

Abstract
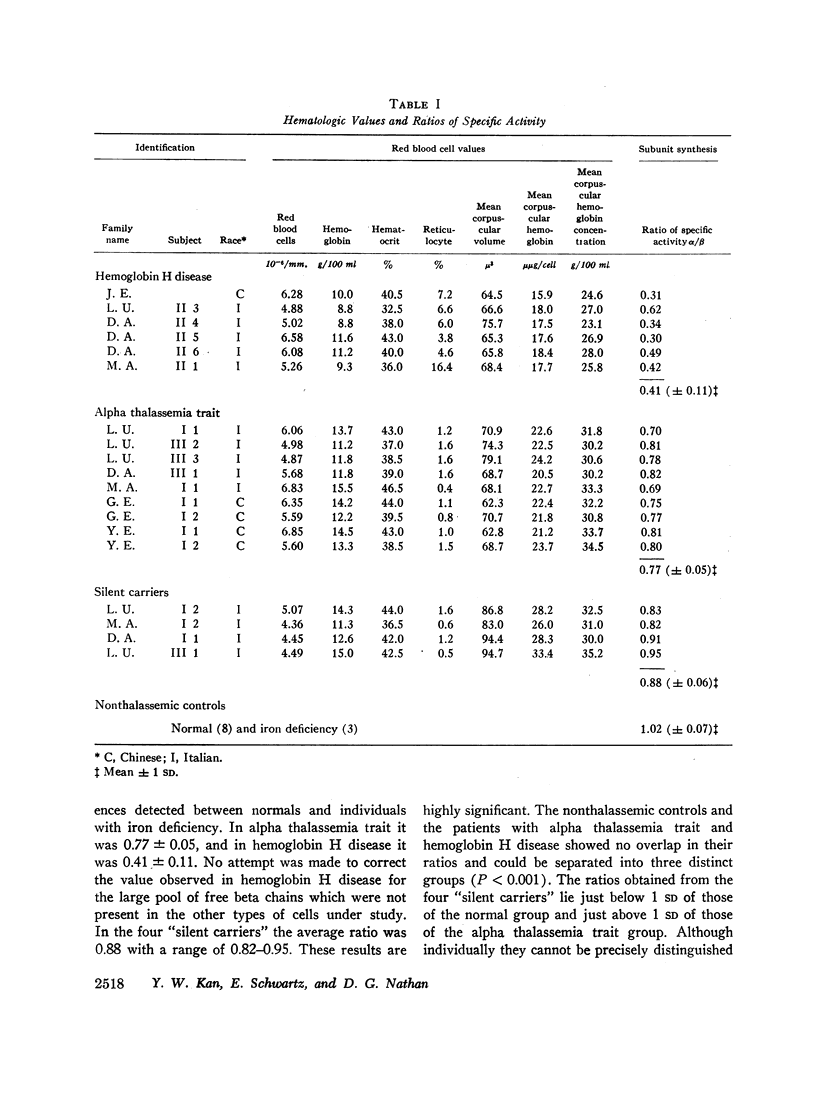
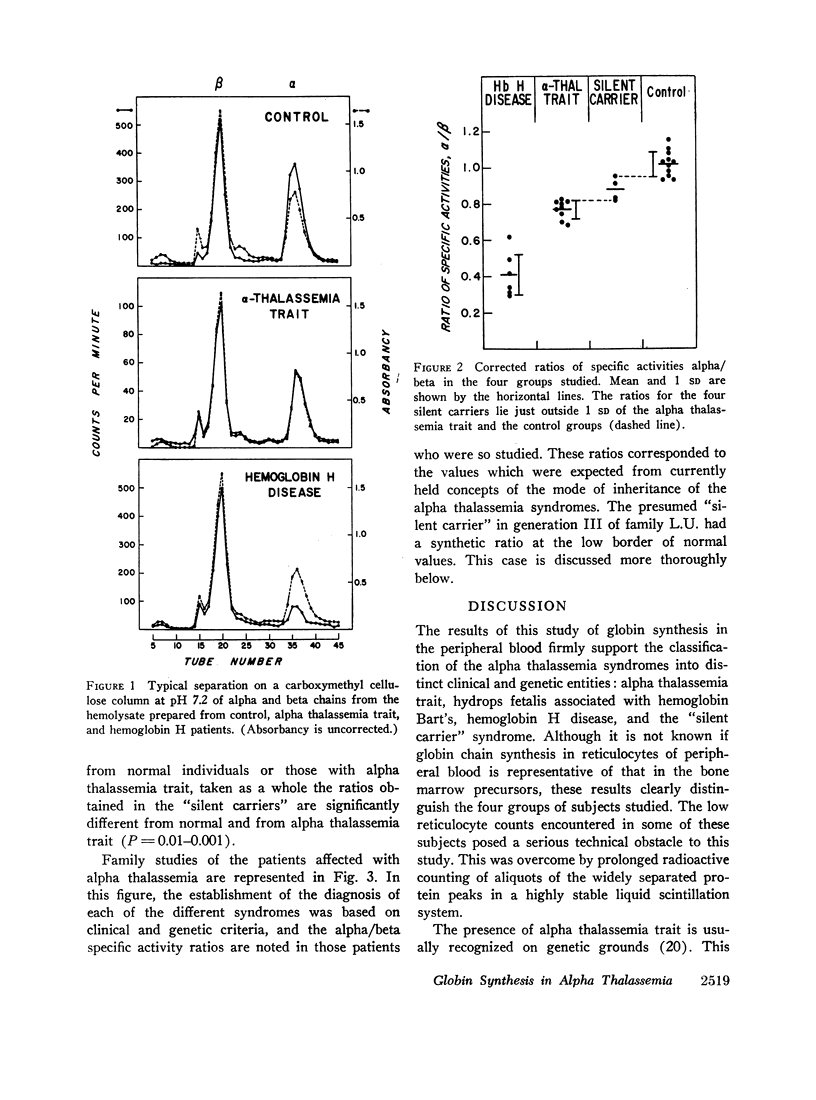
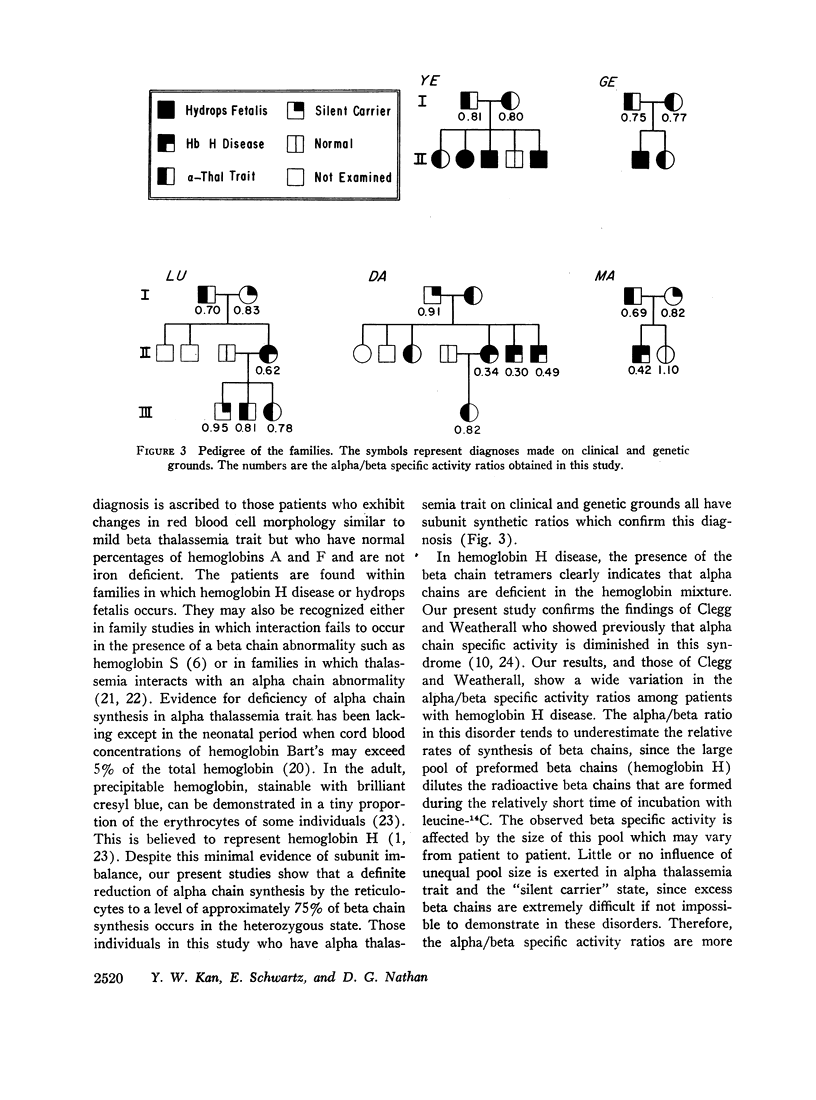
Whole blood samples of patients with various forms of alpha thalassemia including hemoglobin H disease, alpha thalassemia trait, and the “silent carrier” state were incubated with leucine-14C for definition of relative rates of production of alpha and beta chains in these disorders. The chains were separated by carboxymethyl cellulose chromatography in the presence of 8 M urea and dithiothreitol. Their absorptions at 280 mμ were determined and their radioactivities measured in a liquid scintillation spectrometer. After correction for differences in extinction coefficients, the specific activities of the widely separated alpha and beta peaks were determined. In 11 nonthalassemic individuals, the alpha/beta specific activity ratios were found to be 1.02±0.07; in nine patients with alpha thalassemia trait, 0.77±0.05; in six patients with hemoglobin H disease, 0.41±0.11; and in four “silent carriers,” 0.88 with a range of 0.82-0.95. The results show that in peripheral blood, alpha chain production relative to beta chain production is indeed limited in the alpha thalassemia syndromes. Hemoglobin H disease results from doubly heterozygous inheritance of a gene resulting in moderate depression of alpha chain production (alpha thalassemia trait) and a gene resulting in very mild depression of alpha chain production (the “silent carrier” syndrome.”
