


Abstract
The observation of light metal ions in nucleic acids crystals is generally a fortuitous event. Sodium ions in particular are notoriously difficult to detect because their X-ray scattering contributions are virtually identical to those of water and Na+…O distances are only slightly shorter than strong hydrogen bonds between well-ordered water molecules. We demonstrate here that replacement of Na+ by K+, Rb+ or Cs+ and precise measurements of anomalous differences in intensities provide a particularly sensitive method for detecting alkali metal ion-binding sites in nucleic acid crystals. Not only can alkali metal ions be readily located in such structures, but the presence of Rb+ or Cs+ also allows structure determination by the single wavelength anomalous diffraction technique. Besides allowing identification of high occupancy binding sites, the combination of high resolution and anomalous diffraction data established here can also pinpoint binding sites that feature only partial occupancy. Conversely, high resolution of the data alone does not necessarily allow differentiation between water and partially ordered metal ions, as demonstrated with the crystal structure of a DNA duplex determined to a resolution of 0.6 Å.
INTRODUCTION
Despite significant advances in the X-ray analysis of 3D structures of DNA and RNA over the past two decades, reliable detection of light metal ions surrounding nucleic acid molecules remains a challenge. Owing to their regular coordination geometry, earth alkali metal ions such as Mg2+ and Ca2+ can often be located in Fourier electron density maps. Examples include crystal structures of small and medium size RNAs determined at both relatively low (1–4) and high (5,6) resolutions. Crystal structures of oligodeoxynucleotides at atomic resolution revealed the positions of Mg2+ and Ca2+ ions that account for the neutralization of between 50 and 100% of the negative charges of phosphate groups (7–12).
Conversely, there are less than a dozen structures currently deposited in the Nucleic Acid Database (13) that feature an ordered Na+ ion and in most of these the ion displays octahedral coordination geometry. For a list of these structures see Table S1 (Supplementary Material available online). The limited number of examples attests to the difficulties of reliably detecting Na+ ions with less regular coordination geometry. Thus, whether a particular peak in Fourier electron density maps ends up as a water molecule or a Na+ ion is often a matter of personal judgment and the decision to place an ion, although it may be chemically reasonable, lacks solid experimental evidence in many cases. The atomic resolution crystal structure of a parallel-stranded G-tetraplex constitutes an exception in this respect (14). Since monovalent cations affect the stability of the G-tetrad in a crucial manner, the interpretation that electron density peaks in and between the planes formed by four guanines represent sodium ions is unchallenged.
Replacement of Na+ or K+ by heavier alkali ions in crystallization of a nucleic acid fragment or soaking crystals in solutions of the heavier alkali ions may be helpful for locating sites occupied by Na+ or K+. For example, crystals of a B-DNA dodecamer grown from solutions supplemented with either rubidium or cesium chloride in combination with difference Fourier synthesis revealed coordination of a single Rb+ in the minor groove (15). Keeping in mind the differences in ionic radius and relative binding affinities of alkali metal ions for double-stranded DNA (Cs+ > Rb+ > K+ > Na+; 16), we may take this observation as an indication that Na+ can occupy a more or less identical site as Rb+. In a similar fashion, binding of K+ below a so-called AA platform within a domain of group I intron RNA was confirmed by soaking crystals in thallium acetate or cesium hydroxide and examining the corresponding region of difference electron density maps for changes in intensity (17).
To date there are no reports that exploit the anomalous scattering contributions of alkali metal ions to determine their locations in nucleic acid crystals. All these ions with the exception of Na+ exhibit significant anomalous effects (Fig. 1). In combination with a tunable X-ray synchrotron source, the use of anomalous effects for locating metal ions promises to be much more sensitive than difference Fourier synthesis along with monitoring of the Rfree value (18) during the ensuing refinement cycles. Moreover, the presence of anomalously scattering K+, Rb+ or Cs+ ions offers the opportunity to determine nucleic acid crystal structures by the single wavelength anomalous diffraction (SAD) technique.
Figure 1.
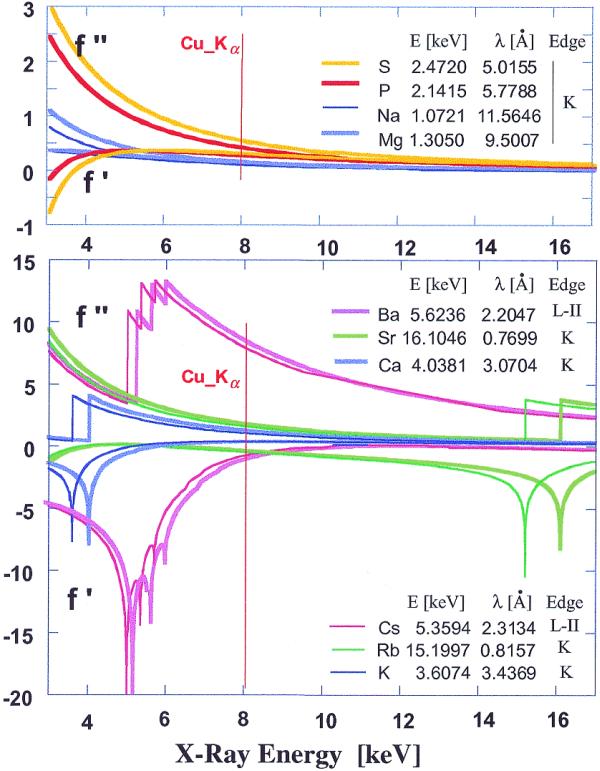
Idealized curves for the real (f′) and imaginary (f′′) anomalous scattering components of alkali and earth alkali metal ions as a function of X-ray energy (taken from 29). Data for elements P, S and the CuKα wavelength (1.5418 Å) are included for comparison. The energy E and the corresponding wavelength λ of absorption edges are shown together with the color code for each ion/element. The wavelength of the beam at the Advanced Photon Source ID beamlines can easily be tuned within the 2–0.66 Å (third harmonic) range. Absorption edges below 6 keV correspond to X-ray wavelengths >2 Å, which present difficulties for use in crystallography. Among alkali metal ions only the K absorption edges of Rb+ and Sr2+, located near the right-hand side border, are suitable for use in high energy SAD experiments. The anomalous signals of Ba2+ and Cs+ as well as of Ca2+ and K+ can be used at low energy.
