


Abstract
Chromosome 7q22 has been the focus of many cytogenetic and molecular studies aimed at delineating regions commonly deleted in myeloid leukemias and myelodysplastic syndromes. We have compared a gene-dense, GC-rich sub-region of 7q22 with the orthologous region on mouse chromosome 5. A physical map of 640 kb of genomic DNA from mouse chromosome 5 was derived from a series of overlapping bacterial artificial chromosomes. A 296 kb segment from the physical map, spanning Ache to Tfr2, was compared with 267 kb of human sequence. We identified a conserved linkage of 12 genes including an open reading frame flanked by Ache and Asr2, a novel cation-chloride cotransporter interacting protein Cip1, Ephb4, Zan and Perq1. While some of these genes have been previously described, in each case we present new data derived from our comparative sequence analysis. Adjacent unfinished sequence data from the mouse contains an orthologous block of 10 additional genes including three novel cDNA sequences that we subsequently mapped to human 7q22. Methods for displaying comparative genomic information, including unfinished sequence data, are becoming increasingly important. We supplement our printed comparative analysis with a new, Web-based program called Laj (local alignments with java). Laj provides interactive access to archived pairwise sequence alignments via the WWW. It displays synchronized views of a dot-plot, a percent identity plot, a nucleotide-level local alignment and a variety of relevant annotations. Our mouse–human comparison can be viewed at http://web.uvic.ca/~bioweb/laj.html. Laj is available at http://bio.cse.psu.edu/, along with online documentation and additional examples of annotated genomic regions.
INTRODUCTION
The Giemsa negative band q22 of human chromosome 7 is an area known to be gene-rich and prone to chromosomal breakage. Aberrations at 7q22 are commonly observed in myeloid leukemias and myelodysplastic syndromes, with critical deleted regions being mapped using fluorescent in situ hybridization (1–4) and polymorphic microsatellite markers (5). Specifically, the segment between CYP3A4 and CUTL1 contains a region deleted in two patients with chronic myeloid leukemia (3). Recent evidence for the association of schizophrenia with 7q22 further establishes the need for a detailed characterization of this region (6).
The fact that the mouse is the leading model for studying disease processes in mammals makes the emerging mouse genomic data a valuable resource for functional studies (7). Mouse–human comparative analyses will facilitate the design and interpretation of mouse knockout studies. Extensive refinements to the cytogenetically-based Mouse Genome Informatics database map and the Davis Human/Mouse homology map have been made by identifying human orthologs to mapped mouse genes (8).
Detailed characterizations of disease regions are increasingly being done through mouse–human genomic comparisons aimed at identifying novel genes and regulatory elements. For example, a comparison of 7q11.23 with its orthologous region on mouse chromosome 5 has identified one new gene and ruled out the existence of two previously proposed genes within the region commonly deleted in patients with Williams–Beuren syndrome (9). This growing number of mouse–human comparisons of increasing size creates a need for new ways to display comparative genomic information. While several workbench programs such as Genotator (10), RUMMAGE (11), PowerBLAST/SEQUIN (12,13) and GESTALT (14) address the demand for effective analysis, annotation and display of large single genomic sequences, new tools for displaying comparative sequence analysis are just beginning to emerge. These programs include CGAT (15), Intronerator (16,17) and Alfresco (18). In this paper we introduce a program called Laj (local alignments with java), which can be used as an interactive WWW-based tool for displaying pairwise sequence alignments and associated annotations.
In addition to the proven value of comparing complete genomic sequences, analysis of the rapidly-generated data from lower redundancy sequencing may also be of use to the biological community (13,19). The PipMaker Web server supports analysis of working draft sequences by permitting one of the two sequences to consist of multiple unoriented contigs (20). Laj provides a graphical way to organize, label and display the results from PipMaker, thereby facilitating the analysis of unfinished sequence.
We extend the sequence of the gene-dense, GC-rich EPO contig on human 7q22, previously described (11) and compare it with ∼380 kb of the corresponding mouse genomic DNA. This genomic comparison involves both complete and partial mouse sequences, which together reveal extensive conservation of gene content and order. Novel transcripts and new splice variants are identified, and the mouse–human map for this region is further refined. Our Web site, http://web.uvic.ca/~bioweb/laj.html, provides an electronic supplement to the results presented in this paper.