Full text
PDF




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- ATWATER J., SCHWARTZ I. R., ERSLEV A. J., MONTGOMERY T. L., TOCANTINS L. M. Sickling of erythrocytes in a patient with thalassemia-hemoglobin-I disease. N Engl J Med. 1960 Dec 15;263:1215–1223. doi: 10.1056/NEJM196012152632402. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Naughton M. A., Weatherall D. J. An improved method for the characterization of human haemoglobin mutants: identification of alpha-2-beta-2-95GLU, haemoglobin N (Baltimore). Nature. 1965 Aug 28;207(5000):945–947. doi: 10.1038/207945a0. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Weatherall D. J. Haemoglobin synthesis in alpha-thalassaemia (haemoglobin H disease). Nature. 1967 Sep 16;215(5107):1241–1243. doi: 10.1038/2151241a0. [DOI] [PubMed] [Google Scholar]
- DORMANDY K. M., LOCK S. P., LEHMANN H. Haemoglobin Q-alpha-thalassaemia. Br Med J. 1961 Jun 3;1(5239):1582–1585. doi: 10.1136/bmj.1.5239.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAMMACK D. B., HUEHNS E. R., SHOOTER E. M., GERALD P. S. Identification of the abnormal polypeptide chain of hemoglobin G-Ib. J Mol Biol. 1960 Dec;2:372–378. doi: 10.1016/s0022-2836(60)80048-4. [DOI] [PubMed] [Google Scholar]
- GOUTTAS A., FESSAS P., TSEVRENIS H., XEFTERI E. Description d'une nouvelle variété d'anémie hémolytique congénitale; etude hématologique, électrophorétique et génétique. Sang. 1955;26(9):911–919. [PubMed] [Google Scholar]
- HILL R. J., KONIGSBERG W., GUIDOTTI G., CRAIG L. C. The structure of human hemoglobin. I. The separation of the alpha and beta chains and their amino acid composition. J Biol Chem. 1962 May;237:1549–1554. [PubMed] [Google Scholar]
- Heywood J. D., Karon M., Weissman S. Studies of the kinetics of alpha and beta hemoglobin chain synthesis. J Lab Clin Med. 1966 Feb;67(2):246–254. [PubMed] [Google Scholar]
- INGRAM V. M., STRETTON A. O. Genetic basis of the thalassaemia diseases. Nature. 1959 Dec 19;184:1903–1909. doi: 10.1038/1841903a0. [DOI] [PubMed] [Google Scholar]
- KOLER R. D., RIGAS D. A. Genetics of haemoglobin H. Ann Hum Genet. 1961 May;25:95–100. doi: 10.1111/j.1469-1809.1961.tb01503.x. [DOI] [PubMed] [Google Scholar]
- Kan Y. W., Allen A., Lowenstein L. Hydrops fetalis with alpha thalassemia. N Engl J Med. 1967 Jan 5;276(1):18–23. doi: 10.1056/NEJM196701052760103. [DOI] [PubMed] [Google Scholar]
- LIE-INJO L. E. Alpha-chain thalassemia and hydrops fetalis in Malaya: report of five cases. Blood. 1962 Nov;20:581–590. [PubMed] [Google Scholar]
- Necheles T. F., Cates M., Sheehan R. G., Meyer H. J. Hemoglobin H disease. A family study. Blood. 1966 Oct;28(4):501–512. [PubMed] [Google Scholar]
- RAMOT B., SHEBA C., FISHER S., AGER J. A., LEHMANN H. Haemoglobin H disease with persistent haemoglobin "Bart's" in an Oriental Jewess and her daughter: a dual alpha-chain deficiency of human haemoglobin. Br Med J. 1959 Dec 5;2(5161):1228–1230. doi: 10.1136/bmj.2.5161.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WASI P., NA-NAKORN S., SUINGDUMRONG A. HAEMOGLOBIN H DISEASE IN THAILAND: A GENETICAL STUDY. Nature. 1964 Nov 28;204:907–908. doi: 10.1038/204907a0. [DOI] [PubMed] [Google Scholar]
- Weatherall D. J., Clegg J. B., Naughton M. A. Globin synthesis in thalassaemia: an in vitro study. Nature. 1965 Dec 11;208(5015):1061–1065. doi: 10.1038/2081061a0. [DOI] [PubMed] [Google Scholar]