

Abstract
Cooperative interactions mediate information transfer between structural domains of a protein molecule and are major determinants of protein function and modulation. The prevalent theories to understand the thermodynamic origins of cooperativity have been developed to reproduce the complex behavior of a global thermodynamic observable such as ligand binding or enzyme activity. However, in most cases the measurement of a single global observable cannot uniquely define all the terms that fully describe the energetics of the system. Here we establish a theoretical groundwork for analyzing protein thermodynamics using site-specific information. Our treatment involves extracting a site-specific parameter (defined as χ value) associated with a structural unit. We demonstrate that, under limiting conditions, the χ value is related to the direct interaction terms associated with the structural unit under observation and its intrinsic activation energy. We also introduce a site-specific interaction energy term (χdiff) that is a function of the direct interaction energy of that site with every other site in the system. When combined with site-directed mutagenesis and other molecular level perturbations, analyses of χ values of site-specific observables may provide valuable insights into protein thermodynamics and structure.
Keywords: cooperativity, ion channels, protein thermodynamics
Biological macromolecules often accomplish diverse functions by undergoing a series of conformational changes that are mediated by cooperative interactions between structural domains (1, 2). A ligand binding domain, for instance, may transfer its stabilization energy upon ligand binding to an effector domain or another ligand binding domain (3, 4). This long-range propagation of energy within a protein molecule has been widely studied in multisubunit allosteric proteins such as hemoglobin (5–7) and the nicotinic acetylcholine receptor (8). Classical theories such as the Monod–Wyman–Changeux (MWC) model (9) and the Koshland–Nemethy–Filmer (KNF) model (10) have been developed to provide a simple mechanistic understanding of biological cooperativity.
The most important difference between the various cooperative theories is their treatment of interaction terms and the microscopic equilibrium constants associated with the individual domains or subunits (11, 12). According to the MWC model, cooperativity arises out of a preexisting equilibrium between two conformational states, which differ in their ligand binding affinities. The binding of a ligand shifts the equilibrium toward the high affinity state(s). The KNF model, on the other hand, is based on step-wise changes in protein conformation on ligand binding. Interactions between neighboring ligand bound subunits induce a conformational change that alters the binding affinity of other unoccupied sites. The main descriptor of protein conformation in these models is a single observable representative of a global conformational change in the protein (global observable) such as enzyme activity or binding site occupancy that often is a state-dependent function of the whole protein. However, this global parameter cannot uniquely define the microscopic thermodynamic terms associated with local conformational transitions (13). In order to make the analysis more tractable, these models impose geometry and/or symmetry constraints thereby reducing the number of independent terms (14). Thus, the interpretation of the underlying thermodynamics is model-specific and, perhaps, restrictive.
In this study, we propose a methodology to identify the energetic effects of mutations in macromolecules, by using site-specific information. Site-specific measurements have increasingly become the method of choice because of their ability to provide information about the local conformation and constrain the values of microscopic terms associated with a structural unit. This approach relies on spectroscopic [fluorescence spectroscopy (15), EPR (16), and NMR (17)], biochemical [cysteine and blocker accessibility (18–20)], and electrophysiological (21, 22) measurements to monitor the time- and stimulus-dependent changes in the local structure. To interpret the effect of protein engineering on the energetics of a macromolecular process, we have formulated an approach based on a set of site-specific parameters referred to as χ values. The macromolecule was modeled as an ensemble of an arbitrary number of interacting structural units where each unit is capable of undergoing a single conformational transition. We have considered that the conformational transitions of the individual units are voltage-dependent. We demonstrate that the χ value analysis can assess the effect of mutations on local thermodynamic parameters of an arbitrarily complex system and, more importantly, allows us to probe the energetic linkage between any two sites (structural domains).
Theory
I. Theory for a Coupled Three-Particle Ensemble.
Let us consider a simple system consisting of three nonidentical particles (Fig. 1). Each of the three particles can exist in two conformations, activated and resting (referred to as the microstate of a particle). The transition between the resting and activated microstates is influenced by an external variable, which here is taken to be voltage. At low voltage, each particle prefers to remain in the resting conformation, whereas at a high voltage, the particles prefer to remain in their activated microstates. The conformational change of each particle is associated with an intrinsic activation constant, 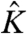 . Because the activation process of each of the particles is driven by voltage, the intrinsic activation constants should be voltage-dependent, and their voltage dependence can be expressed as (23).
