

Abstract
The impairment of hippocampal neurogenesis has been linked to the pathogenesis of neurological disorders from chronic neurodegenerative disease to the progressive cognitive impairment of children who receive brain irradiation. Numerous studies provide evidence that inflammation downregulates neurogenesis, with multiple factors contributing to this impairment. Although mitochondria are one of the primary targets of inflammatory injury, the role of mitochondrial function in the modulation of neurogenesis remains relatively unstudied. In this study, we used neurosphere-derived cells to show that immature doublecortin (Dcx)-positive neurons are uniquely sensitive to mitochondrial inhibition, demonstrating rapid loss of mitochondrial potential and cell viability compared with glial cells and more mature neurons. Mitochondrial inhibition for 24 h produced no significant changes in astrocyte or oligodendrocyte viability, but reduced viability of mature neurons by 30%, and reduced survival of Dcx+ cells by 60%. We demonstrate that protection of mitochondrial function with mitochondrial metabolites or the mitochondrial chaperone mtHsp75/mortalin partially reverses the inflammation-associated impairment of neurogenesis in vitro and in irradiated mice in vivo. Our findings highlight mitochondrial mechanisms involved in neurogenesis and indicate mitochondria as a potential target for protective strategies to prevent the impairment of neurogenesis by inflammation.
Introduction
Neurogenesis continues throughout life, with thousands of new neurons generated daily in the dentate gyrus subgranular zone (Gage, 2000; Cameron and McKay, 2001). Impairment of hippocampal neurogenesis is implicated in aging-associated cognitive decline, Alzheimer's disease, and ionizing radiation damage (Haughey et al., 2002; Monje and Palmer, 2003; Rola et al., 2004; Galvan and Jin, 2007), all settings in which inflammation is thought to play a role (Nelson et al., 2002; Sparkman and Johnson, 2008). The number of activated microglia directly correlates with the impairment of neurogenesis in irradiated animals (Mizumatsu et al., 2003) and treatment with antiinflammatory drugs reversed impairment of neurogenesis caused by inflammation or irradiation (Ekdahl et al., 2003; Monje et al., 2003). Cerebral ischemia stimulates neurogenesis in adult brain (Liu et al., 1998; Türeyen et al., 2004; Tonchev et al., 2005), and the nonsteroidal antiinflammatory drug indomethacin increases survival of these newly generated cells (Takasawa et al., 2002; Sun et al., 2003; Hoehn et al., 2005).
Only activated, not resting, microglia inhibit neurogenesis (Monje et al., 2003). Activated microglia produce proinflammatory factors, including IL-6 and tumor necrosis factor-α (TNF-α) (Pocock and Liddle, 2001; Hanisch, 2002), reactive oxygen species (ROS), and nitric oxide (Rock et al., 2004). TNF-α induces mitochondrial damage through suppression of mitochondrial complexes I and IV and pyruvate dehydrogenase (PDH) activities (Stadler et al., 1992; Zell et al., 1997; Samavati et al., 2008). Exposure to increased ROS impairs oxidative phosphorylation through oxidation of mitochondrial lipids and respiratory enzymes (Wagner et al., 1990; Halliwell, 2006), and IL-6 stimulates increased ROS production in brain (Behrens et al., 2008). Nitric oxide is a potent inhibitor of mitochondrial cytochrome c oxidase (complex IV) (Brown, 1995; Giuffrè et al., 1996).
Inflammation induces mitochondrial dysfunction in vitro and in vivo (Xie et al., 2004; Hunter et al., 2007), and exposure to IL-6, TNF-α, and nitric oxide impair in vitro neurogenesis (Monje et al., 2003; Covacu et al., 2006; Peng et al., 2008), so we postulated that impairment of mitochondrial function by inflammatory mediators is a mechanism by which inflammation impairs neurogenesis.
Two recent studies highlight the involvement of mitochondrial function in neurogenesis. First, mice deficient in α-ketoglutarate dehydrogenase complex activity were shown to have reduced hippocampal neurogenesis with fewer doublecortin-positive (Dcx+) cells (Calingasan et al., 2008). Second, treatment with the mitochondrial antioxidant α-lipoic acid partially reversed the radiation-induced reduction of immature Dcx+ neurons in hippocampus in vivo (Fike et al., 2007). These studies led us to postulate that Dcx+ cells might represent the stage in neurogenesis most susceptible to inhibition by mitochondrial impairment, such as that triggered by inflammation. We therefore tested which cells produced in the course of neural progenitor differentiation were most vulnerable to inhibition of mitochondrial function and identified the Dcx+ early neuron as most susceptible. Several strategies to protect mitochondria, including overexpressing a mitochondrial chaperone, heat shock protein 75 (mtHsp75), or providing mitochondrial fuels and cofactors, were found here to rescue the impairment of neurogenesis caused by activated microglial medium-induced inflammation in vitro and radiation in vivo.
Materials and Methods
Cell culture.
Neural precursor cells were isolated from newborn mice. The brains were removed, freed of meninges, and diced with a sterile razor blade in dissociation buffer [DMEM/F12 medium containing 2.5 U/ml papain (Worthington), 1 U/ml Dispase II (Roche Diagnostics), and 250 U/ml DNase I (Worthington)]. After a 1 h incubation, the cells were washed once with DMEM/F12 supplemented with 10% fetal bovine serum (FBS) (HyClone) and then twice with DMEM/F12. The cells were resuspended in growth medium, Neurobasal A (Invitrogen), with 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen), B-27 without vitamin A (Invitrogen), 20 ng/ml fibroblast growth factor-2 (PeproTech), and 20 ng/ml epidermal growth factor (PeproTech), and plated at a density of 1 brain per six-well plate. Neural precursor cells proliferated and started to form neurospheres in 2–3 d. One-half of the growth medium was changed every 2 d. To prevent attachment of the neurospheres to the bottom, the medium was pipetted to resuspend the plated neurospheres. The neurospheres were passaged when the diameter of neurospheres reached ∼100 μm. For passaging, the floating neurospheres were collected by centrifugation at 400 × g for 5 min. The supernatant was removed and the cells were gently triturated with a 200 μl pipette tip 20–25 times, resuspended in growth medium, and plated in six-well plates after 1:2 dilution. Neural precursor cells were induced to differentiate by plating dissociated neurospheres (second or third passage) into laminin-coated 24-well plates. The plates were coated with 10 μg/well laminin (Invitrogen) in dH2O for 2–3 h, and rinsed two times with dH2O. The floating neurospheres were again collected by centrifugation, and the cell pellet was resuspended in a small volume of differentiation medium, Neurobasal A (Invitrogen), B27 without vitamin A (Invitrogen), 1% FBS, 100 nm all-trans retinoic acid (Sigma-Aldrich), 1 ng/ml fibroblast growth factor-2 (PeproTech), 10 ng/ml brain-derived neurotrophic factor (PeproTech), and 10 ng/ml neurotrophic factor-3 (PeproTech). The neurospheres were gently triturated with a 200 μl pipette tip. The cells were subsequently resuspended into 5 ml of differentiation medium and passed through a 40 μm cell strainer (BD Biosciences). The cells were plated onto laminin-coated 24-well plates at a density of ∼100,000 cells/well. One-half of the differentiation medium was changed every 2 d. For the mitochondrial inhibition experiments, the cells were allowed to differentiate for 8–9 d and treated with 2 μm antimycin A for 24 h. For the microglial conditioned media (CM) experiments (see below), the cells were allowed to differentiate for 5–6 d before the 24 h CM exposure.
BV-2 cells and CM.