MATERIALS AND METHODS
Isolation and characterization of clones
The P1-derived artificial chromosome (PAC) clone H_DJ0138m12 was from the RPCI PAC whole genome library. The cosmid clones cos159d9.L and cos8a5.L were obtained from the chromosome 7-specific cosmid library of the Lawrence Livermore National Laboratory (LL07NCC01). Human bacterial artificial chromosome (BAC) 183H5 was obtained from the BAC library of Genome Systems, Inc. (St Louis, MO) using human probes for zonadhesin. The mouse genomic DNA BAC clones (423o3 and 558e17) were obtained from the Research Genetics CITB-CJ7-B mouse BAC library using PCR probes for the mouse Ache and Epo genes, respectively. A 3 kb gap between BACs 423o3 and 558e17 was bridged by PCR using primers (423sp6-F: 5′-CTGAGAGAGACTGACACCAGAAGG-3′, and 558T7-R: 5′-TGACTCGGGTCAATTCTAAGTT-3′) and mouse genomic DNA. The resulting DNA was T-A cloned into pGEM-T vector (Promega). Additional mouse BAC clones were obtained from the Research Genetics CITB-CJ7-B mouse BAC library using gene- or BAC end-specific primers. Clones from the RPCI-11 Human Male BAC Library were identified by hybridization using BAC end sequence from NH452F23, NH0044M06, NH0264N05 and NH0126L15.
DNA sequencing
A shotgun sequencing strategy was employed for clones 423o3, 558e17, 139n8, 493b1, cos159d9.L, cos8a5.L, H_DJ0138m12, the 3 kb gap clone and the 6357 nt XbaI fragment from BAC clone 183H5. Clones were isolated with NucleoBond Plasmid Maxi Kits (Clontech) and randomly sheared by nebulization. Blunt ends were assured and fragments from 1.5 to 3 kb were size-fractionated by agarose gel electrophoresis. Fragments were ligated into SmaI-cut M13mp19 vector and single-stranded templates were purified with Qiagen M13 plates. Random clones from each sub-library were then run on ABI 373 or 377 automated DNA sequencers using fluorescently labeled primers (Amersham). Clones 423o3, 558e17, cos159d9.L and H_DJ0138m12 were sequenced to 7-fold redundancy. Gaps were filled using a PCR approach and dye-terminator sequencing with unique primers (Amersham and ABI). At the time of analysis, BAC139n8 had been sequenced to 3.75-fold and 493b1 to 1.57-fold redundancy.
Sequence analysis
DNAStar software was used for gel trace analysis, contig assembly and DNA and protein alignments. Repeat elements were characterized using RepeatMasker2 (A.F.A.Smit and P.Green, unpublished results; http://ftp.genome.washington.edu/cgi-bin/RepeatMasker). Comparative sequence alignments displayed in this paper were generated using PipMaker (20; http://bio.cse.psu.edu/). DNA and protein sequences were compared with available public databases using the various BLAST programs available through the network server at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). Putative exons were identified using a variety of programs GENSCAN (21; http://bioweb.pasteur.fr/seqanal/interfaces/genscan-simple.html), FGENES, FGENESH and FGENES-M from the BCM Gene-Finder (V.V.Solovyev, unpublished results; http://dot.imgen.bcm.tmc.edu:9331/gene-finder/gfb.html). Compiled, overlapping sets of expressed sequence tags (ESTs) were downloaded as UniGene clusters (22) and the individual ESTs were assembled using DNAStar software. Additional, overlapping cDNA and EST sequences were then obtained by submitting the consensus sequence of the assembled ESTs to BLAST. This facilitated the verification of splice sites, established the location of novel genes and allowed the extension of previously published cDNA sequences. Differences, including SNPs, alternative splicing events and predictions not supported by EST data, were further investigated by inspecting the trace data from the original genomic sequence assembly.
