mRNA capping enzyme and mRNA (guanine-7-)-methyltransferase cDNAs from Xenopus laevis. Biochem. Biophys. Res. Commun., 268, 617–624. [DOI] [PubMed] [Google Scholar]
- 34.Yagi Y., Mizumoto,K. and Kaziro,Y. (1984) Limited tryptic digestion of messenger RNA capping enzyme from Artemia salina: isolation of domains for guanylyltransferase and RNA 5′-triphosphatase. J. Biol. Chem., 259, 4695–4698. [PubMed] [Google Scholar]
- 35.Gross C.H. and Shuman,S. (1998) Characterization of a baculovirus-encoded RNA 5′ triphosphatase. J. Virol., 72, 7057–7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takagi T., Hao,L., Charbonneau,H. and Buratowski,S. (1998) A protein-tyrosine phosphatase-like protein from baculovirus has RNA 5′-triphosphatase and diphosphatase activities. Proc. Natl Acad. Sci. USA, 95, 9808–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deshpande T., Takagi,T., Hao,L., Buratowski,S. and Charbonneau,H. (1999) Human PIR1 of the protein-tyrosine phosphatase superfamily has RNA 5′-triphosphatase and diphosphatase activities. J. Biol. Chem., 274, 16590–16594. [DOI] [PubMed] [Google Scholar]