BV-2 murine microglia were plated on uncoated plastic tissue culture plates and grown in DMEM supplemented with 10% FBS, and 100 U/ml penicillin and 100 μg/ml streptomycin. BV-2 cells were treated with fresh differentiation medium with added lipopolysaccharide (LPS) (1 μg/ml; Sigma-Aldrich) to prepare CM from activated microglia, or without LPS for control medium, for 6–8 h. The medium was collected, sterile filtered, and applied to neural progenitor cells as described above. For in vitro mitochondrial protection experiments, lactate (Sigma-Aldrich), pyruvate (Sigma-Aldrich), thiamine-HCl (Sigma-Aldrich), glutathione (GSH) (Sigma-Aldrich), glutathione ethyl ester (GSH-EE) (Sigma-Aldrich), cyclosporin A (CsA) (Sigma-Aldrich), α-lipoic acid (Geronova Research), and NIM811 (Novartis) were added to CM for the duration of treatment.
In vitro immunocytochemistry.
Fluorescence immunocytochemistry was performed on cell cultures in 24-well plates. The cultures were washed with PBS and then fixed in 4% paraformaldehyde (PFA) for 30 min at room temperature. The cells were then washed twice with PBS, and nonspecific binding was blocked with 3% bovine serum albumin, 0.3% Triton X-100 in PBS for 1 h. The cells were subsequently incubated with primary antibodies diluted in blocking buffer overnight at 4°C. The cell-specific antibodies used were Dcx for immature neurons (1:500; sc-8066; Santa Cruz Biotechnology); glial fibrillary acid protein (GFAP), an intermediate filament protein expressed by astrocytes (1:3; 22522; Immunostar); NG2, a marker expressed by immature oligodendrocytes (1:200; AB5320; Millipore Bioscience Research Reagents); microtubule-associated protein 2 (MAP2), a marker for mature neurons (1:500; AB5622; Millipore Bioscience Research Reagents); and CD11b, a surface marker expressed by microglial cells (1:200; MCA74G; Serotec). Cells were subsequently incubated with the appropriate secondary Alexa Fluor 488- or 594-conjugated secondary antibodies (1:200; Invitrogen). To identify proliferating cells that had incorporated 5-bromodeoxyuridine (BrdU), the cultures were fixed with 4% PFA after the cell-specific labeling, rinsed two times in saline, and incubated in 2 m HCl in saline for 30 min at 37°C. The BrdU staining was performed using primary anti-BrdU antibody (1:500; OBT0030; Accurate Chemical and Scientific) and Alexa Fluor 594-conjugated secondary antibody (1:200; Invitrogen). Cell nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (0.5 μg/ml; Sigma-Aldrich). The immunofluorescence was visualized with an epifluorescence microscope (Zeiss Axiovert 200M) as previously described (Voloboueva et al., 2008).
Live imaging.
To monitor changes in mitochondrial membrane potential, cells were incubated with the mitochondrial membrane potential sensitive dye tetramethylrhodamine ethyl ester (TMRE) (50 nm). Cells were illuminated at 535 nm, and fluorescence emission was observed at 590 nm. The location of the collected images was marked on the cell culture wells. The cells were subsequently fixed and stained with anti-Dcx antibody as described above, and the images of Dcx staining were taken in the same areas where TMRE imaging was performed. Subsequently, the red and green channels corresponding to TMRE and Dcx fluorescent signals were overlapped using Adobe Photoshop 6.0. Mitochondrial-produced ROS were evaluated after 1 h of antimycin A treatment using MitoSox staining (Invitrogen) according to the manufacturer's instructions. Staining for apoptotic cells was performed using Magic Red caspase 3 and 7 detection kit (Immunochemistry Technologies) according to the manufacturer's instructions.
Dcx–green fluorescent protein construct and lentiviral preparation.
The mouse Dcx promoter (MmDCXp) was cloned from C57BL/J6 mouse genomic DNA by PCR using the following primers: CTCGAGATATTCTTATCGCCGCACATC and GGATCCTTGGTGGAACCACAGCAACCTGA. The product was cloned into pCR2.1 using a TOPO-TA kit (Invitrogen) and excised by XhoI and BamHI. It was subcloned into pL_UGIN (Iain Frasier, Caltech, Pasadena, CA), after removing the ubiquitin promoter with the same enzymes. To produce virus, the resulting plasmid, pL_DCXpGIN, was cotransfected with pMD.G and pCMVDR8.91 (Ory et al., 1996; Zufferey et al., 1997) into 293FT cells (Invitrogen) using FuGene6 (Roche Diagnostics) in a T75 flask with 12 ml of 50% DMEM plus 8% FBS, 50% OptiMEM (Invitrogen). Forty-eight hours after transfection, 293 FT cells were fusing and expressing green fluorescent protein (GFP). The culture medium was harvested, filtered through a 0.45 μm syringe filter (Millipore), and concentrated from 12 to ∼0.5 ml using a Vivaspin 20 with 100,000 kDa molecular weight cutoff (Sartorius). Viral titers were typically in the range 106 cfu/ml after concentration. The entire concentrated viral supernatant was mixed with dissociated neurospheres suspended in 40 ml of the differentiation medium. The cells were plated onto 24-well laminin-coated plates as described above and allowed to differentiate for 7–8 d.
Mitochondrial mtHsp75 transfection.
mtHsp75/mortalin is an essential mitochondrial chaperone that has been shown to protect mitochondrial function both in vitro and in vivo (Voloboueva et al., 2008; Williamson et al., 2008; Xu et al., 2009). For mtHsp75 transfection experiments, the neurospheres were grown, passaged, and dissociated for plating onto laminin-coated 24-well plates to induce differentiation as described above. Immediately before plating, the cells were cotransfected with either LXSN-mtHsp75 (Voloboueva et al., 2008) and pDsRed2-N1 (Clontech) plasmids, or empty LXSN and pDsRed2-N1 plasmids for control conditions, using FuGENE 6 (Roche Diagnostics). pDsRed2-N1 plasmid was used as a fluorescent marker to label the transfected cells. After 3 d of differentiation, the cells were again transfected with the same plasmids using FuGENE HD (Roche Diagnostics). The cells were allowed to differentiate for a total of 5–6 d before 24 h exposure to microglial conditioned media. The cells were fixed and immunostained for Dcx as described above. Four 400 × 300 μm2 areas per well, in three wells per transfection, were quantified in three independent experiments. Only the transfected Dcx cells demonstrating DsRed2 fluorescence were counted.
Thiamine treatment and x-ray irradiation.
Two-month-old male C57BL/J6 mice (∼20 g; The Jackson Laboratory) were used in all studies. Mice were housed in a temperature- and light-controlled environment with a 12 h light/dark cycle and provided food and water ad libitum. All protocols were approved by the Stanford Panel on Laboratory Animal Care and Use. The mice were split into two groups 5 d before the irradiation procedure, with the thiamine-treated group receiving 1% thiamine in the drinking water for the remainder of the experiment. All mice, including sham-irradiated animals, were anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg). Irradiation was performed with a Phillips orthovoltage x-ray system operated at 200 kVp and 20 mA. A single dose of 5 Gy was limited by lead shielding to a 1 cm diameter column that included left and right hippocampal formations. The remaining body, neck, eyes, ears, and snout were shielded. Dosimetry and field dimensions were confirmed using TLD dosimeters (K&S Associates) buried in the hippocampi and surrounding brain areas of killed mice. The dose rate was ∼78.0 cGy/min. To label dividing cells, animals were injected intraperitoneally with halogenated thymidine analogs 5-iodo-2′-deoxyuridine (IdU) (Sigma-Aldrich) or 5-chloro-2′-deoxyuridine (CldU) (Sigma-Aldrich) at a daily dose of 50 mg/kg using a 10 mg/ml solution in saline. IdU injections were given on days 1–3. CldU injections were given on days 4–7.
Tissue preparation.
Animals were anesthetized and killed 8 d after irradiation. Brains were fixed by transcardial perfusion with 4% PFA, removed from the skull, and postfixed overnight in 4% PFA, and then equilibrated in phosphate-buffered 30% sucrose. Free-floating 40 μm sections were collected on a freezing sledge microtome and stored in tissue cryoprotectant solution at −20°C until used.
Immunohistochemistry, cell counting, and unbiased stereology.