Expression studies, cloning and partial cDNAs
Mouse and human expression studies of ASR2 and CIP1 were carried out on Clontech multiple tissue panels using HotstarTaq (Qiagen). A 50 µl vol was used for all expression studies, and the cycling parameters were as follows: 15 min at 95°C for Taq activation, and eight cycles of 94°C for 30 s; 65°C for 1 min; 72°C for 1 min; followed by 30–33 cycles of 94°C for 30 s; 58°C for 1 min; 72°C for 1 min; and a final extension of 5 min at 72°C. Primers used for expression studies are available from the authors upon request. Partial mouse cDNAs for Asr2, Cip1 and Perq1 were amplified from mouse brain cDNA (Clontech). CIP1 was amplified from human brain cDNA (Clontech). The PCR products from the cDNA were T-A cloned into pGEM-T vector (Promega). Asr2 and CIP1 were completely sequenced by primer walking. Perq1 and Cip1 clones were end sequenced.
RESULTS
We obtained a minimal tiling path of BACs representing ∼640 kb of mouse chromosome 5 orthologous to a gene-dense, GC-rich isochore from the EPO region of 7q22 (Fig. 1). Mouse BACs 423o3 and 558e17 were completely sequenced along with a 3 kb segment that links the two clones, producing a gap-free contig of 296 kb. Two overlapping human clones (cosmid 159d9 and PAC 138m12) were also sequenced completely, yielding a 118 kb contig. This contig was used to fill in a 94 kb gap linking the ACHE-containing genomic clone AF002993 (23) to the 228 kb EPO contig AF053356 previously described (11; Fig. 1).
Figure 1.

Overview of the refined chromosome 7q22 physical map for the region spanning the genes PAI1 and CYP3A4, with a direct comparison with 640 kb of orthologous genomic sequence from mouse chromosome 5. Black rectangles represent completely sequenced regions, gray rectangles represent partially sequenced or PCR-mapped clones and gray hatches represent parts of clones whose overlapping distances are not known. (A) Representative genes and clones provide further refinement to the human gene map previously described (11). A minimal tiling path determined by hybridization, PCR and in silico methods clearly establishes MUC3 on the telomeric side of ACHE and links the HRBL end of AF053356 to the CYP3A4 locus (see http://www.genet.sickkids.on.ca/chromosome7/ for a more detailed physical map including additional genes, further clones and experimental details). (B) Comparative human–mouse genomic map showing conserved synteny of at least 22 genes. The composite gene map shows human genes (upper case letters), all of which have a mouse ortholog, as well as the mouse gene Cyp3a13, whose human ortholog is yet to be determined. The orientation of the mouse BAC clones in relation to chromosome 5 was inferred from the location of the mouse Cyp3a gene locus on a refined chromosome 5 map (http://genome.nhgri.nih.gov/chr7/comparative; 8). PCR mapping determined that a TSC-22-like gene, THG-1, is next to HRBL in both human and mouse followed by ZE03F02 and FLJ20257 (Ze03f02 and D5Wsu46e) whose order has not yet been determined. None of the genes shown between FLJ20257 and CYP3A4 has been identified in our partial mouse sequence data for BACs 139n8 and 493b1 (D5Wsu46e to Cyp3a13).