Here we present protocols for the determination of alkali metal ion-binding sites in nucleic acid crystals and demonstrate that the anomalous diffraction components of Rb+ and Cs+ are suitable for SAD-type structure determination. In the first application of the SAD technique for crystallographic structure determination, diffraction data of a protein–peptide complex were phased using a single iodine atom (19). The use of SAD to locate sulfur atoms present in almost all proteins shows promise as a general method for solving the phase problem of native crystals of macromolecules (20). Use of the SAD technique successfully demonstrated here for locating alkali metal ions in crystals of oligodeoxynucleotides should be of general applicability with DNA and RNA.
MATERIALS AND METHODS
Oligonucleotide synthesis and purification
To investigate DNA–ion interactions in crystals we chose A-DNA decamers of sequence GCGTATACGC. This particular DNA sequence was used previously to analyze the conformations of DNA–RNA chimeras (21), DNA hydration (22) and the structural origins of the enhanced RNA affinity (23,24) and exonuclease resistance (25) displayed by 2′-O-modified oligonucleotide analogs. Four DNA decamers with a single 2′-O-aminooxyethyl (AOE)-, 2′-O-methyl-3′-methylenephosphonate (MEP)-, 2′-O-fluoroethyl (FET)- or 2′-O-methyl-[tri(oxyethyl)] (TOE)-thymine in place of T6 were included in the crystallization experiments. We found that chemical modification of the T6 sugar moiety in this particular A-DNA decamer leads to crystals of exceptional quality (22). The conformational features of the 2′-O-substituent in TOE were reported before (23) and more detailed accounts of the syntheses, structures and hydration for the other modifications will be described elsewhere. The Z-DNA hexamer CGCGCG was synthesized on the 3 µmol scale (Pharmacia Expedite), deprotected following established procedures and purified by HPLC. All other oligonucleotides were purified by HPLC and ion exchange chromatography to ≥98%.
Crystallizations
Three different ion concentrations were used to grow crystals with Na+, K+, Rb+ or Cs+: 40 (low salt), 230 (medium salt) and 450 mM (high salt) (Table 1). Hanging drops (4 µl) containing 1 mM DNA, 40 mM alkali metal ions (chloride salt), 20 mM alkaline cacodylate buffer, pH 6.0, 6 mM spermine and 10 mM MgCl2 were equilibrated against 1 ml 35% 2-methyl-2,4-pentanediol (MPD). Low salt crystals appeared within 2 weeks and were used for data collection [Mg-MEP (22) and Mg-TOE (23); only Mg2+ and spermine were located]. The phase diagram technique (26,27) was used to grow medium and high salt crystals. Thus, 1 (medium salt) and 3 µl (high salt) 1 M alkali chloride solutions, respectively, were added to untouched droplets left from low salt crystallizations. Initially crystals in the droplets dissolved, but appeared again within 2 weeks after the MPD concentration in the reservoir was raised to 40%. Ba2+-containing crystals of AOE were grown from 4 µl hanging drops containing 1 mM DNA, 20 mM sodium cacodylate, pH 6.0, 2 mM spermine and 5 mM BaCl2, equilibrated against 1 ml 35% MPD. Z-DNA crystals were grown from sitting droplets that contained 1.5 mM DNA, 20 mM rubidium cacodylate, pH 7.0, and 10 mM spermine and were equilibrated against a reservoir of 20% MPD.
Table 1. Data collection and refinement statistics of A-DNA decamers GCGTATACGC in space group P212121.