. Because the activation process of each of the particles is driven by voltage, the intrinsic activation constants should be voltage-dependent, and their voltage dependence can be expressed as (23).
| [1] |
where 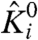 is voltage independent and represents the contribution of chemical interactions to the intrinsic activation constant (referred to as the intrinsic chemical activation constant) of the ith particle, qiindicates the voltage dependence of its activation, V is the voltage, F is the Faraday constant, and β = 1/RT. For positive qi,
is voltage independent and represents the contribution of chemical interactions to the intrinsic activation constant (referred to as the intrinsic chemical activation constant) of the ith particle, qiindicates the voltage dependence of its activation, V is the voltage, F is the Faraday constant, and β = 1/RT. For positive qi, 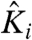 increases with voltage.
increases with voltage.
Fig. 1.

Coupled model for a three-particle system. A system of three particles, 1, 2, and 3, each of which can exist in two conformations: i0 (resting state of i) and i1 (activated state of i). Vertical double-arrowed lines indicate the intrinsic activation constants of the particles. All three particles are thermodynamically linked to each other via four state-dependent pairwise coupling factors, as described in the text. These are depicted by the horizontal and diagonal lines connecting the two particle microstates.
The coupling between any two particles of the system is assumed to be mediated by pairwise state-dependent interactions or “coupling factors,” which are specified as:
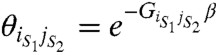 |
[2] |
where GiS1jS2 indicates the free-energy of interaction between the ith particle in microstate s1 and jth particle in microstate s2 (s1 and s2 can be 0 or 1 corresponding to the resting or activated microstate respectively). Thus for each pair of particles we have four interaction terms, which may be nonidentical, describing the coupling between them. We assume that these coupling interaction energies are independent of the external stimulus (i.e., voltage in our case).
This three-particle system can exist in eight possible macrostates. Each macrostate of the system is a unique combination of individual particles in different microstates. The energy of each macrostate is the sum of the intrinsic energies of all the particles in their given microstates and the net coupling energy between them. The canonical partition function for this system, 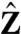 , is:
, is:
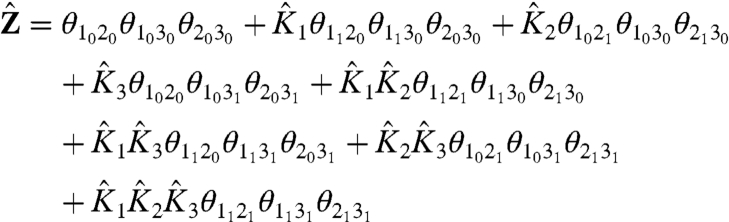 |
[3] |
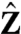 can be normalized by dividing Eq. 3 with θ1020θ1030θ2030 and substituting the following normalized parameters:
can be normalized by dividing Eq. 3 with θ1020θ1030θ2030 and substituting the following normalized parameters:
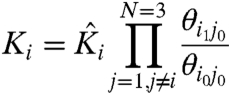 |
[4.1] |
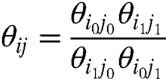 |
[4.2] |
Ki is the apparent activation constant of particle i (hereafter referred to simply as i), θij is the macroscopic coupling factor between i and j. The normalized canonical partition function can now be expressed as:
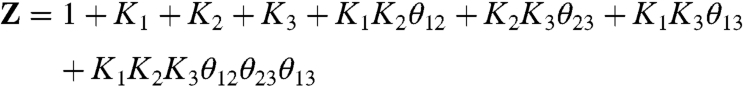 |
[5] |
Such normalized parameters simplify the partition function describing a coupled system and have been widely used in Wyman’s linkage theory and other theoretical descriptions of coupled systems (24). In addition, the thermodynamic terms have exact correspondence to the number of free parameters that can be extracted experimentally. However, Ki is  normalized by the interaction energies of i with the resting state of other particles and is not the true intrinsic equilibrium constant of the particle. Perturbations that alter the ground state interaction energies would affect all the normalized thermodynamic parameters. This may be incorrectly interpreted as the mutation having varied effects. Thus, it is important to consider the explicit interaction terms that contribute to the apparent equilibrium constants when interpreting the effects of mutation. Both Ki and
normalized by the interaction energies of i with the resting state of other particles and is not the true intrinsic equilibrium constant of the particle. Perturbations that alter the ground state interaction energies would affect all the normalized thermodynamic parameters. This may be incorrectly interpreted as the mutation having varied effects. Thus, it is important to consider the explicit interaction terms that contribute to the apparent equilibrium constants when interpreting the effects of mutation. Both Ki and 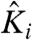 have the same voltage dependence because the coupling factors are voltage-independent:
have the same voltage dependence because the coupling factors are voltage-independent:
| [6] |
where 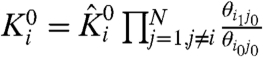 .
.
The θij terms in Eq. 5 are measures of the energy difference between “like-state” interactions between i and j (θi0j0 and θi1j1) and the “cross-interactions” between them (θi1j0 and θi0j1). It is a measure of coupling energy and reflects the stability of the two particles when they are in the “like” microstates, relative to their “unlike” microstates.