We detected a sequence transposition within the previously reported human genomic sequence AF053356 (11) when we compared it with our mouse genomic sequence in the 5′ region of the zonadhesin (Zan) locus and with the mouse zonadhesin cDNA (MMU97068) (24). Inspecting our partial human sequence from cosmid 8a5 (Fig. 1) reinforced the possibility of sequence transposition, as cosmid 8a5 did not contain any sequence from positions 38341 to 45778 of AF053356 but did match positions 1–38340 and 45779–48328 of AF053356. Position 38340 of AF053356 corresponds to the one sequence gap in the EPO contig that could not be filled using a PCR approach (11). Furthermore, alignments with our complete sequence of the human zonadhesin (ZAN) cDNA (T.Cheung, M.Wassler, G.Cornwall and D.Hardy, manuscript in preparation) revealed that ZAN exon 7 was missing and exons 1–6 were downstream of exons 8–11 in AF053356. The positioning of exons 1–6 (including the putative promoter region) downstream of exons 8–11 alters the conserved domain order of the zonadhesin protein observed in mouse, human and pig, strongly suggesting that the ZAN locus on AF053356 was incorrectly assembled and does not represent a natural variant of the gene. To resolve this we obtained human BAC 183H5 from Genome Systems and utilized unfinished high throughput genomic (HTG) data from The Washington University sequencing project (AC009488; Fig. 1). The sequence of a 1253 bp PCR product amplified from a human placental genomic DNA template confirmed the accuracy of AC009488_3ctg3 from positions 24030–25292 (sense primer: CTCTATCCGCCGGGGCTCCTGTA; antisense primer: CACGCCTGGCTTTCCTGATGACC). In addition, the sequence of a 2268 bp PCR product bridged a gap between exons 11 and 12 in AF053356 (sense primer: ACACTGGGCCCTCGGACATAAAA; antisense primer: GACTGGCCTGCGTGGGAGAAC). We also identified a 6357 bp XbaI fragment from BAC clone 183H5 that spans exons 7–8 by Southern blotting using positions 809–1103 of the zonadhesin cDNA as a probe. The sequence of this fragment merged two contigs of AC009488, and, together with the sequences of the PCR products, properly ordered exons 1–6 and 8–11, which are transposed in AF053356. Thus, collectively these sequences resolved discrepancies between AC009488 and AF053356 and corrected the sequence transposition in AF053356.
We directly compared 267 kb of the corrected 365 kb human contig to 296 kb of the contiguous mouse sequence, revealing the genomic structure of a conserved linkage of 12 genes (Ache to Tfr2; Fig. 2). The data are shown as a traditional dot-plot (Fig. 2A) and percent identity plot (PIP; Fig. 2B). New information regarding specific genes is provided in Tables 1 and 2. We also compared adjacent incomplete sequence data from BAC 139n8 to the orthologous region on AF053356 (PCOLCE to HRBL; Fig. 1). This revealed that the mouse orthologs to PCOLCE, FBXO24, LRN, IRS3L and HRBL are all present on BAC 139n8 (Fig. 1). In addition to the above tables and figures, we utilize our new program Laj to interactively display both our complete and partial genomic alignments (http://web.uvic.ca/~bioweb/laj.html). This electronic supplement displays the alignments themselves, hyperlinks to reference cDNA sequences, UniGene clusters (22), GeneCards (25), LocusLinks (26) and ESTs not linked to any cDNA, along with PubMed links to relevant literature (Fig. 3).
Figure 2.



(A) Dot-plot display of mouse (x-axis) and human (y-axis). (B) PIP. Nucleotide positions are shown for the mouse sequence on the x-axis, and percent sequence identity with the corresponding human sequence (50–100%) is shown on the y-axis. The purple and orange shading on the upper and lower halves of the PIP represent the locations of exons on the + and – strands, respectively, while the pink and light orange areas represent UTRs. Red stripes indicate regulatory regions that have been functionally characterized. Genes are labeled based on cDNA, EST and comparative genomic information. RepeatMasker2 (A.F.A.Smit and P.Green, unpublished results) was used to locate repeat elements.
Table 1. Mouse and human genes in the ACHE/TFR2 region.