| Compound
Salt form |
|
Ba-AOE |
Cs-MEP high |
Cs-TOE medium |
Rb-FET high |
Rb-TOE medium |
K-MEP high |
K-TOE medium |
Na-TOE medium |
Na-FET medium |
Z-DNA |
| Unit cell | a, Å | 24.99 | 24.82 | 24.87 | 24.82 | 25.16 | 24.65 | 25.13 | 25.13 | 24.96 | 18.32 |
| b, Å | 44.57 | 44.33 | 44.68 | 45.26 | 44.83 | 44.61 | 44.53 | 44.53 | 43.57 | 30.68 | |
| c, Å | 45.12 | 44.82 | 44.69 | 43.97 | 44.82 | 44.16 | 45.13 | 45.13 | 45.40 | 42.49 | |
| B factora (Å2) | DNA | 21 | 8 | 13 | 12 | 16 | 25 | 15 | 15 | 19 | 6 |
| Site 1 | – | 10 | 15 | 15 | 25 | 30 | 33 | – | – | – | |
| Site 2 | 35 (Ba) | 25 | 35 | 30 | 40 | 35 | 32 | 21 | 50 (Mg) | – | |
| Site 3 | – | 60 | 65 | 69 | 70 | 35 | 34 | 25 | 33 | – | |
| High resolution X-ray datab | λ, Å | 1.5418 | 0.8151 | 0.9500 | 0.9500 | 0.9500 | 0.8151 | 0.8151 | 0.8151 | 0.6630 | |
| D, Å | 1.70 | 1.06 | 1.05 | 1.05 | 1.30 | 1.30 | 1.30 | 1.45 | 0.60 | ||
| N ref | 6913 | 22 132 | 19 840 | 22 802 | 12 230 | 12 024 | 12 024 | 8695 | 57 623 | ||
| % | 97.5 | 97.2 | 95.1 | 99.8 | 99.5 | 98.9 | 98.9 | 99.7 | 96.0 | ||
| Rmerge | 0.068 | 0.057 | 0.041 | 0.046 | 0.044 | 0.041 | 0.041 | 0.058 | 0.062 | ||
| Rwork/free | 19.0/21.2 | 15.0/16.9 | 16.1/17.8 | 15.4/17.0 | 16.5/19.0 | 20.2/24.6c | 16.1/17.9 | 16.1/18.1 | 15.9/19 | 16.0/18.5 | |
| Anomalous X-ray datad | λ, Å | 1.5418 | 1.6531 | 1.7970 | 0.8151 | 0.8151 | 1.6513 | ||||
| D, Å | 2.50 | 2.0 | 2.0 | 1.50 | 1.50 | 2.0 | |||||
| % | 97.5 | 99.3 | 98.7 | 98.8 | 99.1 | 98.4 | |||||
| Rmerge | 0.065 | 0.082 | 0.079 | 0.059 | 0.044 | 0.092 | |||||
| ΔF/F | 0.0469 | 0.0784 | 0.0384 | 0.0620 | 0.021 | 0.019 | |||||
| PP | 1.20 | 1.68 | 1.35 | 2.66 | – | – | |||||
| FOM | 0.33 | 0.41 | 0.35 | 0.51 | – | – |
aRefinement with ion occupancy equal to 1.
bR factor = Σhkl|F(hkl)o – F(hkl)c|/ΣhklF(hkl)o. Rmerge = ΣhklΣi|I(hkl)i – <I(hkl)>|/ΣhklΣi<I(hkl)i>. λ, D, the wavelength and resolution of the X-ray data; N ref, %, the number of unique reflections and completeness.
cR factors based on 2.0 Å anomalous data.
dΔF/F =Σhkl<|F(+)| – |F(–)|>/<1/2(|F(+)| + |F(–)|>. PP, FOM, phasing power and figure of merit.
X-ray data collection and processing
Crystals suitable for data collection were mounted in nylon loops and frozen and stored in liquid nitrogen. Data sets with crystals containing Ba2+ were collected on an in-house rotating anode generator/image plate set-up (Table 1). All other data collections were conducted at the Advanced Photon Source, using the insertion device beamlines (ID-B) of the DND and IMCA collaborative access teams, located at sectors 5 and 17, respectively. Both are equipped with 165 mm MARCCD detectors. Data to the maximum resolution limits of the individual crystals were collected at wavelengths <1 Å. To improve completeness and to avoid overloads in the lower resolution bins, separate data sets were measured for the low and high resolution ranges in each case. Anomalous data were collected at a wavelength of 0.8151 Å for Rb+-containing crystals (third harmonic range) and in the low energy range (≥1.54 Å) for all other crystals (Fig. 1). All data were integrated and scaled either in the DENZO/SCALEPACK or HKL2000 suites (28) and selected crystal data and data collection parameters are summarized in Table 1.
Structure determination, Patterson and electron density map calculation and refinement
Heavy atom searches, Patterson map as well as electron density map calculations and SAD phasing were performed with the program CNS (29) and maps were displayed with the program TURBO FRODO (30). For all structures initial refinements were performed with CNS. To calculate the Rfree values (18) 10% of the data were set aside prior to the refinements. All anisotropic refinements with high resolution data were conducted with the program SHELX-97 (31). Selected refinement parameters for all structures based on high resolution data are listed in Table 1.
Coordinates
Final coordinates and structure factors for all structures were deposited in the Protein Data Bank (PDB). PDB codes: 1I0F (Ba-AOE), 1I0J (Cs-MEP), 1I0K (Cs-TOE), 1I0M (Rb-FET), 1I0N (Rb-TOE), 1I0O (K-MEP), 1I0P (K-TOE), 1I0Q (Na-TOE), 1I0G (Na-FET) and 1I0T (Z-DNA).
RESULTS AND DISCUSSION
Overall strategy
In order to explore the feasibility of using the anomalous diffraction of alkali metal ions to locate their binding sites in nucleic acid crystals and to provide phase information via SAD, we conducted an extensive set of X-ray diffraction experiments at the Advanced Photon Source. The study included a variety of A-form DNA decamer crystals for which multiple data sets at different wavelengths were collected so as to optimize both the magnitude of the anomalous signal and the resolution (Table 1). The structures of a Ba2+-form crystal (strong anomalous signal) and of the Z-DNA duplex (resolution 0.6 Å) were used as ‘anomalous diffraction’ and ‘high resolution’ references.
Anomalous data were collected in the low energy range for K+, Cs+ and Ba2+ or, in the case of Rb+, at the K absorption edge (Fig. 1). Preliminary diffraction experiments were also conducted with decamer crystals containing Sr2+ and confirmed the usefulness of this ion for SAD-based structure determination (data not shown). While anomalous data were typically collected to resolutions of ∼2 Å, data to high resolution (up to 1.05 Å) were also collected at a shorter wavelength for each crystal (Table 1). The benefits of atomic resolution data for analyzing ion and solvent interactions in crystals are obvious and an approach that combines the precision of high resolution with the sensitivity of anomalous diffraction can be expected to be superior to all previous crystallographic methods to locate alkali metal ions. In addition, to potentially gain an insight into both low and high occupancy DNA binding sites for these metal ions their concentrations in the crystallization solutions were varied (see Materials and Methods).