From the linkage theory (25) we see that the probability of i to be in microstate s (Pis) will depend on the Boltzmann weights of all the macrostates where i is in the microstate s. Thus, P11 will be determined by all the macrostates where particle 1 is in the activated conformation:
 |
[7] |
where z(s1, s2, s3) is the Boltzmann weight of the macrostate (s1, s2, s3). However, at very low voltages P11 would be dominated by the Boltzmann weight of the macrostate (1,0,0) and it would become z(1,0,0)/Z. A simple physical interpretation of this behavior is that at very low voltages the probability of finding two (or more) particles simultaneously in activated microstate is much smaller than the probability of a single activated particle (SI Text I). In contrast, at high voltages P11 would be simplified to z(1,1,1)/Z. When similar approximations are applied to the probability of particle 1 being in the resting conformation, we find that at very low voltages, P10 is z(0,0,0)/Z, whereas at very high potentials, P10 is z(0,1,1)/Z.
We define a new parameter, εi (referred to as the “coupled equilibrium constant”) as:
 |
[8] |
which is the ratio of the probability of i in the activated microstate to its probability in the resting microstate. At intermediate potentials, the coupled equilibrium constant depends on all the parameters of the system (SI Text I). At extreme voltages where only few macrostates are thermodynamically relevant, the expression for εi(i = 1,2,3) greatly simplifies. For example, the coupled equilibrium constant for particle 1 at very low and high voltages would be given by:
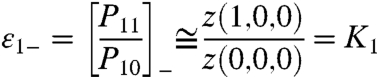 |
[9.1] |
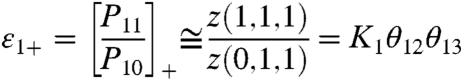 |
[9.2] |
These equations show that at extreme voltages ε1+ and ε1- are essentially single exponential functions of voltage and thus a plot of ln ε vs. V would be linear at extreme voltages.
The above treatment for a coupled three-particle system can be applied to multidomain proteins. Each particle represents a structural or functional domain, whose voltage-dependent conformational dynamics can be monitored using site-specific measurements (SI Text II). We can calculate the probability of activation of the domain and compute the extremal ε values for each of the three structural units. Combining Eqs. 6 and 9.1 and 9.2 the extremal ε values for particle 1 in a three-particle system can be expressed as:
| [10] |
for low voltages. For very high voltages, the equation is:
| [11] |
From the above equations, we see that the linear plots of ln ε1+ and ln ε1- with respect to V have the same slope, q1Fβ, which reflects the intrinsic voltage-dependence of activation of particle 1. The two plots, however, differ in their constant terms 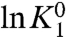 and
and 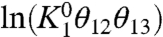 , which we hereafter refer to as χ1- and χ1+,respectively (Fig. 2A).
, which we hereafter refer to as χ1- and χ1+,respectively (Fig. 2A).
Fig. 2.

Simulations of the ln ε vs. V plots. (A) The plot of ln ε vs. V for a three-particle system in the presence (▪) or absence (+) of interactions between the three particles. The extrapolation of the linear segments at low and high voltages to the V = 0 axis (vertical dashed line) gives the respective χ values. Difference between these intercepts (χ values) is the χdiff parameter. (B) The ln ε vs. V for a three-particle system where χdiff for a specific particle is zero (▪). Although the χdiff is zero, perturbation of the intrinsic equilibrium constant of another particle alters ln ε vs. V plot (+). (C–F) ln ε1 vs. V plots for the DI voltage sensor (particle 1) of the model of the voltage-dependent sodium channel (Fig. S1) in response to perturbation of different parameters of the model. The intrinsic chemical activation constant of particles 1, 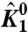 (C), and 5,
(C), and 5, 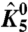 (E), were varied over 6 orders of magnitude:
(E), were varied over 6 orders of magnitude: 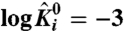 (∘), 0 (+), 3(▪) (i = 1 or 5). Interaction between resting states of particles 1 and 5, θ1050 (D), and that between resting states of particles 2 and 5, θ2050 (F), were varied over 6 orders of magnitude: log θi050 = -3 (∘), 0 (+), 3(▪) (i = 1 or 2). Vertical dashed line corresponds to the voltage axis at 0 mV. Extrapolating the linear segments of the traces (at high and low voltages) to this axis gives the corresponding χ values.
(∘), 0 (+), 3(▪) (i = 1 or 5). Interaction between resting states of particles 1 and 5, θ1050 (D), and that between resting states of particles 2 and 5, θ2050 (F), were varied over 6 orders of magnitude: log θi050 = -3 (∘), 0 (+), 3(▪) (i = 1 or 2). Vertical dashed line corresponds to the voltage axis at 0 mV. Extrapolating the linear segments of the traces (at high and low voltages) to this axis gives the corresponding χ values.