| ACHE Ache | AChE (acetylcholinesterase) has multiple molecular forms (28) (Fig. 2B displays the dominant E4-E6 hydrophilic variant). A highly conserved region just 5′ of the transcription start site, highlighted by a red stripe in Figure 2B (positions 7445–7516), contains transcription factor binding sites previously described (55). A second region containing an alternative promoter that acts synergistically with the proximal promoter in enhancing Ache expression in neuroblastoma cells (29) is also highly conserved, and highlighted by a red stripe in Figure 2B (positions 6374 –7139). The conserved regions found upstream of the alternative promoter have yet to be explored for regulatory elements. |
| Hs.157779 Mm.21836 | Using our comparative genomic data we predict a 217 amino acid product in mouse just downstream of Ache. In human we can predict a 227 amino acid product from AC002993 that is 79.2% identical to a putative 217 amino acid product in mouse (Table 2). These products show similarity to the C-terminal end of the putative proteins CG16979 (Drosophila), CAB41160 (Arabidopsis), F38A5.1 (C.elegans) and BAA92064 (human). However, on our human cosmid 159d9 and clone NH0126L15 sequenced by Washington University in St Louis (AC011895), the start codon has a C instead of an A, making the largest ORF 146 amino acids. |
| ASR2 Asr2 (Also known as: Ars2) | Asr2 (arsenite resistance protein 2) was named for its ability to confer arsenite resistance to an arsenite-sensitive Chinese hamster ovary cell line (56). We characterized the complete genomic structure of ASR2 (Table 2 and Fig. 2B) and found it transcribed in all tissues tested in both human and mouse (data not shown). The 876 amino acid human product differs in both size and amino acid content from that predicted by a full-length cDNA clone AL096723. This can be explained by two separate, single nucleotide differences in AL096723 that cause a frameshift error. These differences are resolved by our human genomic sequence and were probably due to sequence compressions in AL096723. Alternative splicing of the Asr2 mRNA was revealed by sequencing two different PCR-derived Asr2 cDNA clones that contain the full coding region of the mouse Asr2 cDNA (less the first methionine and the last eight codons). By comparing these cDNA clones with genomic and EST sequences we found three alternative splice events that decreased the coding size by 12, 21 or 33 (12 plus 21) bp (Table 2). An examination of human ESTs revealed only the 12 bp alternative splice event which, as in mouse, uses a non-canonical splice site (TG instead of AG in the AG–GT splice rule). |
| CIP1 Cip1 | We identified and characterized a novel cation-chloride cotransporter-like gene in both human and mouse. Recently, Caron et al. (57) independently characterized a 3.2 kb transcript that they named CIP1 (cation-chloride cotransporter-interacting protein) as it coimmunoprecipitates with endogenous NKCC (Na-K-Cl cotransporter) and in Xenopus laevis oocytes, it inhibits NKCC1-mediated cation transport. CIP1 is 100% identical to our putative cation-chloride cotransporter-like protein and is predicted by PSORT (58) to have 12 transmembrane helices. To confirm our gene predictions we cloned and sequenced a partial cDNA, which spans all 13 exon/intron boundaries, from a human brain cDNA pool. This cDNA, combined with human and mouse genomic sequence comparisons, allowed us to predict the mouse exon structure, of which exons 7–13 have subsequently been confirmed by sequencing cloned mouse brain cDNA. We screened Clontech mouse and human multiple tissue cDNA panels using PCR, and found that this gene is ubiquitously transcribed in all tissues tested (data not shown). Alternative splice variants were apparent in all mouse tissues tested, while human expression studies using primers designed for the corresponding positions on human cDNA showed no evidence of alternative splice products. However, human EST AW675796 reveals an alternative splicing event that results in a 627 amino acid isoform of CIP1 that is predicted to have a truncated C-terminus, yet is still predicted to contain 12 transmembrane spanning regions. CIP1 shows higher similarity to Drosophila (60%; CG10413), C.elegans (54%; T04B8.5) and putative yeast (49%; NP009794.1) proteins, than it does to other members of the two established branches (Na+-K+-Cl– and K+-Cl–) of the cation-chloride cotransporter family. This evolutionary conservation combined with preliminary functional studies place CIP1 in a third branch of the cation-chloride cotransporter family and suggest that it may be part of a new family of proteins that modify the activity or kinetics of other cation-chloride transporters (57). |