SAD phasing
Data collection at the appropriate wavelengths with DNA crystals containing alkali or earth alkali metal ions demonstrates the merits of some of these ions for phasing (Table 1). The heavy atom search procedure in the program CNS was applied to anomalous difference Patterson maps calculated at 2.5, 2.0 and 1.5 Å based on data sets for DNA crystals containing Ba2+, Cs+ and Rb+, respectively. An example of an anomalous Patterson map is depicted in Figure S1 (Supplementary Material available online). One ion-binding site was located both in Cs-TOE (medium salt) and Ba-AOE crystals (sites 1 and 2, respectively, Fig. 2A). Ions at both sites 1 and 2 were located in Cs-MEP and Rb-FET crystals grown under high salt conditions. In the Harker sections peak 1 was twice as high as peak 2. Based on the sites determined in the Patterson maps, SAD phasing was then performed using data to 2.5, 2.0 and 1.8 Å resolution for Ba2+, Cs+ and Rb+, respectively. Values for phasing power and figure of merit are listed in Table 1. However, the heavy atom search procedure was not successful in the case of the Rb-TOE (medium salt) and K-MEP (high salt) structures.
Figure 2.
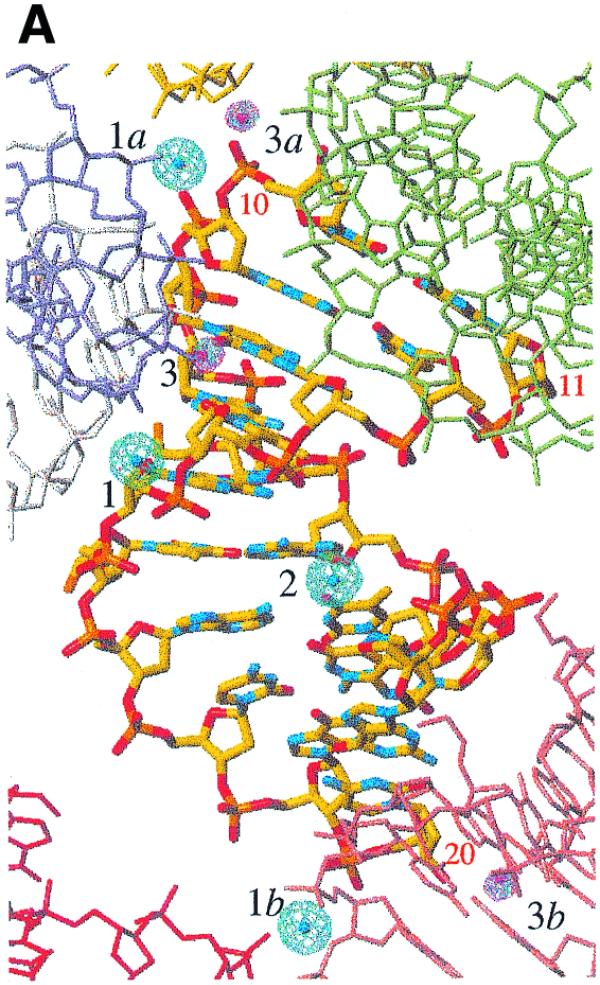
Maps calculated at 1.8 Å resolution with SAD phases for the Rb-FET structure and superimposed on the final structure (selected DNA residues are numbered). (A) The anomalous difference Fourier (blue) and double-difference (red) maps contoured at the 4σ level. Ion-binding sites 1 and 2 (shown with their symmetry mates, designated by lower case letters) were located in the anomalous difference Patterson map and used for phasing. Ion-binding site 3 was found at this stage and is characterized by a significantly greater peak height than the remaining peaks in the double-difference density map. (B) Stereo diagram of the density-modified electron density map contoured at the 1σ level and superimposed on the central TAT portion of the structure. DNA atoms are colored yellow, red, cyan, orange and green for carbon, oxygen, nitrogen, phosphorus and fluorine, respectively. Electron density for the FET substituent of T6 is clearly visible in this map.
In order to find additional binding sites the above SAD phases were used in combination with the anomalous difference structure factors to compute anomalous difference Fourier and double-difference (or log likelihood gradient) maps. Examples of anomalous difference Fourier and double-difference maps for the Rb-FET structure are depicted in Figure 2A. In this manner a third ion-binding site was retrieved in the Rb-FET and Cs-MEP structures (site 3). Moreover, a second site (site 2) previously absent in the Cs-TOE structure was also found in this fashion. The resulting phases were subsequently improved by density modification.
As expected, Ba2+ present in the reference structure proved very potent for phasing and a single site allowed structure determination via SAD. Similarly, Cs+- (high and medium salt forms) and Rb+-form (high salt form) crystals were used for SAD-type phasing. Electron density for two thirds of the duplex was clearly visible with the Ba-AOE, Cs-MEP and Rb-FET structures and a portion of the Rb-FET experimental electron density map is depicted in Figure 2B.