Next, we extended our analysis to a system of 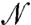 nonidentical particles (SI Text III). The general expressions for the χ values of i in an
nonidentical particles (SI Text III). The general expressions for the χ values of i in an 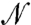 -particle system are:
-particle system are:
| [12] |
 |
[13] |
We introduce another parameter, 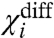 , which is the difference of the two χ values:
, which is the difference of the two χ values:
 |
[14] |
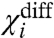 is a measure of the energy difference between all the like-state direct interactions associated with i and its cross-interactions. Because
is a measure of the energy difference between all the like-state direct interactions associated with i and its cross-interactions. Because 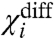 depends only on the coupling factors directly associated with i, it can be used to deduce the thermodynamic effect of a site-specific mutation in an allosteric system. If a mutation perturbs any of the coupling factors directly associated with i, then
depends only on the coupling factors directly associated with i, it can be used to deduce the thermodynamic effect of a site-specific mutation in an allosteric system. If a mutation perturbs any of the coupling factors directly associated with i, then 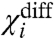 changes, whereas if the mutation alters any of the intrinsic chemical activation constants or coupling factors between other particles then
changes, whereas if the mutation alters any of the intrinsic chemical activation constants or coupling factors between other particles then 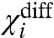 remains unchanged. Thus by direct measurement of a single experimental parameter one could potentially identify whether a perturbation specifically alters the coupling or the intrinsic stability of the structural unit. In addition, the free energy of perturbation on coupling, ΔGp is:
remains unchanged. Thus by direct measurement of a single experimental parameter one could potentially identify whether a perturbation specifically alters the coupling or the intrinsic stability of the structural unit. In addition, the free energy of perturbation on coupling, ΔGp is:
| [15] |
It is important to note that 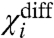 is the net macroscopic interaction energy. If
is the net macroscopic interaction energy. If  is greater than zero, then i is positively coupled to system as a whole, whereas if it is negative then i is negatively coupled to the system. Its absence, however, does not necessarily establish that i is independent of other particles in the system. It signifies that the positive interaction of i with some particles in the system equals its negative interaction with others. But i may still be coupled to the rest of the system. This can be easily illustrated by considering the three-particle system where θ12 equals to 1/θ13 and therefore the
is greater than zero, then i is positively coupled to system as a whole, whereas if it is negative then i is negatively coupled to the system. Its absence, however, does not necessarily establish that i is independent of other particles in the system. It signifies that the positive interaction of i with some particles in the system equals its negative interaction with others. But i may still be coupled to the rest of the system. This can be easily illustrated by considering the three-particle system where θ12 equals to 1/θ13 and therefore the  is zero. Fig. 2B shows that the ln ε vs. voltage plot is not linear (except at the extremities) indicating that particle 1 is coupled to the other particles in the system. Perturbation of the intrinsic stability of particle 2 alters the behavior of particle 1 at intermediate potentials, as expected in a coupled system.
is zero. Fig. 2B shows that the ln ε vs. voltage plot is not linear (except at the extremities) indicating that particle 1 is coupled to the other particles in the system. Perturbation of the intrinsic stability of particle 2 alters the behavior of particle 1 at intermediate potentials, as expected in a coupled system.
Apart from identifying whether a mutation affects the coupling associated with a specific structural unit, this approach could also provide useful information about state-dependence of the interactions. Remembering that the parameters in Eqs. 12 and 13 are normalized (Eqs. 4.1 and 4.2), we expand them to arrive at the following expressions:
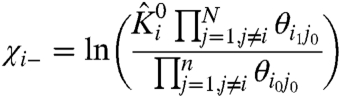 |
[16] |
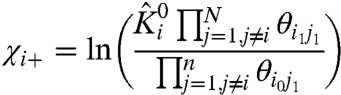 |
[17] |
These generalized expressions help us observe some important features of the χ values. First, both the χ values of a particle depend on its intrinsic chemical activation constant. Second, the χ values of a particle depend only on the interactions directly associated with it—the coupling factors between two particles have no effect on the χ values of a third particle. Third, the χ values do not depend on the same coupling factors—χi- depends on the interactions of i with the resting state of the other particles while χi+ depends on its interactions with the activated state of the other particles. Suppose we monitor the conformational dynamics of the ith structural unit of the protein and extract its χ values (at high and low voltages). A generalized equation for χ value (SI Text IV) in terms of the probability of activation of a specific structural unit (PA) is:
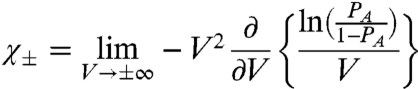 |
[18] |
where V is the voltage. If upon mutation, 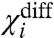 changes then the mutation affects a coupling factor directly associated with i. If the change in
changes then the mutation affects a coupling factor directly associated with i. If the change in 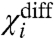 is due to only χi- changing, then we can attribute the effect of the mutation to the alteration of a coupling factor associated with i and the resting state of another particle. Conversely, if only the χi+ changes it can be concluded that the mutation affects only the coupling factor(s) associated with i and the activated state of other particles. If, however, both χi- and χi+ values change and the changes are equal (ΔX1- = ΔX1+ and ΔXdiff1 = 0) then the primary effect of the perturbation is on the intrinsic chemical activation constant of the particle.