For confocal imaging, the sections were immunostained as described previously (Monje et al., 2003) using the following primary antibodies: anti-NeuN (1:200; MAB377; Millipore Bioscience Research Reagents), anti-Dcx (1:500; sc-8066; Santa Cruz Biotechnology), and anti-Iba-1 (1:1000; Wako). To detect IdU+ and CldU+ nuclei, the sections were labeled with rat anti-BrdU (1:300; 347580; BD Biosciences) and mouse anti-BrdU (1:500; ab6326-250; Abcam). For the purpose of the study, IdU and CldU labeling was not discriminated by using secondary antibodies with the same detection marker, Cy3-conjugated donkey anti-mouse and Cy3-conjugated donkey anti-rat (1:500; 715-165-151 and 712-165-153, respectively; Jackson ImmunoResearch). The total number of dividing cells was estimated as the sum of IdU+ and CldU+ cells, and thus dividing cells are denoted as “IdU++CldU+” in all experiments. Confocal microscopy was performed using a Zeiss 510 confocal microscope as described previously (Monje et al., 2003). In stereological studies, all counts were limited to the hippocampal granule cell layer proper and a 50 μm border along the hilar margin that included the neurogenic subgranular zone. The total number of Dcx- or IdU++CldU+-labeled cells per hippocampal granule cell layer and subgranular zone was determined using immunodetection of Dcx or IdU++CldU+ followed by HRP-coupled secondary antibodies and diaminobenzadine stain (Vector Laboratories). Stained Dcx- and IdU++CldU+-positive cells were scored under light microscopy using MicroBrightField Stereo Investigator software and a modification of the dissector method in which random grid placement provided systematic and unbiased sampling of Dcx+ cell density within the dentate gyrus. All analyses were performed by investigators blinded to sample identity and treatment group.
Statistics.
Statistical differences between two groups were determined using unpaired two-tailed Student's t test with Welch's correction. Comparisons between multiple groups were performed with ANOVA followed by Bonferroni's test for selected groups. Analysis of protection by different mitochondrial compounds was performed with ANOVA followed by Dunnet's posttest for comparison between CM-treated group versus all other treatment groups.
Results
Mitochondrial inhibition promotes rapid loss of mitochondrial potential in Dcx+ cells
The effect of inhibition of mitochondrial electron transport on the mitochondrial membrane potential depends on cell type (Beltrán et al., 2000; Almeida et al., 2001) and cellular energetic demands (Voloboueva et al., 2007). We used the mitochondrial membrane potential-sensitive dye TMRE to investigate changes induced by mitochondrial inhibition in neural cultures after 8–9 d of differentiation. Immature neurons express Dcx, a reliable and specific marker of neurogenesis (Couillard-Despres et al., 2005). Figure 1A shows bright TMRE staining of Dcx+ and surrounding cells in control vehicle-treated cultures. Treatment with the mitochondrial inhibitor antimycin A promoted rapid loss of mitochondrial membrane potential in Dcx+ cells, as evidenced by loss of TMRE signal, whereas the surrounding cells retained strong TMRE staining, as shown in Figure 1B. We verified that antimycin A treatment inhibited mitochondrial complex III activity and increased mitochondrial-produced ROS, using MitoSox, a mitochondrial superoxide indicator (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Because collapse of the mitochondrial potential is often associated with release of proapoptotic factors and cell death (Liu et al., 1996; Susin et al., 1999), we investigated changes in viability of different cell types with mitochondrial inhibition.
Figure 1.

Treatment with the mitochondrial inhibitor antimycin A promotes loss of mitochondrial membrane potential in Dcx+ cells. A, Dcx+ cells (arrows; green staining) in control vehicle-treated cultures demonstrate normal mitochondrial potential by bright mitochondrial TMRE red staining. Cell nuclei are counterstained with DAPI (blue). B, Treatment for 4 h with antimycin A promotes strong selective loss of mitochondrial potential in Dcx+ cells (arrows), whereas nearby cells retain TMRE staining. Scale bar, 50 μm.
Effects of mitochondrial inhibition on differentiating cell viability
Neural stem cells allowed to differentiate for 8–9 d were treated with antimycin A for 24 h, and the numbers of surviving cells were assessed. At 8–9 d of differentiation, the majority of cells express cell type-specific markers, allowing us to compare vulnerability of different cell types to mitochondrial inhibition. As shown in the top panels of Figure 2, 24 h mitochondrial inhibition promoted morphological changes in the treated cultures compared with control vehicle-treated cultures. However, the antimycin A treatment promoted no significant change in the fraction of cells expressing the astrocyte-specific marker GFAP (mean ± SEM, 58 ± 2.5% GFAP+ cells in control vs 56 ± 3% GFAP+ in antimycin A-treated cultures; p > 0.05). Comparison of the bright-field and immunostained images of the same field (Fig. 2, control conditions) indicates that the majority of the small process-bearing cells with extended thin processes in bright-field stain for Dcx. These cells either disappeared or demonstrated markedly changed, shrunken morphology after 24 h of mitochondrial inhibition. In parallel, Dcx staining was markedly decreased in the antimycin A-treated cultures. Figure 3 shows that the 24 h antimycin A treatment promoted no significant change in the number of cells expressing immature oligodendrocyte marker NG2 (mean ± SEM, 12 ± 1.2% NG+ cells in control vs 13 ± 1% NG+ in antimycin A-treated cultures; p > 0.05). Again, significant changes were observed in morphology and Dcx staining of the antimycin A-treated cultures. Only a very small fraction of cells (<0.5%) were immunoreactive for the microglial specific marker CD11b. More severe stress caused by chronic antimycin treatment from the beginning of differentiation resulted in complete absence of Dcx cells, and reduced astrocyte differentiation (data not shown), whereas the more limited duration of inhibition only affected Dcx cells, evidence of their greater sensitivity.
Figure 2.

Mitochondrial inhibition does not induce significant changes in the number of astrocytes. Control (left panels) and antimycin A-treated (2 μm for 24 h) (right panels) cultures stained with the astrocyte marker GFAP (green) and the immature neuron marker Dcx (red). Cell nuclei are counterstained with DAPI (blue). Antimycin A treatment promoted marked loss of Dcx+ cells but did not significantly change GFAP staining cells. Scale bar, 50 μm.
Figure 3.

Mitochondrial inhibition does not induce a significant change in the number of immature oligodendrocytes. Control (left panels) and antimycin A-treated (2 μm for 24 h) (right panels) cultures stained with NG2 (green) and Dcx (red) markers. Cell nuclei are counterstained with DAPI (blue). Antimycin A treatment promoted marked loss of Dcx+ cells but did not significantly change NG2 staining. Scale bar, 50 μm.
Effects of mitochondrial inhibition on immature Dcx+ and mature MAP2+ neuronal cells
As described above, mitochondrial inhibition for 24 h resulted in a sharp decrease in the number of Dcx+ cells. Interestingly, we found that Dcx+ cells that also coexpressed MAP2, a marker of more mature neurons, demonstrated better preservation of cell morphology and Dcx+ staining. As shown in Figure 4, Dcx+-single-positive cells retained very weak Dcx labeling and demonstrated a shrunken cellular morphology after 16 h of antimycin A treatment (Fig. 4A, arrowheads). In contrast, Dcx+ cells that colabeled with MAP2 retained a relatively strong Dcx signal and better preserved cell morphology (Fig. 4A, arrows). As demonstrated in Figure 4B, 24 h of mitochondrial inhibition caused a 60% loss of Dcx+ cells, whereas MAP2 staining was decreased by only 30%. These results indicate that single-positive Dcx+ cells are particularly vulnerable to inhibition of mitochondrial function compared with glial cells and more mature neurons. To investigate whether the high vulnerability of Dcx+ cells is related to their proliferative state, we treated cells with 10 μm BrdU during the 24 h antimycin A treatment to label proliferating cells. Less than 10% of Dcx+ cells were labeled with BrdU after 8–9 d of differentiation (Fig. 4C), and the fraction of BrdU-labeled cells was not changed by antimycin A treatment (mean ± SEM, 7 ± 1.3% BrdU+ cells in control vs 8 ± 2% BrdU+ in antimycin A-treated cultures, relative to the total number of Dcx+ cells; p > 0.05).