Ion-binding sites and coordination modes
Our experiments reveal up to three alkali metal ion-binding sites in the A-DNA decamer crystals (Fig. 2A). Sites 1 and 3 are exclusively occupied by alkali ions, whereas site 2 can accommodate both alkali and earth alkali metal ions (Fig. 3). Site 1 involves phosphate groups from three adjacent duplexes and the coordination modes for individual alkali metal ions are depicted in Figure 4. For a superposition of this binding site from seven different structures see Figure S2. Site 2 is located in the major groove and comprises three adjacent base pairs (Fig. 5). Sites 3 and 1 are fairly closely spaced and at the former ions are engaged in direct contact to backbone atoms at the minor groove–terminal base pair interface formed by three neighboring duplexes (Fig. 6). Summaries of the distances to DNA atoms and water molecules for individual alkali ions at sites 1, 2 and 3 are given in Tables S2–S4, respectively.
Figure 3.

(A) Superposition of A-DNA decamer duplexes crystallized in the presence of different alkali or earth alkali metal ions. The bottom halves of duplexes were used in the superposition (r.m.s.d. <0.5 Å). The view depicts binding site 2 in the major groove common for all ions and demonstrates similar kinks for duplexes at the T4pA5 base-pair steps. Binding site 1 involving phosphate groups is exclusively occupied by alkali metal ions and is shown along with symmetry-related sites (1a and 1b). DNA duplexes listed in Table 1 are drawn as stick models with color codes indicated. The low salt Mg-MEP (21) and Mg-TOE (22) forms were also included in the superposition and are colored red and magenta, respectively. Mg2+ and Ba2+ coordinated in the major groove are shown as spheres with larger radius. Alkali metal ions are drawn as smaller spheres. (B) Schematic of the major groove of the A-DNA decamer with sequence GCGTATACGC. A green circle indicates ions binding at the G3pT4 step. No ion was found at the chemically identical G13pA14 step. Asterisks indicate 2′-O-modified nucleotides.
Figure 4.
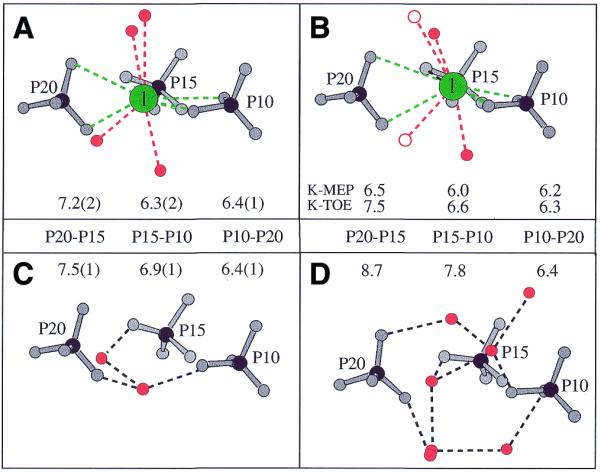
Coordination geometry of alkali metal ions bound at site 1, involving phosphate groups from three adjacent duplexes. (A) Cs-MEP, Rb-FET (high salt), Cs-TOE and Rb-TOE (medium salt) structures. (B) K-MEP (high salt) and K-TOE (medium salt) structures. Ordered ions (green) are arranged in the plane defined by P10, P15 and P20 (A and B) and ion–DNA (green) and ion–water contacts (red) are represented by dashed lines. (C) Na-TOE and Na-FET structures (medium salt). (D) Mg-MEP (low salt). No ordered ions were found in the structures depicted in (C) and (D). P…P distances are in Å and correspond to average values with standard deviations in parentheses (A and C). Water molecules are red and open circles in (B) are waters absent in K-MEP due to limited resolution.
Figure 5.
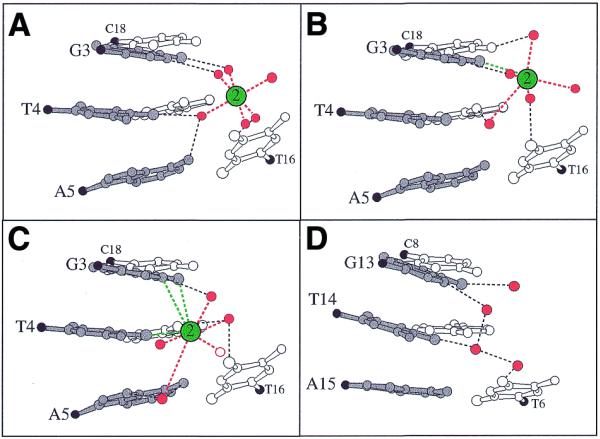
Major groove solvation around GpTpA portions. (A) G3pT4pA5: Mg-MEP and Mg-TOE (low salt). (B) G3pT4pA5: Ba-AOE. (C) G3pT4pA5: consensus geometry of site 2 in the Cs-MEP and Rb-FET (high salt) as well as in Cs-, Rb-, K- and Na-TOE (medium salt) crystals. The water drawn with an open circle is absent in all medium salt forms. (D) Hydration of the chemically equivalent G13pT14pA15 portion. Residues from strands one and two are drawn with solid and open bonds, respectively, C1′ atoms are black and ion–DNA (green) and ion–water contacts (red) are represented by dashed lines.
Figure 6.