is due to only χi- changing, then we can attribute the effect of the mutation to the alteration of a coupling factor associated with i and the resting state of another particle. Conversely, if only the χi+ changes it can be concluded that the mutation affects only the coupling factor(s) associated with i and the activated state of other particles. If, however, both χi- and χi+ values change and the changes are equal (ΔX1- = ΔX1+ and ΔXdiff1 = 0) then the primary effect of the perturbation is on the intrinsic chemical activation constant of the particle.
Although Eq. 18 describes a system-independent mathematical expression relating the χ values to the probability of activation of a particle, it may not always be practical because it involves taking derivatives of noisy data traces. A better approach may be to fit the data points at extremities in the ln ε vs. V plots (Fig. 2) to a straight line and extrapolating it to 0 mV. How do we ascertain that we have reached such extreme conditions? According to Eqs. 10 and 11, the slopes at both extremities should be identical and are related to intrinsic voltage dependence of the structural unit. This criterion can be used as a practical method to identify the linear regimes suitable for extracting the χ values.
II. Systems with Identical Structural Units.
Suppose in the N-particle system, there are multiple (Ni) copies of particle i (labeled as i through i + Ni - 1). To illustrate the χ value approach for this system, we can show that:
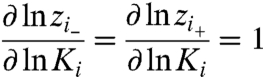 |
[19] |
where zi- is the Boltzmann weight of the macrostate where all particles but a single copy of i are resting and zi+ is the Boltzmann weight of the macrostate where all particles but a single copy of i are activated. The probability of activation of structural unit i in this case is:
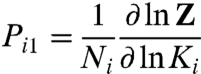 |
[20] |
At very low potentials, Z can be approximated as:
| [21] |
where z(0,0,…,0) is the Boltzmann weight of the macrostate where all particles are resting. In Eq. 21 we have assumed that all macrostates where any one of the Ni copies of i is activated, while all the remaining particles are resting, are identical in energy. Using Eq. 21 in Eq. 20, along with Eq. 19, we get, Pi1 = Zi-/Z. Furthermore, at very low voltages, Pi0 = z(0,0,…,0)/Z and εi- turns out to be zi-/z(0,0,…,0). Thus εi- is essentially Ki. Similarly, at very high voltages, by approximating Z as:
| [22] |
we can show that εi+ would remain the same as before and the resultant χ value can still be expressed by Eq. 13. It is important to note that in the expression for χi+ (for this system), the running variable j samples all the remaining particles of the system—the ones that are different from i as well as its Ni - 1 replicates. Consequently 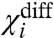 would depend on all the macroscopic coupling factors associated with i—interaction terms with its identical copies as well as all nonidentical particles of the system. Thus the χ value analysis can be applied to completely or partially homomeric systems as well as heteromeric systems.
would depend on all the macroscopic coupling factors associated with i—interaction terms with its identical copies as well as all nonidentical particles of the system. Thus the χ value analysis can be applied to completely or partially homomeric systems as well as heteromeric systems.
III. Numerical Simulations to Demonstrate the χ-Value Approach.
To illustrate our theory, we carried out numerical simulations for a model that approximates a voltage-dependent sodium channel (Fig. S1). The channel consists of four nonidentical voltage-sensing domains (labeled as particles 1 to 4). The four pore segments associate to form the ion permeation pathway, which is represented as a single structural unit (particle 5). Voltage-dependent conformational dynamics of these discrete structural units can be tracked using a site-specific probe. For example, in voltage-gated sodium channels, fluorescence probes have been used to measure the dynamics of each of the four voltage sensors, while ionic current measurements have long been used to probe the conformational status of the pore domain (26). In the model, that we use for our simulations, we assume that each of the voltage sensors is coupled to the pore domain through four state-dependent interaction terms (θi050, θi150, θi051, θi151—where i represents a voltage sensor, i = 1, 2, 3, or 4). The four voltage sensors do not directly interact with each other. Numerical simulations were performed using mostly arbitrary values of the thermodynamic parameters whose initial values are listed in Table S1. The initial values of 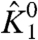 and
and 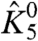 , the intrinsic chemical activation constants of domain I (DI) voltage sensor and the pore, respectively, and the coupling factors between them were chosen to be close to that obtained from our previous modeling data (27). The remaining thermodynamic parameters were randomly distributed around these values. Each of the explicit thermodynamic terms in the system were separately varied over several orders of magnitude, and the ε values of DI voltage sensor (ε1) were calculated over a large range of voltage, from which both its χ values were obtained. The numerical simulations were performed using MATLAB 2008b (The Mathworks). The simulations were independently verified by calculating the χdiff values using Eq. 14. The plots were generated using Origin (Microcal).