| TRIP6 Trip6 | TRIP6 (thyroid receptor interacting protein 6) is a human LIM domain-containing protein previously mapped to 7q22 (59) whose proposed function is to transmit signals from the cell surface to the nucleus (60). We have characterized the genomic structure of the mouse and human Trip6 genes (Table 2). |
| EPHB4 Ephb4 | EPHB4 (ephrin type-B receptor 4 precursor), originally named HTK (hepatoma transmembrane kinase), is a receptor tyrosine kinase expressed in a variety of tissues, with its hematopoietic expression localized to the monocytic lineage (61,62). EPHB4 is preferentially expressed in veins (63) and recent studies have clearly linked Ephb4 to angiogenesis in X.laevis (64). The mouse ortholog, Ephb4, was originally designated Myk1 (mouse tyrosine kinase; 65) and later called Mdk2 (mouse developmental kinase 2; 66). The EST sequence AI049017 extends the 5′ UTR of the Ephb4 cDNA and suggests that an alternative splicing event occurs in this region of the gene. While our genomic data confirm both the coding region and the overall transcript size previously described (66), we do not find a match for the 3′ end (positions 3756–4473) of that sequence (Z49085); however, this sequence matches perfectly to positions 781–1496 of the Cctg gene (Z31556), which has been mapped to mouse chromosome 3 (67). We thus conclude that Z49085 contains sequence from two separate genes. |
|
ZAN Zan |
ZAN (Zonadhesin)
codes for a sperm membrane protein that binds to the zona pellucida
of the egg in a species-specific manner (68). In mice, the Zan product
is a large (5374 amino acid) testes-specific protein with a complex
mosaic structure consisting of MAM (meprin, A5 receptor, protein
tyrosine phosphatase mu), mucin, VWD (von Willebrand factor type
D), EGF (epidermal growth factor), transmembrane and intracellular
domains (24,69). By comparing our mouse genomic sequence with the
mouse Zan mRNA MMU97068 (16.1 kb) we find that
the mouse Zan gene contains 88 exons and spans
98.2 kb of genomic DNA. In contrast, the human ZAN gene
contains 48 exons, spans 61 kb and codes for a protein of 2724 amino
acids (T.Cheung, M.Wassler, G.Cornwall and D.Hardy, manscript in
preparation). The expansion of the mouse Zan gene
stands out in the dot-plot (Fig. 2A) as
a 39.5 kb elongation along the x-axis (mouse; positions
108788–147614 on Fig. 2B), and
is primarily due to multiple duplication of the partial D3 domain
sequence. Each of the 20 partial D3 domains (exons 38–77)
consists of a segment containing two exons of ∼218
and 142 bp in length. |
| ACHE Ache | AChE (acetylcholinesterase) has multiple molecular forms (28) (Fig. 2B displays the dominant E4-E6 hydrophilic variant). A highly conserved region just 5′ of the transcription start site, highlighted by a red stripe in Figure 2B (positions 7445–7516), contains transcription factor binding sites previously described (55). A second region containing an alternative promoter that acts synergistically with the proximal promoter in enhancing Ache expression in neuroblastoma cells (29) is also highly conserved, and highlighted by a red stripe in Figure 2B (positions 6374 –7139). The conserved regions found upstream of the alternative promoter have yet to be explored for regulatory elements. |
| EPO | Erythropoietin (EPO) regulates the production of red blood cells by promoting the proliferation and differentiation of erythroid precursor cells (reviewed in 70). The mouse and human genomic structures for EPO have previously been described in detail, with considerable progress being made in characterizing the regulatory elements (reviewed in 49). The major proximal regulatory elements are a promoter at PIP positions 205140–205195 in the mouse sequence and a 3′ enhancer at 201210–201252 (71). These elements, marked by red stripes in Figure 2B, are substantially more conserved than the surrounding sequence. While initial clues for their location were provided by careful inspection of human–mouse alignments between much shorter sequences, Figure 2B verifies that they also stand out clearly in a completely automated comparison of long genomic regions. In particular, the promoter corresponds very closely to a 48 bp gap-free segment with 94% nucleotide identity (PIP positions 205144–205191), and the enhancer is contained within a 149 bp gap-free segment with 79% identity (PIP positions 201141–201289). Two additional elements regulating EPO transcription have been mapped to the region 14 kb upstream of the transcription start site in humans by inserting large DNA fragments into transgenic mice (summarized in 49). These results suggest that the conserved regions around positions 209–214 kb may be worthy of investigation. |