Figure 4.
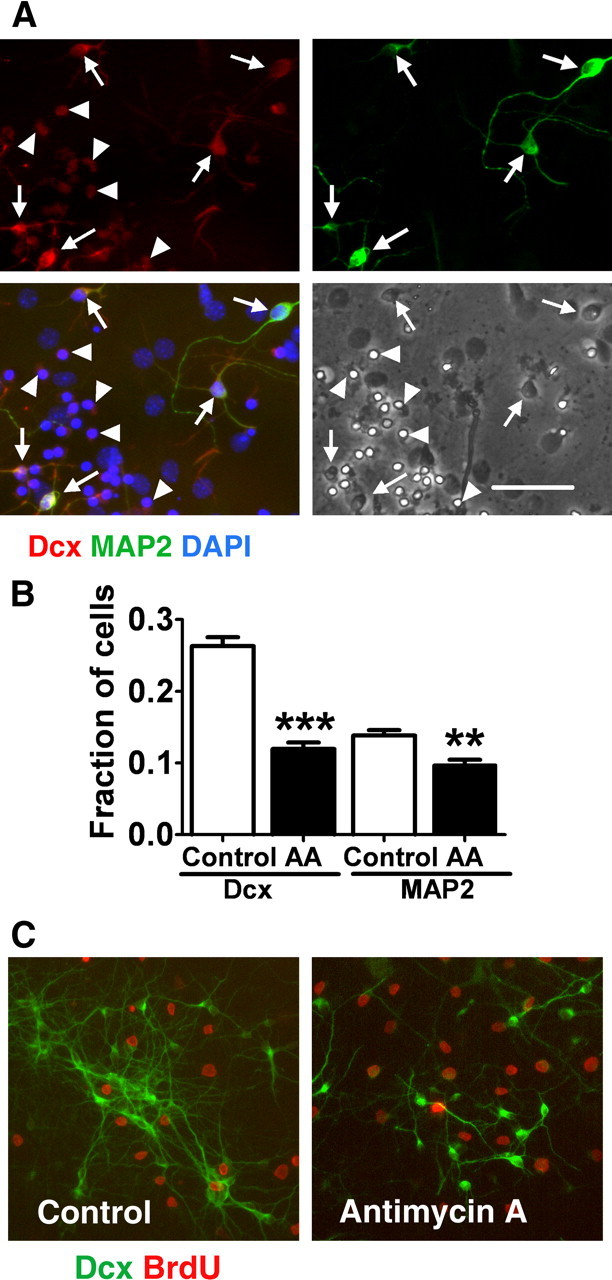
Immature Dcx+ neurons are selectively vulnerable to mitochondrial inhibition. A, Cultures treated with antimycin A (2 μm for 16 h) stained with an immature neuron marker Dcx (red) and a marker for more mature neurons MAP2 (green). Cell nuclei are counterstained with DAPI (blue). The cultures were stained after 16 h of antimycin A treatment, compared with the 24 h time point used for quantification to capture the residual Dcx staining that is essentially gone after 24 h. Cells expressing both Dcx and MAP2 cell markers (arrows) showed better preserved morphology and Dcx staining compared with Dcx-only expressing cells (arrowheads). Scale bar, 50 μm. B, Fractions of Dcx+ and MAP2+ cell (relative to the total cell number) in control- and antimycin A (2 μm, 24 h)-treated cultures. The data are representative of three independent experiments with at least 700 cells per condition in each experiment. Shown are mean ± SEM. **p < 0.01 compared with control MAP2; ***p < 0.001 compared with control Dcx. C, Only a small fraction of Dcx+ cells (green) were proliferating after 8–9 d of differentiation, as demonstrated by colabeling with BrdU (red). The fraction of proliferating BrdU cells was not significantly changed by 24 h antimycin A treatment.
Mitochondrial inhibition promotes apoptotic death in Dcx+ cells
Mitochondrial impairment has been shown to promote apoptotic cell death with depolarization associated with release of apoptosis-inducing factor and cytochrome c, and activation of procaspases (Liu et al., 1996; Susin et al., 1999). To investigate the mode of Dcx+ cell death induced by mitochondrial inhibition, the cultures were infected with a lentiviral vector in which GFP was placed under the control of the mouse Dcx promoter to label Dcx+ cells (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This allowed us to monitor caspase 3 and 7 activity in live cells with a fluorescent caspase substrate. As demonstrated in Figure 5, mitochondrial inhibition induced caspase activation in Dcx+ cells.
Figure 5.

Dcx+ cells (green GFP signal) demonstrate signs of apoptotic cell death (Magic Red live caspase 3 and 7 staining) after 12 h of antimycin A treatment. Scale bar, 50 μm.
Protection of mitochondrial function reduces inflammation-associated Dcx+ cell death in vitro
In our previous study, we demonstrated that treatment with media preconditioned by LPS-activated microglia (CM) significantly decreased the number of Dcx+ cells in vitro (Monje et al., 2003). Activated microglia release a variety of cytokines and reactive species that can impair mitochondrial function, either directly or through cellular pathways. As described above, Dcx+ cells are particularly sensitive to mitochondrial impairment. To investigate the effect of inflammatory mediators released by microglia on mitochondrial potential of Dcx+ cells, cultures were treated with CM medium for 6 h. Figure 6A shows that CM treatment promoted loss of mitochondrial membrane potential in Dcx+ cells, whereas the surrounding cells retained mitochondrial potential, as observed above with direct mitochondrial inhibition with antimycin A (Fig. 1B). We hypothesized that mitochondrial inhibition is at least partially responsible for the inflammation-associated decrease in the number of Dcx+ cells. To test this, we studied the effect of different mitochondrial metabolites and substrates that might protect mitochondrial function and thus inhibit the death of Dcx+ cells induced by CM exposure. In these experiments, cultures were differentiated for 5–6 d before the CM treatment to reduce the number of more mature MAP2+ neuronal cells that were less sensitive to mitochondrial inhibition (Fig. 4).
Figure 6.
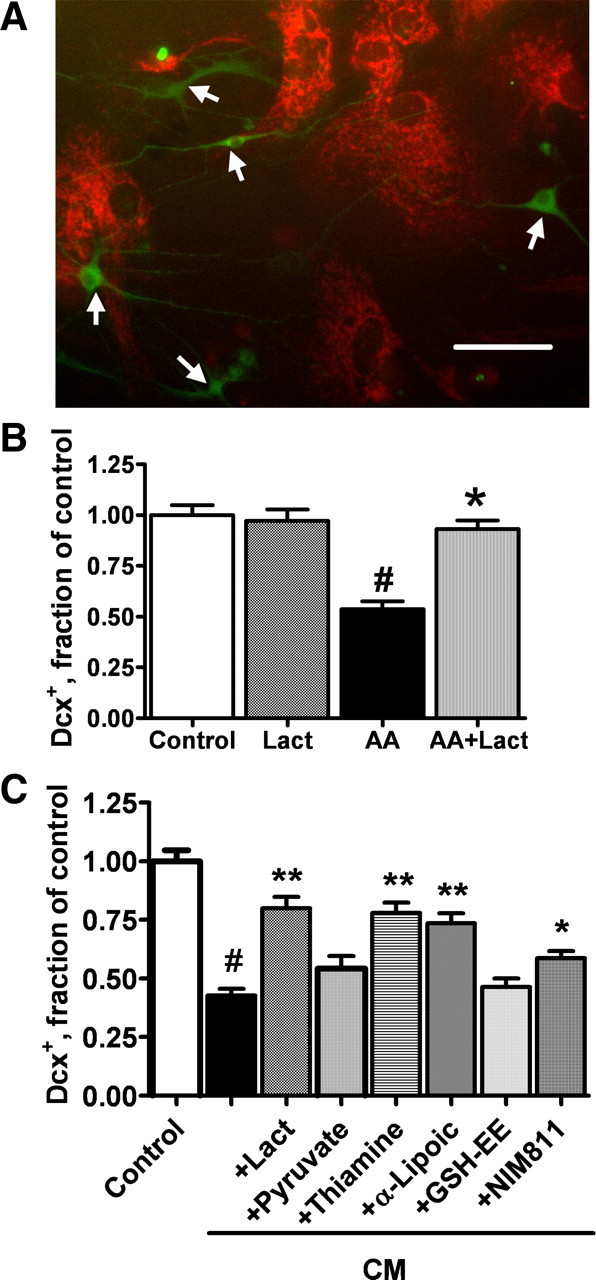
Cotreatment with mitochondrial metabolites and protective compounds attenuate loss of Dcx+ cells induced by exposure to LPS-treated microglial CM. A, CM treatment promoted rapid loss of mitochondrial membrane potential in Dcx+ cells (arrows) as evidenced by selective disappearance of TMRE staining after 6 h of CM treatment. Scale bar, 50 μm. B, Changes in the number of Dcx+ cells induced by 24 h of 10 nm antimycin A (AA) exposure, with and without 5 mm lactate (Lact). C, Changes in the number of Dcx+ cells induced by CM treatment alone or with the addition of mitochondrial metabolites and compounds: lactate (5 mm), pyruvate (5 mm), thiamine (4 mm), α-lipoic acid (100 μm), GSH-EE (1 mm), and NIM811 (2 μm). The data are representative of three independent experiments with at least 150 Dcx+ cells per control condition in each experiment. Shown are mean ± SEM. #p < 0.001 compared with control; *p < 0.05 compared with CM treatment; **p < 0.01 compared with CM treatment.