, the intrinsic chemical activation constants of domain I (DI) voltage sensor and the pore, respectively, and the coupling factors between them were chosen to be close to that obtained from our previous modeling data (27). The remaining thermodynamic parameters were randomly distributed around these values. Each of the explicit thermodynamic terms in the system were separately varied over several orders of magnitude, and the ε values of DI voltage sensor (ε1) were calculated over a large range of voltage, from which both its χ values were obtained. The numerical simulations were performed using MATLAB 2008b (The Mathworks). The simulations were independently verified by calculating the χdiff values using Eq. 14. The plots were generated using Origin (Microcal).
Representative plots for ln ε1 vs. V in response to perturbation of different thermodynamic parameters of the system are shown in Fig. 2 C–F. Three phases are clearly visible in these plots—at very low voltages the plot is linear, at intermediate voltages it becomes nonlinear, and finally at very high voltages the plot is linear again. χ1- and χ1+ are extracted from these traces by extrapolating the respective linear segments to the y-axis (V = 0). Clearly, as 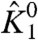 is increased (Fig. 2C) both χ values increase, maintaining a constant
is increased (Fig. 2C) both χ values increase, maintaining a constant  . When θ1050, the interaction between the resting DI voltage sensor and the resting pore is increased (Fig. 2D), and the χ1+ value remains unchanged, as seen by the convergence of the different traces at very high voltages. However, χ1- shows a graded decrease (indicated by a decrease in the saturating values of ln ε1) and as a result,
. When θ1050, the interaction between the resting DI voltage sensor and the resting pore is increased (Fig. 2D), and the χ1+ value remains unchanged, as seen by the convergence of the different traces at very high voltages. However, χ1- shows a graded decrease (indicated by a decrease in the saturating values of ln ε1) and as a result,  also increases. Fig. 2 E and F show the ln ε1 vs. V traces in response to perturbation of
also increases. Fig. 2 E and F show the ln ε1 vs. V traces in response to perturbation of 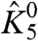 and θ2050 (the interaction between resting DII voltage sensor and the resting pore), respectively. While there are some deviations between the different traces (in both figures) at intermediate voltages, they all converge at saturating voltages, indicating that these perturbations affect neither χ1- nor χ1+ (and thus not even
and θ2050 (the interaction between resting DII voltage sensor and the resting pore), respectively. While there are some deviations between the different traces (in both figures) at intermediate voltages, they all converge at saturating voltages, indicating that these perturbations affect neither χ1- nor χ1+ (and thus not even 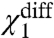 ). These simulations provide a clear picture of the χ-value analysis—χ1+ depends only on
). These simulations provide a clear picture of the χ-value analysis—χ1+ depends only on 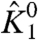 and the direct interactions of particle 1 with the activated state of other particles, and χ1- depends only on
and the direct interactions of particle 1 with the activated state of other particles, and χ1- depends only on 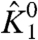 and the direct interactions of particle 1 with the resting state of other particles.
and the direct interactions of particle 1 with the resting state of other particles. 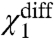 depends only on the direct interactions of particle 1 with the other particles of the system (Fig. S2). Thus experimentally observing the changes in these three parameters in response to a mutation, we can identify whether the mutation affects the intrinsic stability of a structural unit or its interaction with other structural units of the protein, and in the case of the latter we could obtain useful information about the state-dependence of the interaction as well.
depends only on the direct interactions of particle 1 with the other particles of the system (Fig. S2). Thus experimentally observing the changes in these three parameters in response to a mutation, we can identify whether the mutation affects the intrinsic stability of a structural unit or its interaction with other structural units of the protein, and in the case of the latter we could obtain useful information about the state-dependence of the interaction as well.