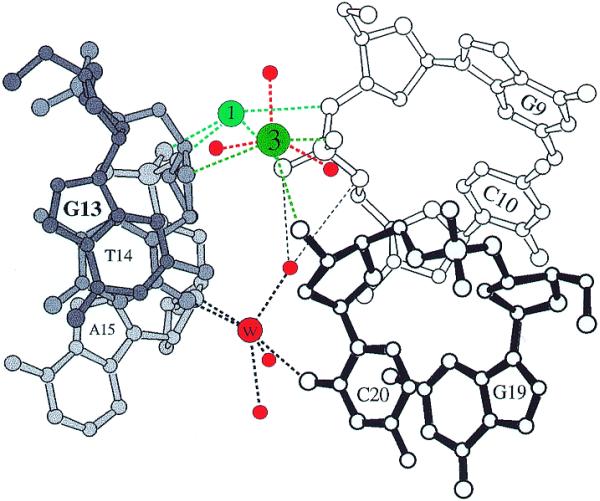
Coordination geometry of Cs+ and Rb+ at binding site 3 in the Cs-MEP and Rb-FET (high salt) structures. Residues from three adjacent duplexes at the minor groove–terminal base pair interface are drawn with solid, gray and open bonds and are numbered. Ion–DNA and ion–water contacts are drawn with green and red dashed lines, respectively. An ion bound at site 1 and the water molecule (W) corresponding to the peak indicated by red arrows and lines in Figures 7 and 8, respectively, are included. In the Rb-, K- and Na-TOE (medium salt) structures, the peak at site 3 was found to be shifted from its position in the Cs-MEP and Rb-FET structures (see Table S4)..
All analyzed duplexes exhibit a kink of between 12° and 15° into the major groove at the T4pA5 step and ions bound at site 2 stabilize this kink (Fig. 3). Numerical details for helical parameters of all duplexes are depicted in Figure S3. Interestingly, the magnitude of the kink appears to be independent of the nature of the bound ion. The packing mode of decamers in the orthorhombic lattice involves stacking of terminal base pairs from two duplexes into the minor groove of a third. The observed metal ions either stabilize lattice interactions (sites 1 and 3) or the kink of duplexes (site 2), the latter likely being caused by the particular packing mode. Thus, each duplex is in contact with seven ions; three symmetry-related ions each at the type 1 and 3 sites and one ion at site 2.
Occupancies of binding sites
In addition to B-factor refinements (values for individual ions are listed in Table 1), analyses of ion–ligand distances in crystals may furnish insight into the occupancy of a particular site. For example, comparisons of alkali metal ion–ligand distances at sites 1–3 as a function of the metal ion concentration demonstrate that site 1 can contract to accommodate the smaller ions (Fig. 7). For the high salt Cs+, Rb+ and K+ crystal forms the site appears to be fully occupied, while medium ion concentrations seem to lower the occupancies for K+ and Rb+. On the other hand, site 2 appears to be ideal for Cs+, independent of the ion concentration, while the geometric constraints of the binding site may not be ideal for Rb+ and K+. Based on an analysis of ion–ligand distances, site 3 appears to be well suited for coordination by Cs+ and Rb+. However, the high B factors of these ions (Table 1) plus the fact that they were not visible in Patterson maps suggest relatively low occupancies. The reduced occupancies at site 3 compared with site 1 are probably due to the different numbers of phosphates present at the two sites (three versus one at sites 1 and 3, respectively). These data are in line with the observation that sites 1 and 3 are exclusively occupied by alkali metal ions and that site 2 is optimal for the larger alkali and all earth alkali metal ions. Taken together, it appears that analyses of ion–ligand distances in DNA crystals can give qualitative information about ion occupancy.
Figure 7.
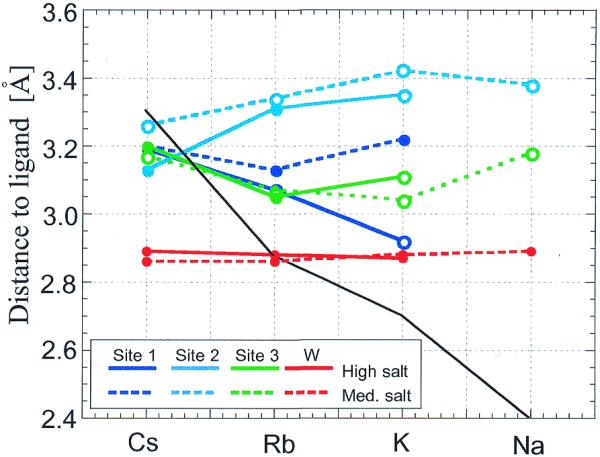
Average distances between alkali metal ions and oxygen atoms (DNA or water) plotted as a function of ion type for high and medium salt concentrations. The black solid line indicates ideal coordination distances for individual alkali metal ions. Open circles indicate coordination spheres that lack water molecules compared with fully occupied sites (high salt Cs-MEP and Rb-FET; see Tables S2–S4).
Anomalous diffraction versus high resolution data
Do resolution-dependent Fourier electron density maps and anomalous maps provide a consistent picture concerning the locations of alkali metal ions in the DNA crystals? We examined this question by comparing the putative identities of peaks in both types of maps (Fig. 8). Accordingly, we found only one case (Cs-MEP, high salt form) where the top three peaks in the electron density map coincide with the three strongest peaks in the anomalous map, independent of the resolution of the former. In all other cases the locations of at least two of the peaks identified as ions based on the anomalous data deviate from those hinted at as ions based on electron density peak lists (Fig. 8). In general, the two techniques provide a more consistent picture with high resolution data based on crystals grown from solutions containing relatively high concentrations of alkali metal ion salts (Rb-FET and Cs-TOE, Fig. 8).
Figure 8.