We investigated whether Dcx+ cell death induced by direct mitochondrial inhibition with antimycin A could be alleviated by supplementation with lactate, a major mitochondrial energy substrate (Schurr, 2006; Herrero-Mendez et al., 2009). Because antimycin A is a potent inhibitor of mitochondrial complex III, the antimycin A concentration was titrated down to a concentration that caused only partial impairment of mitochondrial activity. Treatment with 10 nm antimycin A for 24 h resulted in 49% Dcx+ cell loss, compared with 67% loss in cultures treated with 2 μm antimycin A. The loss induced by 10 nm antimycin A treatment was completely prevented by cotreatment with 5 mm lactate (Fig. 6B).
Treatment with CM for 24 h resulted in 60% loss of Dcx cells compared with control cultures treated with media preconditioned by resting microglia (Fig. 6C). Cotreatment with mitochondrial metabolites and substrates that might protect mitochondrial function showed that lactate (5 mm) reduced CM-associated cell death such that 80% of Dcx+ cells survived compared with cultures treated with control CM. Interestingly, cotreatment with pyruvate (5 mm), another direct substrate of mitochondrial metabolism, was less effective, with protection not reaching significance (p = 0.06). Thiamine and α-lipoic acid are both cofactors of mitochondrial pyruvate and α-ketoglutarate dehydrogenases that have been shown to be protective against brain mitochondrial damage (Sheline et al., 2002; Liu, 2008). Cotreatment with thiamine (4 mm) or α-lipoic acid (100 μm) partially protected against CM toxicity, resulting in survival of 78 and 74% of Dcx+ cells, respectively, compared with control. Because α-lipoic acid is also a potent antioxidant, we investigated the effect of antioxidant treatment on Dcx+ viability. Treatment with different concentrations of the antioxidant GSH (0.2–5 mm) (data not shown) or with the membrane-permeable GSH-EE (0.2–5 mm; 1 mm shown) did not provide significant protection against CM injury. Treatment with NIM811 (2 μm), an inhibitor of mitochondrial permeability transition pore opening that is devoid of calcineurin-inhibiting activity, resulted in survival of 59% of Dcx+ cells compared with control. Treatment with CsA, a more common inhibitor of the mitochondrial permeability transition, did not protect Dcx+ cells against the CM toxicity (data not shown), likely because of its additional activity as a calcineurin inhibitor (Nazareth et al., 1991).
As an alternative strategy to target mitochondrial protection, we investigated the effect of mitochondrial mtHsp75 overexpression on Dcx+ cell viability. Our previous studies demonstrated the protective properties of mtHsp75 overexpression against ischemic stress both in vitro and in vivo (Voloboueva et al., 2008; Xu et al., 2009). Figure 7 shows Dcx staining in control and mtHsp75-overexpressing cultures. mtHsp75 overexpression resulted in a significant 1.3-fold increase in the number of Dcx+ cells in untreated cultures. This suggests that mitochondrial function limits Dcx cell survival under baseline culture conditions. In this series of experiments, CM treatment for 24 h caused an 80% loss of Dcx+ cells in control plasmid-transfected cells compared with a 60% Dcx+ cell loss in mtHsp75-transfected cultures, significantly better survival.
Figure 7.
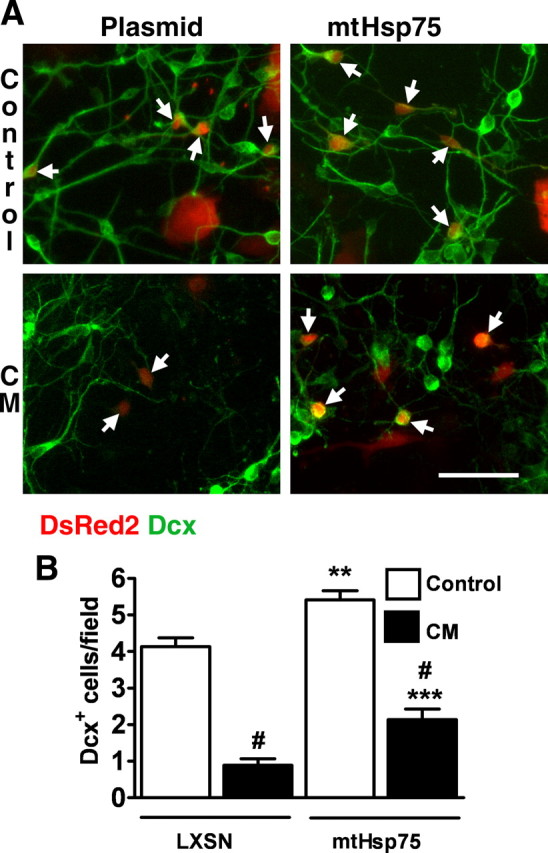
mtHsp75 overexpression attenuates the loss of Dcx+ cells induced by CM exposure. A, Examples of Dcx staining in control plasmid- and mtHsp75-transfected cultures. Transfected cells were identified by DsRed2 coexpression (see Materials and Methods) and are indicated by arrows. Scale bar, 50 μm. B, Changes in the number of transfected Dcx+ cells in control (LXSN empty plasmid) and mtHsp75-overexpressing cultures induced by 24 h CM exposure (black columns). The data are representative of three independent experiments with a total of ∼150 Dcx+ cells per vector control condition. Shown are mean ± SEM. #p < 0.001 compared with respective plasmid- or mtHsp75-transfected cultures without CM treatment; **p < 0.01 compared with plasmid without CM treatment; ***p < 0.001 compared with plasmid plus CM.
Protection of Dcx+ cells in vivo
To test whether this strategy of providing mitochondrial protection would be effective in vivo, we used thiamine oral supplementation in the setting of brain radiation. Thiamine treatment has been considered in several clinical settings (Ozdemir et al., 2002; Haupt et al., 2005; Gibson and Blass, 2007), and studies in animals have shown that thiamine protects against disorders involving mitochondrial dysfunction (Sheline and Wei, 2006). Thiamine is water soluble and absorbed in the intestine by both active and passive transport. Thiamine treatment nearly doubled survival of Dcx+ cells in hippocampi after irradiation from 26% in control-irradiated animals, to 51% in animals receiving thiamine (Fig. 8A,B). We next investigated the effect of thiamine treatment on proliferating Dcx+ cells. As shown in Figure 8C, irradiation promoted a 50% decrease in the total number of dividing IdU+ and CldU+ cells in hippocampus, with thiamine treatment resulting in no significant change in IdU+ and CldU+ cell number in either control or irradiated animals. The percentage of proliferating IdU+ and CldU+ cells demonstrating Dcx+ colabeling was decreased by 60% after irradiation. Thiamine treatment did not significantly change the Dcx+ percentage of proliferating IdU+ and CldU+ in nonirradiated animals, but doubled the Dcx+ fraction of IdU+ and CldU+ cells in irradiated animals (Fig. 8D). The results indicate that thiamine protection doubled the surviving numbers of both total and proliferating Dcx+ cells. Figure 8E shows that, although irradiation caused marked proliferation of microglia, 20% of dividing IdU+ and CldU+ cells coexpressed the microglial marker Iba-1 in irradiated animals, thiamine treatment had no significant effect on this aspect of irradiation-induced inflammation (Fig. 8E).