Discussion
The above analysis shows that a site-specific observable in an allosteric protein can be described, under limiting conditions, by a small set of thermodynamic parameters. These thermodynamic terms are directly related to the structural unit under observation. This approach, which we refer to as the χ value analysis, relies on measuring the probability of activation of individual structural units under limiting conditions where the system dynamics can be approximated to a two-state process. This allows us to determine the effects of site-specific mutations in macromolecular systems without a priori assumptions about the nature of allosteric connectivity. χ values of a structural unit of a protein can be obtained experimentally by probing its conformational changes using site-specific measurements such as EPR or fluorescence spectroscopy. Our theory allows us to incorporate such site-specific observables to gain valuable insight into the thermodynamics of elementary conformational changes in a protein, as opposed to a single global observable (e.g., fraction of ligand bound sites). This approach is reminiscent of the analysis used to study the allosteric thermodynamics of calcium and voltage-activated potassium channels, within the framework of specific cooperative models (28). In that case, driving the system to extreme conditions greatly simplifies the equations, making it possible to assign values to specific parameters upon fitting the experimental data to the model.
The term χdiff introduced in Eq. 14 has an important physical meaning in relation to Wyman’s linkage theory. It reflects how easily a domain can activate when all the other domains are in their activated conformations relative to the case when all the remaining domains are resting. Wyman showed that the difference between the final and initial asymptotes of a Hill plot represents the net global cooperativity parameter. This parameter is a measure of the total interaction energy between different allosterically linked sites of a macromolecule. Estimates of such macroscopic interaction parameters have been frequently obtained using experimental observables that report the global states of a macromolecule. The ln ε vs. V plot is similar to the Hill plot and hence χdiff is related in significance to the net global cooperativity parameter. But, ln ε vs. V plots are obtained using site-specific measurements. Thus χdiff would yield an estimate for net stabilization energy associated with a local site of a macromolecule as opposed to a global parameter that has contributions from several different sites embedded within it. Obtaining energetic parameters such as χdiff would be indispensible in gaining a thorough understanding of thermodynamic principles governing local conformational dynamics—for example, χdiff for a specific voltage sensor of the voltage-gated sodium channels can tell us what fraction of the net free-energy change associated with the voltage-sensor activation is derived from interaction with other structural units in the protein.
Protein thermodynamics and structure are intimately related. Inferences can be drawn about protein structure, if we are able to relate the effect of mutations to perturbations of specific thermodynamic terms. Our theory posits that if a subunit does not form a direct energetic contact with a second subunit, then no amount of perturbation of the second subunit would affect the χ parameters of the first. In our simulations the χ values of the DI voltage sensor remained invariant when the thermodynamic parameters of all the indirectly linked segments were varied over many orders of magnitude (Fig. S2). Therefore, if in an actual experiment the χ values of the DI voltage sensor remain unaltered in response to systematic perturbations throughout the DII voltage sensor, then we can conclude that the DI and DII voltage sensors are unlikely to share a common structural interface through which they interact directly.
In order to deconstruct the thermodynamic perturbation caused by a mutation using χ value analysis it is necessary that we are able to make measurements under conditions where the ln ε vs. V plots become linear. If the voltage dependence of the constituent domains is small, the extreme voltages necessary to evoke a linear response might become experimentally infeasible. Again, if the coupling factors are voltage-dependent, then the approximations that simplify the expressions for coupled equilibrium constants may not be valid. Also, the limiting slopes at high and low voltages may be different if the coupling terms have different voltage dependencies. Extraction of the χ values might become difficult in these situations. Also in cases where two structural units are extremely tightly coupled, it might not be possible to experimentally probe one structural unit separately from the other. The two structural units need to be treated as a single macro-unit, and the χ value analysis could be used to probe the effect of point mutations on such a macro-unit. In our description, the conformational dynamics of every structural unit is treated as a single-step transition. In many instances, the activation of specific structural units may involve multiple transitions (29–31). In such cases, if the site-specific probe monitors the final conformational state, then the χ value analysis becomes valid because the intermediate nonactivated microstates can be lumped together, rendering an effective single-step transition. However, it is important to remember that the energetics extracted using the χ-value analysis is relative to these intermediate states rather than the absolute ground state.
Despite these limitations, the χ value is a powerful analytical tool to dissect the thermodynamic parameters using site-specific measurements. When combined with site-directed mutagenesis, this approach could help us understand the role of protein structure in determining thermodynamics and function. Other methods based on measurements of global observable such as double mutant cycle analysis have been used to extract interaction energies between specific sites in proteins, but their applicability is limited by strict constraints (32). While our analysis was based on the assumption that the transitions are voltage-dependent, similar equations can be developed if the conformational transitions depend on other stimuli such as ligand. Therefore, χ value analysis can be applied to wider class of proteins including proteins activated by ligand and other cofactors.
Supplementary Material
Acknowledgments.
We thank Drs. M. Jones, M. Goldschen, M. Jackson, and Q. Cui for their valuable comments. The Matlab code used for simulations of N-particle system is available upon request. This project was supported by funds from the National Institutes of Health (GM084140) and the Shaw Scientist award to B.C.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1003609107/-/DCSupplemental.