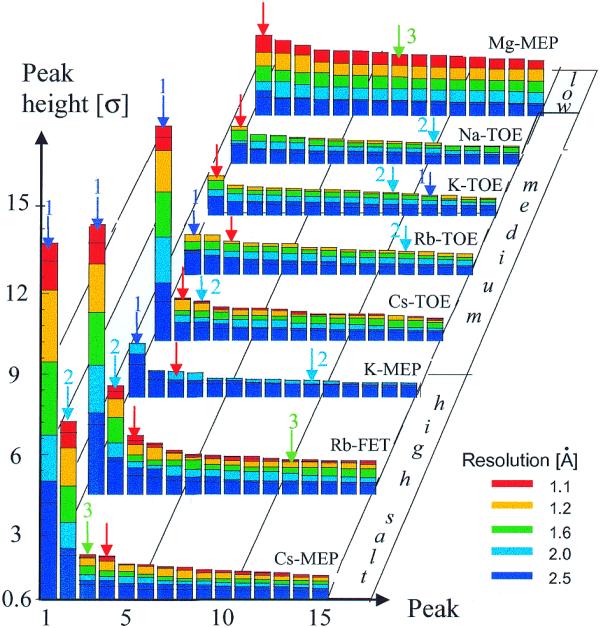
Dependence of ‘solvent’ peak height in electron density maps of selected DNA decamers as a function of X-ray data resolution, nature of alkali metal ion and ion concentration. Blue, cyan and green arrows represent ions coordinated at sites 1, 2 and 3, respectively, and red arrows represent hydration site W (Fig. 6).
A particular peak in the electron density maps that is among the four highest in all structures (Fig. 8, red arrows) is absent in anomalous maps. Without the availability of anomalous data one may have been tempted to interpret the peak as an ion, particularly when taking into account the fact that its primary coordination sphere involves only oxygen atoms (Fig. 6, site W, and Table S5). The interpretation that this conserved peak is indeed a water molecule and not an ion is borne out by its distance geometry, which is independent of the nature and concentration of the ion in the individual crystals (Fig. 7). Thus, anomalous and high resolution data provide a basis for reliable identification of K+, Rb+ and Cs+ ions around DNA. Sites that bind these three types of ions are likely to also harbor Na+, albeit with different occupancies and coordination geometries (Figs 4–6). Conversely, high resolution electron density maps alone are not sufficient to unambiguously differentiate between water and partially ordered ions.
Provided data to ultra-high resolution were available, could one differentiate between light alkali metal ions and water molecules based on the presence of difference electron density for hydrogen atoms around the latter? To answer this question and examine if hydrogen atoms of water molecules can indeed be visualized in DNA crystals at ultra-high resolution, we refined the structure of a Z-DNA hexamer duplex to 0.6 Å (Table 1). However, the vast majority of ‘solvent’ peaks (refined as oxygen atoms) did not exhibit the expected lobes of difference electron density expected for hydrogen atoms of water (data not shown). Therefore, electron density maps even at this level do not provide insight into the exact nature of a peak, apart from the fact that most crystals will not yield diffraction data to such a resolution. Conversely, the use of both anomalous diffraction and atomic resolution data for nucleic acid crystals grown from solutions of different ionic strengths allows a refined understanding of alkali metal ion coordination to DNA and should benefit nucleic acid crystallography in general.
SUPPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Supplementary Material
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Mr Guillermo Vasquez, Mr Martin Casper and Dr Haoyun An (Isis Pharmaceuticals Inc., Carlsbad, CA) for providing the 2′-O-modified AOE, FET and MEP decamers, Dr M.Teplova for help with the crystallizations and Dr Lucy V.Malinina for discussions. This work was supported by NIH grant GM-55237 (M.E.) and C.J.W. acknowledges fellowship support by the Natural Sciences and Engineering Research Council of Canada. The DuPont-Northwestern-Dow Collaborative Access Team Synchrotron Research Center at the Advanced Photon Source (Sector 5) is supported by E. I. DuPont de Nemours & Co., The Dow Chemical Company, the National Science Foundation and the State of Illinois.
References
- 1.Quigley G.J., Teeter,M.M. and Rich,A. (1978) Structural analysis of spermine and magnesium ion binding to yeast phenylalanine transfer RNA. Proc. Natl Acad. Sci. USA, 75, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott W.G., Finch,J.T. and Klug,A. (1995) The crystal structure of an all-RNA hammerhead ribozyme: a proposed mechanism for RNA catalytic cleavage. Cell, 81, 991–1002. [DOI] [PubMed] [Google Scholar]
- 3.Cate J.H. and Doudna,J.A. (1996) Metal-binding sites in the major groove of a large ribozyme domain. Structure, 4, 1221–1229. [DOI] [PubMed] [Google Scholar]
- 4.Cate J.H., Hanna,R.L. and Doudna,J.A. (1997) A magnesium core at the heart of a ribozyme domain. Nature Struct. Biol., 4, 553–558. [DOI] [PubMed] [Google Scholar]
- 5.Correll C.C., Freeborn,B., Moore,P.B. and Steitz,T.A. (1997) Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell, 91, 705–712. [DOI] [PubMed] [Google Scholar]
- 6.Ippolito J.A. and Steitz,T.A. (1998) A 1.3-Å resolution crystal structure of the HIV-1 trans-activation response region RNA stem reveals a metal-ion dependent bulge conformation. Proc. Natl Acad. Sci. USA, 95, 9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gessner R.V., Frederick,C.A., Quigley,G.J., Rich,A. and Wang,A.H.-J. (1989) The molecular structure of Z-DNA at high resolution. J. Biol. Chem., 264, 7921–7935. [DOI] [PubMed] [Google Scholar]
- 8.Tereshko V., Minasov,G. and Egli,M. (1999) The Dickerson-Drew B-DNA dodecamer revisited at atomic resolution. J. Am. Chem. Soc., 121, 470–471. [Google Scholar]