Figure 8.

Thiamine treatment attenuates the irradiation-associated loss of hippocampal Dcx+ cells. A, Representative Dcx staining of control and thiamine-treated hippocampi 8 d after x-irradiation. B, Quantification of hippocampal Dcx+ cells shows that thiamine treatment significantly reduced irradiation-induced Dcx+ cell loss. C, Thiamine treatment promotes no significant change in the irradiation-induced loss of proliferating IdU++CldU+ cells in hippocampus. D, The percentage of proliferating IdU++CldU+ cells that coexpress Dcx is markedly decreased after irradiation. Thiamine treatment significantly attenuates this decrease in the Dcx+ fraction of IdU++CldU+ cells. E, Microglial proliferation (fraction of IdU++CldU+ cells coexpressing the microglial marker Iba-1) is strongly increased by irradiation, and thiamine treatment does not change the number of proliferating microglia. The data are representative of five animals per each treatment group. Shown are mean ± SEM. #p < 0.001 compared with corresponding nonirradiated group; **p < 0.01, ***p < 0.001 compared with control-irradiated group.
Discussion
Different cell types have different sensitivities to interventions targeting mitochondrial function. It has been shown that, although various cell lines, like HeLa, XP30RO, and GM10115, can tolerate mitochondrial DNA (mtDNA) depletion for extended periods of time, neural precursor cells die within a short period of time after mtDNA depletion (Fike et al., 2009). Inhibition of mitochondrial electron transport in astrocytes strongly upregulates glycolysis. Glycolytically generated ATP is used to support mitochondrial membrane potential through the reversed action of mitochondrial F1F0-ATPase in astrocytes (Almeida et al., 2001). Inhibition of mitochondrial electron transport in neurons promotes rapid loss of mitochondrial membrane potential (Bolaños and Almeida, 2006), although some studies indicate a limited ability of neurons to maintain mitochondrial potential through F1F0-ATPase reversal (Anderson et al., 2002). In our previous study, we demonstrated that changes in energetic demands can affect the ability of the cell to maintain mitochondrial potential in the face of mitochondrial inhibition. In that study, energetic demands imposed on astrocytes by coculture with neurons or by added metabolic load during mitochondrial inhibition prevented maintenance of mitochondrial membrane potential through F1F0-ATPase reversal and resulted in loss of mitochondrial potential and astrocytic death (Voloboueva et al., 2007).
Loss of mitochondrial membrane potential and mitochondrial permeability transition can initiate both apoptotic and necrotic cell death (Kroemer et al., 1998; Lemasters et al., 1998). Because of this, the ability of cells to maintain mitochondrial membrane potential closely correlates with their viability under conditions promoting mitochondrial dysfunction. Here, we observed rapid loss of mitochondrial membrane potential in Dcx+ cells on inhibition of mitochondrial electron transport. This rapid loss of mitochondrial potential correlated with a significant drop in Dcx+ cell survival, in striking contrast to astrocytes and oligodendrocytes, which exhibited no change in viability. Even comparing Dcx single-positive cells to slightly more mature Dcx and MAP2 double-positive cells viability was cut in one-half. This indicates that Dcx+ single-positive immature neurons are particularly vulnerable to conditions impairing mitochondrial metabolism, including the presence of inflammatory mediators.
Previous studies demonstrated that microglial CM exposure resulted in decreased neurogenesis in vitro, estimated by the number of Dcx+ cells (Monje et al., 2003). Activated microglia release cytokines and reactive species, including TNF-α, IL-1α, IL-6, ROS, and nitric oxide, that promote mitochondrial dysfunction (Giuffrè et al., 1996; Zell et al., 1997; Halliwell, 2006; Behrens et al., 2008). Here, we found that treatment of differentiating neurons with lactate, a major mitochondrial substrate (Schurr, 2006; Herrero-Mendez et al., 2009), during exposure to CM provided significant protection of Dcx+ cells, whereas cotreatment with pyruvate, a direct substrate of the mitochondrial PDH complex, was less effective. These results are consistent with several previous studies that demonstrated that lactate was a more efficient stimulus of mitochondrial metabolism compared with pyruvate (Yang et al., 1999; Ainscow et al., 2002). Conversion of lactate to pyruvate also generates NADH, whereas excessive pyruvate concentrations stimulate lactate dehydrogenase to operate in reverse mode and consume NADH, thus promoting a more oxidized cytosolic environment (Zhao and Rutter, 1998; Ainscow et al., 2002). This suggests that maintenance of a sufficiently reduced cytosol is important in the protection of Dcx+ cells by lactate. Also, cytosolic NADH generated from lactate conversion can promote increased mitochondrial NADH levels through the activity of mitochondrial redox shuttles (i.e., the glycerol phosphate shunt or malate–aspartate shunt), resulting in higher mitochondrial electron transport activity.
Thiamine is a coenzyme of mitochondrial PDH and α-keto glutarate dehydrogenase (KGDH). Thiamine treatment has been shown to enhance PDH and KGDH activities (Rokutan et al., 1987; Strumilo and Markiewicz, 1995) and protect mitochondrial function both in vitro and in vivo (Sheline and Choi, 2004; Depeint et al., 2006). Cotreatment with thiamine during exposure to inflammatory CM resulted in a 1.8-fold increase in the number of Dcx+ cells compared with CM-only-treated cultures. α-Lipoic acid is also a coenzyme of mitochondrial PDH and KGDH complexes. Supplementation with α-lipoic acid has been shown to protect against oxidative and ischemic injury in vitro (Packer et al., 1997) and attenuate pathology-associated mitochondrial and cognitive dysfunction of the brain (Liu, 2008). In our experiments, cotreatment with α-lipoic acid during the CM exposure resulted in 1.7-fold increase in the number of Dcx+ cells compared with CM-only-treated cultures. Lipoic acid is also a potent antioxidant. However, we failed to observe protection of CM-treated Dcx+ cells by glutathione. This is in agreement with a recent study demonstrating that overexpression of the antioxidant enzyme glutathione peroxidase did not attenuate the decrease in neurogenesis associated with traumatic brain injury (Potts et al., 2009).
Since mitochondrial damage promotes the permeability transition and can trigger both apoptotic and necrotic cell death (Kroemer and Reed, 2000), we also investigated the protective potential of agents that inhibit mitochondrial permeability transition. One commonly used inhibitor is CsA. In our experiments, CsA treatment during CM exposure exacerbated the loss of Dcx+ cells induced by inflammatory CM (data not shown). CsA toxicity has been observed in several studies and might be attributable to CsA inhibition of calcineurin (Nazareth et al., 1991; Qian et al., 1997). NIM811, a CsA analog that does not inhibit calcineurin but inhibits permeability transition without cytotoxic effects (Waldmeier et al., 2002; McEwen et al., 2007), was effective in improving survival of Dcx+ cells compared with CM-treated cultures. This suggests that Dcx+ cell death induced by inflammatory CM is mediated by opening of the permeability transition pore and is consistent with our observation that caspases are activated in the Dcx+ cells.
mtHsp75 is the mitochondrial localized member of the HSP70 family and is an essential mitochondrial chaperone. Several studies have demonstrated the protective potential of mtHsp75 overexpression against mitochondrial dysfunction and cell death (Liu et al., 2005; Voloboueva et al., 2008; Williamson et al., 2008; Xu et al., 2009). In the present study, mtHsp75 overexpression resulted in improved survival of Dcx+ cells in untreated cultures, as well as additional improved survival of inflammatory CM exposed Dcx+ cells. This suggests that the survival of Dcx+ cells under these in vitro differentiation conditions is already limited to some extent by mitochondrial impairment.