References
- 1.Pauling L. The oxygen equilibrium of hemoglobin and its structural interpretation. Proc Natl Acad Sci USA. 1935;21(4):186–191. doi: 10.1073/pnas.21.4.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monod J, Jacob F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb Sym Quant Biol. 1961;26:389–401. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- 3.Perutz MF. Stereochemistry of cooperative effects in haemoglobin. Nature. 1970;228(5273):726–739. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 4.Perutz MF, Greer J. Stereochemical effects of amino acid substitution in abnormal human haemoglobin. Biochem J. 1970;119(5):31P. doi: 10.1042/bj1190031pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perutz MF, Wilkinson AJ, Paoli M, Dodson GG. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct. 1998;27:1–34. doi: 10.1146/annurev.biophys.27.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Szabo A, Karplus M. A mathematical model for structure-function relations in hemoglobin. J Mol Biol. 1972;72(1):163–197. doi: 10.1016/0022-2836(72)90077-0. [DOI] [PubMed] [Google Scholar]
- 7.Eaton WA, Henry ER, Hofrichter J, Mozzarelli A. Is cooperative oxygen binding by hemoglobin really understood? Nat Struct Biol. 1999;6(4):351–358. doi: 10.1038/7586. [DOI] [PubMed] [Google Scholar]
- 8.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308(5727):1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 9.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 10.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5(1):365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 11.Hammes GG, Wu CW. Relaxation spectra of aspartate transcarbamylase. Interaction of the native enzyme with carbamyl phosphate. Biochemistry. 1971;10(11):2150–2156. doi: 10.1021/bi00787a030. [DOI] [PubMed] [Google Scholar]
- 12.Edelstein SJ. Extensions of the allosteric model for haemoglobin. Nature. 1971;230(5291):224–227. doi: 10.1038/230224a0. [DOI] [PubMed] [Google Scholar]
- 13.Di Cera E. Site-specific thermodynamics: Understanding cooperativity in molecular recognition. Chem Rev. 1998;98(4):1563–1592. doi: 10.1021/cr960135g. [DOI] [PubMed] [Google Scholar]
- 14.Jackson MB. Molecular and Cellular Biophysics. Cambridge: Cambridge Univ Press; 2006. pp. 111–141. [Google Scholar]
- 15.Weber G. Fluorescence in biophysics: Accomplishments and deficiencies. Methods Enzymol. 1997;278:1–15. doi: 10.1016/s0076-6879(97)78003-0. [DOI] [PubMed] [Google Scholar]
- 16.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7(9):735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 17.Tochtrop GP, et al. Energetics by NMR: Site-specific binding in a positively cooperative system. Proc Natl Acad Sci USA. 2002;99(4):1847–1852. doi: 10.1073/pnas.012379199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong CM. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J Gen Physiol. 1969;54(5):553–575. doi: 10.1085/jgp.54.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258(5080):307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- 20.Stauffer DA, Karlin A. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 1994;33(22):6840–6849. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong CM, Bezanilla F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J Gen Physiol. 1974;63(5):533–552. doi: 10.1085/jgp.63.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandler WK, Schneider MF, Rakowski RF, Adrian RH. Charge movements in skeletal muscle. Philos T Roy Soc B. 1975;270(908):501–505. doi: 10.1098/rstb.1975.0026. [DOI] [PubMed] [Google Scholar]
- 23.Stevens CF. Interactions between intrinsic membrane protein and electric field. An approach to studying nerve excitability. Biophys J. 1978;22(2):295–306. doi: 10.1016/S0006-3495(78)85490-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyman J. Allosteric Linkage. J Am Chem Soc. 1967;89(9):2202–2218. [Google Scholar]
- 25.Wyman J. The binding potential, a neglected linkage concept. J Mol Biol. 1965;11:631–644. doi: 10.1016/s0022-2836(65)80017-1. [DOI] [PubMed] [Google Scholar]
- 26.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120(5):629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muroi Y, Arcisio-Miranda M, Chowdhury S, Chanda B. Molecular determinants of coupling between the domain III voltage sensor and pore of a sodium channel. Nat Struct Mol Biol. 2010;17(2):230–237. doi: 10.1038/nsmb.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horrigan FT, Cui J, Aldrich RW. Allosteric voltage gating of potassium channels I. Mslo ionic currents in the absence of Ca(2+) J Gen Physiol. 1999;114(2):277–304. doi: 10.1085/jgp.114.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecar H, Larsson HP, Grabe M. Electrostatic model of S4 motion in voltage-gated ion channels. Biophys J. 2003;85(5):2854–2864. doi: 10.1016/S0006-3495(03)74708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J Gen Physiol. 1994;103(2):321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoppa NE, Sigworth FJ. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J Gen Physiol. 1998;111(2):313–342. doi: 10.1085/jgp.111.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214(3):613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.