- 9.Minasov G., Tereshko,V. and Egli,M. (1999) Atomic-resolution crystal structures of B-DNA reveal specific influences of divalent metal ions on conformation and packing. J. Mol. Biol., 291, 83–99. [DOI] [PubMed] [Google Scholar]
- 10.Soler-Lopez M., Malinina,L., Liu,J., Hyunh-Dinh,T. and Subirana,J.A. (1999) Water and ions in a high resolution structure of B-DNA. J. Biol. Chem., 274, 23683–23686. [DOI] [PubMed] [Google Scholar]
- 11.Kielkopf C.L., Ding,S., Kuhn,P. and Rees,D.C. (2000) Conformational flexibility of B-DNA at 0.74 Å resolution: d(CCAGTACTGG)2. J. Mol. Biol., 296, 787–801. [DOI] [PubMed] [Google Scholar]
- 12.Chiu T.K. and Dickerson,R.E. (2000) 1 Å crystal structures of B-DNA reveal sequence-specific binding and groove-specific bending of DNA by magnesium and calcium. J. Mol. Biol., 301, 915–945. [DOI] [PubMed] [Google Scholar]
- 13.Berman H.M., Olson,W.K., Beveridge,D.L., Westbrook,J., Gelbin,A., Demeny,T., Hsieh,S.-H., Srinivasan,A.R. and Schneider B. (1992) The nucleic acid database: a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J., 63, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phillips K., Dauter,Z., Murchie,A.I.H., Lilley,D.M.J. and Luisi,B. (1997) The crystal structure of a parallel-stranded guanine tetraplex at 0.95 Ångstrom resolution. J. Mol. Biol., 273, 171–182. [DOI] [PubMed] [Google Scholar]
- 15.Tereshko V., Minasov,G. and Egli,M. (1999) A “hydrat-ion” spine in a B-DNA minor groove. J. Am. Chem. Soc., 121, 3590–3595. [Google Scholar]
- 16.Bleam M.L., Anderson,C.F. and Record,M.T.,Jr (1980) Relative binding affinities of monovalent cations for double-stranded DNA. Proc. Natl Acad. Sci. USA, 77, 3085–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu S., Rambo,R.P., Strauss-Soukup,J., Cate,J.H., Ferré-D’Amaré,A.R., Strobel,S.A. and Doudna,J.A. (1998) A specific monovalent metal ion integral to the AA platform of the RNA tetraloop receptor. Nature Struct. Biol., 5, 986–992. [DOI] [PubMed] [Google Scholar]
- 18.Brünger A.T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 19.Chen L.Q., Rose,J.P., Breslow,E., Yang,D., Chang,W.R., Furey,W.F., Sax,M. and Wang,B.C. (1991) Crystal structure of a bovine neurophysin-II dipeptide complex at 2.8 Å determined from the single-wavelength anomalous scattering signal of an incorporated iodine atom. Proc. Natl Acad. Sci. USA, 88, 4240–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dauter Z., Dauter,M., de La Fortelle,E., Bricogne,G. and Sheldrick,G.M. (1999) Can anomalous signal of sulfur become a tool for solving protein crystal structures? J. Mol. Biol., 289, 83–92. [DOI] [PubMed] [Google Scholar]
- 21.Egli M., Usman,N. and Rich,A. (1993) Conformational influence of the ribose 2′-hydroxyl group: crystal structures of DNA-RNA chimeric duplexes. Biochemistry, 32, 3221–3237. [PubMed] [Google Scholar]
- 22.Egli M., Tereshko,V., Teplova,M., Minasov,G., Joachimiak,A., Sanishvili,R., Weeks,C.M., Miller,R., Maier,M.A., An,H., Cook,P.D. and Manoharan,M. (2000) X-ray crystallographic analysis of the hydration of A- and B-form DNA at atomic resolution. Biopolymers, 48, 234–252. [DOI] [PubMed] [Google Scholar]
- 23.Tereshko V., Portmann,S., Tay,E., Martin,P., Natt,F., Altmann,K.-H. and Egli,M. (1998) Structure and stability of DNA duplexes with incorporated 2′-O-modified RNA analogues. Biochemistry, 37, 10626–10634. [DOI] [PubMed] [Google Scholar]
- 24.Teplova M., Minasov,G., Tereshko,V., Inamati,G.B., Cook,P.D., Manoharan,M. and Egli,M. (1999) Crystal structure and improved antisense properties of 2′-O-(2-methoxyethyl)-RNA. Nature Struct. Biol., 6, 535–539. [DOI] [PubMed] [Google Scholar]
- 25.Teplova M., Wallace,S.T., Minasov,G., Tereshko,V., Symons,A., Cook,P.D., Manoharan,M. and Egli,M. (1999) Structural origins of the exonuclease resistance of a zwitterionic RNA. Proc. Natl Acad. Sci. USA, 96, 14240–14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malinina L.V., Makhaldiani,V.V., Tereshko,V.A., Zarytova,V.F. and Ivanova,E.M. (1987) Phase diagrams for DNA crystallization systems. J. Biolmol. Struct. Dyn., 5, 405–433. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez L.G., Subirana,J.A., Verdaguer,N., Pyshnyi,D., Campos,L. and Malinina,L.V. (1997) Structural variability of A-DNA in crystals of the octamer d(pCpCpCpGpCpGpGpG). J. Biomol. Struct. Dyn., 15, 233–245. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 29.Brünger A.T. (1998) Crystallography & NMR System (CNS), Version 0.5. Yale University, New Haven, CT.
- 30.Cambillau C. and Roussel,A. (1997) Turbo Frodo, Version OpenGL.1. Université Aix-Marseille II, Marseille, France.
- 31.Sheldrick G.M. and Schneider,T.R. (1997) SHELXL: High-resolution refinement. Methods Enzymol., 276, 319–343. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.