Mitochondrial inactivation has been suggested to be a critical mechanism of cellular damage in several neurodegenerative disorders (Fukui and Moraes, 2008; Niizuma et al., 2009). Thiamine supplementation ameliorated mitochondrial dysfunction and reduced neuronal death induced by copper supplementation or transient middle cerebral artery occlusion (Sheline and Choi, 2004; Sheline and Wei, 2006). Thiamine has been demonstrated to show benefits in treating several mitochondrial disorders including mitochondrial encephalomyopathy, West syndrome, lactic acidemia, and Leigh syndrome (Tanaka et al., 1997; Naito et al., 1998, 1999) In our experiments, oral thiamine supplementation resulted in significantly improved survival of Dcx+ cells in experimental animals 8 d after irradiation treatment. Cranial radiation is known to be associated with cognitive decline in both children and adults (Crossen et al., 1994; Roman and Sperduto, 1995; Duffner, 2004); thus, the possibility of using thiamine to ameliorate this devastating process deserves consideration, especially in light of the long history of the use of thiamine clinically, and its relative lack of side effects.
The results of recent in vivo studies suggest that mitochondrial function might play an important role in the process of neurogenesis both during development and in adulthood. Mice deficient in mitochondrial KGDH complex had fewer Dcx+ neuronal progenitor cells (Calingasan et al., 2008), and thiamine deficiency impaired hippocampal neurogenesis and caused cognitive dysfunction in adult mice (Zhao et al., 2008). Thiamine deficiency has long been appreciated to cause Wernicke–Korsakoff syndrome, a form of encephalopathy and psychosis, which is characterized by reduced activity of the KGDH complex in the brain (Gibson et al., 1984). It is plausible that the memory impairment seen in Korsakoff psychosis is related in part to reduced hippocampal neurogenesis.
Neurogenesis is decreased in several inflammation-associated conditions including aging, age-associated pathologies, and irradiation (Mizumatsu et al., 2003; Rola et al., 2004; Rao et al., 2006; Verret et al., 2007). Although inflammation has been shown to downregulate neurogenesis in vitro and in vivo (Ekdahl et al., 2003; Monje et al., 2003), the mechanisms of this downregulation remain poorly understood. Understanding the mechanisms that suppress neurogenesis during inflammation may help identify therapeutic targets for treatment relevant to multiple neurodegenerative disorders. Our findings shed light on mitochondrial mechanisms involved in neurogenesis, identify the immature Dcx+ neuron as exquisitely vulnerable to mitochondrial impairment, and reinforce the concept of targeting mitochondria to prevent death of newborn neurons and enhance neurogenesis under conditions of inflammation.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grant GM49831 (R.G.G.), NIH Grant R01 MH071472 and California Institute of Regenerative Medicine Grant RC1-00134 (T.D.P.), and National Science Foundation Graduate Research Fellowship 2004016504 (S.W.L.).
References
- Ainscow EK, Mirshamsi S, Tang T, Ashford ML, Rutter GA. Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: evidence for ATP-independent control of ATP-sensitive K+ channels. J Physiol. 2002;544:429–445. doi: 10.1113/jphysiol.2002.022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Almeida J, Bolaños JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Norquist BA, Vesce S, Nicholls DG, Soine WH, Duan S, Swanson RA. Barbiturates induce mitochondrial depolarization and potentiate excitotoxic neuronal death. J Neurosci. 2002;22:9203–9209. doi: 10.1523/JNEUROSCI.22-21-09203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 2008;28:13957–13966. doi: 10.1523/JNEUROSCI.4457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci U S A. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolaños JP, Almeida A. Modulation of astroglial energy metabolism by nitric oxide. Antioxid Redox Signal. 2006;8:955–965. doi: 10.1089/ars.2006.8.955. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Ho DJ, Wille EJ, Campagna MV, Ruan J, Dumont M, Yang L, Shi Q, Gibson GE, Beal MF. Influence of mitochondrial enzyme deficiency on adult neurogenesis in mouse models of neurodegenerative diseases. Neuroscience. 2008;153:986–996. doi: 10.1016/j.neuroscience.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol. 2001;435:406–417. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Covacu R, Danilov AI, Rasmussen BS, Hallén K, Moe MC, Lobell A, Johansson CB, Svensson MA, Olsson T, Brundin L. Nitric oxide exposure diverts neural stem cell fate from neurogenesis towards astrogliogenesis. Stem Cells. 2006;24:2792–2800. doi: 10.1634/stemcells.2005-0640. [DOI] [PubMed] [Google Scholar]
- Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627–642. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- Depeint F, Bruce WR, Shangari N, Mehta R, O'Brien PJ. Mitochondrial function and toxicity: role of the B vitamin family on mitochondrial energy metabolism. Chem Biol Interact. 2006;163:94–112. doi: 10.1016/j.cbi.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Duffner PK. Long-term effects of radiation therapy on cognitive and endocrine function in children with leukemia and brain tumors. Neurologist. 2004;10:293–310. doi: 10.1097/01.nrl.0000144287.35993.96. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fike JR, Rola R, Limoli CL. Radiation response of neural precursor cells. Neurosurg Clin N Am. 2007;18:115–127. x. doi: 10.1016/j.nec.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Fike JR, Rosi S, Limoli CL. Neural precursor cells and central nervous system radiation sensitivity. Semin Radiat Oncol. 2009;19:122–132. doi: 10.1016/j.semradonc.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Moraes CT. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci. 2008;31:251–256. doi: 10.1016/j.tins.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Galvan V, Jin K. Neurogenesis in the aging brain. Clin Interv Aging. 2007;2:605–610. doi: 10.2147/cia.s1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, Blass JP. Thiamine-dependent processes and treatment strategies in neurodegeneration. Antioxid Redox Signal. 2007;9:1605–1619. doi: 10.1089/ars.2007.1766. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Ksiezak-Reding H, Sheu KF, Mykytyn V, Blass JP. Correlation of enzymatic, metabolic, and behavioral deficits in thiamin deficiency and its reversal. Neurochem Res. 1984;9:803–814. doi: 10.1007/BF00965667. [DOI] [PubMed] [Google Scholar]
- Giuffrè A, Sarti P, D'Itri E, Buse G, Soulimane T, Brunori M. On the mechanism of inhibition of cytochrome c oxidase by nitric oxide. J Biol Chem. 1996;271:33404–33408. doi: 10.1074/jbc.271.52.33404. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- Haupt E, Ledermann H, Köpcke W. Benfotiamine in the treatment of diabetic polyneuropathy—a three-week randomized, controlled pilot study (BEDIP study) Int J Clin Pharmacol Ther. 2005;43:71–77. doi: 10.5414/cpp43071. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem. 2007;100:1375–1386. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Liu J. The effects and mechanisms of mitochondrial nutrient alpha-lipoic acid on improving age-associated mitochondrial and cognitive dysfunction: an overview. Neurochem Res. 2008;33:194–203. doi: 10.1007/s11064-007-9403-0. [DOI] [PubMed] [Google Scholar]
- Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu W, Song XD, Zuo J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol Cell Biochem. 2005;268:45–51. doi: 10.1007/s11010-005-2996-1. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Springer JE. Pretreatment with the cyclosporin derivative, NIM811, improves the function of synaptic mitochondria following spinal cord contusion in rats. J Neurotrauma. 2007;24:613–624. doi: 10.1089/neu.2006.9969. [DOI] [PubMed] [Google Scholar]
- Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res. 2003;63:4021–4027. [PubMed] [Google Scholar]
- Monje ML, Palmer T. Radiation injury and neurogenesis. Curr Opin Neurol. 2003;16:129–134. doi: 10.1097/01.wco.0000063772.81810.b7. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Kuroda Y. Thiamine-responsive lactic acidaemia: role of pyruvate dehydrogenase complex. Eur J Pediatr. 1998;157:648–652. doi: 10.1007/s004310050903. [DOI] [PubMed] [Google Scholar]
- Naito E, Ito M, Yokota I, Saijo T, Chen S, Maehara M, Kuroda Y. Concomitant administration of sodium dichloroacetate and thiamine in West syndrome caused by thiamine-responsive pyruvate dehydrogenase complex deficiency. J Neurol Sci. 1999;171:56–59. doi: 10.1016/s0022-510x(99)00250-6. [DOI] [PubMed] [Google Scholar]
- Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J Mol Cell Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Soma LA, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem. 2009;109(Suppl 1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci U S A. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir MA, Akcakus M, Kurtoglu S, Gunes T, Torun YA. TRMA syndrome (thiamine-responsive megaloblastic anemia): a case report and review of the literature. Pediatr Diabetes. 2002;3:205–209. doi: 10.1034/j.1399-5448.2002.30407.x. [DOI] [PubMed] [Google Scholar]
- Packer L, Tritschler HJ, Wessel K. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic Biol Med. 1997;22:359–378. doi: 10.1016/s0891-5849(96)00269-9. [DOI] [PubMed] [Google Scholar]
- Peng H, Whitney N, Wu Y, Tian C, Dou H, Zhou Y, Zheng J. HIV-1-infected and/or immune-activated macrophage-secreted TNF-alpha affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia. 2008;56:903–916. doi: 10.1002/glia.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock JM, Liddle AC. Microglial signalling cascades in neurodegenerative disease. Prog Brain Res. 2001;132:555–565. doi: 10.1016/S0079-6123(01)32103-9. [DOI] [PubMed] [Google Scholar]
- Potts MB, Rola R, Claus CP, Ferriero DM, Fike JR, Noble-Haeusslein LJ. Glutathione peroxidase overexpression does not rescue impaired neurogenesis in the injured immature brain. J Neurosci Res. 2009;87:1848–1857. doi: 10.1002/jnr.21996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol. 1997;273:C1783–C1792. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Shetty AK. The window and mechanisms of major age-related decline in the production of new neurons within the dentate gyrus of the hippocampus. Aging Cell. 2006;5:545–558. doi: 10.1111/j.1474-9726.2006.00243.x. [DOI] [PubMed] [Google Scholar]
- Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokutan K, Kawai K, Asada K. Inactivation of 2-oxoglutarate dehydrogenase in rat liver mitochondria by its substrate and t-butyl hydroperoxide. J Biochem. 1987;101:415–422. doi: 10.1093/oxfordjournals.jbchem.a121926. [DOI] [PubMed] [Google Scholar]
- Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, Morhardt DR, Fike JR. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol. 2004;188:316–330. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Roman DD, Sperduto PW. Neuropsychological effects of cranial radiation: current knowledge and future directions. Int J Radiat Oncol Biol Phys. 1995;31:983–998. doi: 10.1016/0360-3016(94)00550-8. [DOI] [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Hüttemann M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008;283:21134–21144. doi: 10.1074/jbc.M801954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A. Lactate: the ultimate cerebral oxidative energy substrate? J Cereb Blood Flow Metab. 2006;26:142–152. doi: 10.1038/sj.jcbfm.9600174. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Choi DW. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004;55:645–653. doi: 10.1002/ana.20047. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Wei L. Free radical-mediated neurotoxicity may be caused by inhibition of mitochondrial dehydrogenases in vitro and in vivo. Neuroscience. 2006;140:235–246. doi: 10.1016/j.neuroscience.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Choi EH, Kim-Han JS, Dugan LL, Choi DW. Cofactors of mitochondrial enzymes attenuate copper-induced death in vitro and in vivo. Ann Neurol. 2002;52:195–204. doi: 10.1002/ana.10276. [DOI] [PubMed] [Google Scholar]
- Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008;15:323–330. doi: 10.1159/000156474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler J, Bentz BG, Harbrecht BG, Di Silvio M, Curran RD, Billiar TR, Hoffman RA, Simmons RL. Tumor necrosis factor alpha inhibits hepatocyte mitochondrial respiration. Ann Surg. 1992;216:539–546. doi: 10.1097/00000658-199211000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumilo S, Markiewicz J. Thiamine pyrophosphate as an effector of 2-oxoglutarate dehydrogenase complex from European bison heart. Biochem Mol Biol Int. 1995;37:101–106. [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prévost MC, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasawa K, Kitagawa K, Yagita Y, Sasaki T, Tanaka S, Matsushita K, Ohstuki T, Miyata T, Okano H, Hori M, Matsumoto M. Increased proliferation of neural progenitor cells but reduced survival of newborn cells in the contralateral hippocampus after focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2002;22:299–307. doi: 10.1097/00004647-200203000-00007. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Nagai T, Arai H, Inui K, Yamanouchi H, Goto Y, Nonaka I, Okada S. Treatment of mitochondrial encephalomyopathy with a combination of cytochrome c and vitamins B1 and B2. Brain Dev. 1997;19:262–267. doi: 10.1016/s0387-7604(97)00573-1. [DOI] [PubMed] [Google Scholar]
- Tonchev AB, Yamashima T, Sawamoto K, Okano H. Enhanced proliferation of progenitor cells in the subventricular zone and limited neuronal production in the striatum and neocortex of adult macaque monkeys after global cerebral ischemia. J Neurosci Res. 2005;81:776–788. doi: 10.1002/jnr.20604. [DOI] [PubMed] [Google Scholar]
- Türeyen K, Vemuganti R, Sailor KA, Bowen KK, Dempsey RJ. Transient focal cerebral ischemia-induced neurogenesis in the dentate gyrus of the adult mouse. J Neurosurg. 2004;101:799–805. doi: 10.3171/jns.2004.101.5.0799. [DOI] [PubMed] [Google Scholar]
- Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C. Alzheimer's-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J Neurosci. 2007;27:6771–6780. doi: 10.1523/JNEUROSCI.5564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Duan M, Ouyang Y, Emery JF, Stoy C, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J Cereb Blood Flow Metab. 2008;28:1009–1016. doi: 10.1038/sj.jcbfm.9600600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Kleinholz M, Myers RE. Delayed decreases in specific brain mitochondrial electron transfer complex activities and cytochrome concentrations following anoxia/ischemia. J Neurol Sci. 1990;100:142–151. doi: 10.1016/0022-510x(90)90025-i. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- Williamson CL, Dabkowski ER, Dillmann WH, Hollander JM. Mitochondria protection from hypoxia/reoxygenation injury with mitochondria heat shock protein 70 overexpression. Am J Physiol Heart Circ Physiol. 2008;294:H249–H256. doi: 10.1152/ajpheart.00775.2007. [DOI] [PubMed] [Google Scholar]
- Xie Z, Smith CJ, Van Eldik LJ. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004;45:170–179. doi: 10.1002/glia.10314. [DOI] [PubMed] [Google Scholar]
- Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Kow LM, Funabashi T, Mobbs CV. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes. 1999;48:1763–1772. doi: 10.2337/diabetes.48.9.1763. [DOI] [PubMed] [Google Scholar]
- Zell R, Geck P, Werdan K, Boekstegers P. TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem. 1997;177:61–67. doi: 10.1023/a:1006896832582. [DOI] [PubMed] [Google Scholar]
- Zhao C, Rutter GA. Overexpression of lactate dehydrogenase A attenuates glucose-induced insulin secretion in stable MIN-6 beta-cell lines. FEBS Lett. 1998;430:213–216. doi: 10.1016/s0014-5793(98)00600-0. [DOI] [PubMed] [Google Scholar]
- Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou G, Xu T, Hong Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiol Dis. 2008;29:176–185. doi: 10.1016/j.nbd.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]