| Epo | |
| RPP20 | RPP20 codes for a 20 kDa protein that co-purifies with human ribonuclease P (Rnase P), a tRNA processing ribonucleoprotein (72). The Rpp20 gene consists of at least two exons, one of which accounts for the entire 140 amino acid product, which is 94.3% identical between human and mouse (Table 2). |
| Rpp20 | |
| PERQ1 | PERQ1 codes for a novel protein rich in the amino acids proline, glutamate, arginine and glutamine (P, E, R and Q), with a glycine-tyrosine-phenylalanine (GYF) domain. The GYF domain is part of a unique helix–bulge–helix domain that binds to proline-rich motifs (73). Our mouse–human comparison, EST data and partial cDNA sequencing predict a putative Perq1 mouse protein and an orthologous PERQ1 human protein that differ somewhat in structure from the putative ORF2 (11). In order to predict the 1004 amino acid product in human, we had to assume that a run of nine cytosine bases (position 88127 of AF053356) actually contained eight or ten, as the reading frame shift produced did not correspond to our mouse cDNA sequence or genomic data. While the cDNAs are still not fully characterized, we predict a minimum 3 kb transcript with 24 exons in both species (Table 2). Considering the mouse–human sequence similarity and the presence of a CpG island found within the region 8.8 kb 5′ of the annotated Perq1 gene (Fig. 2B; PIP positions 229–231 kb), it is possible that part of the upstream 5′ UTR and promoter region has not been characterized. At the 3′ end of the gene, several EST sequences from a variety of species match significantly with the region between Perq1 and the 3′ end of Gnb2. PERQ1 shows 58% identity to two putative human proteins (AK001739 and AB014542) and 55% identity to a putative chicken protein (U90567). These three proteins are also rich in P, E, R and Q, and contain conserved GYF domains. |
| Perq1 | |
| GNB2 | GNB2 (guanine nucleotide binding protein, β polypeptide 2) encodes a β subunit of guanine nucleotide-binding proteins (G-proteins). G-proteins are heterotrimeric, containing α, β and γ subunits, and are involved in a multitude of cellular signaling processes (for a recent review see 74). We characterized the mouse genomic structure for Gnb2. Part of the highly conserved region found in the 5′ UTR (PIP positions 250459–252757) appears to be transcribed in various mouse and human tissues (see http://web.uvic.ca/~bioweb/laj.html). |
| Gnb2 | |
| BAF53B | The human ACTL6-like gene is more closely related to actin genes of lower organisms than to those of vertebrates (11). Since its discovery, ACTL6-like has confusingly been called ACTL6 (BAF53A). However, it is clear from comparing our mouse genomic sequence to AF053356 that the ACTL6-like gene is actually BAF53B. The mouse ortholog Baf53b encodes a 426 amino acid putative product that is 84.1% identical to the putative 475 amino acid human BAF53B (Table 2). Baf53b is 84% identical to mouse Baf53a, whose role in chromosome remodeling has previously been described (75). |
| Baf53b | |
| TFR2 | Transferrin receptors mediate iron uptake by binding and internalizing the carrier protein transferrin. The transferrin receptor 2 gene (TFR2) contains 18 exons, is 2.9 kb in length and is primarily expressed in the liver (11,76). We have determined the genomic structure of the first six exons of mouse Tfr2 and identified an alternative splice variant occurring in the 5′ UTR region of the gene (EST AA537969). |
| Tfr2 |
An expanded version of this table and links to specific sequences can be found at http://web.uvic.ca/~bioweb/laj.html.
Table 2. Comparative gene information.
| Genes, UniGene cluster(s) | Gene structure | Protein | Conservation | ||||
| |
|
mRNA lengtha |
Exons |
Representative alternative spliced
form |
Length
(amino acids) |
% Amino acid identity |
% nt (ORF) |
| Ache | mouse | 2089 | 6 | 614 | 88.4 | 84.8 | |
| human | 2218 | 6 | NM000665, NM015831 | 614 | |||
| Mm.21836 | mouse | 1001 | 1 | 217 | 79.2 | 80.9 | |
| Hs.157779 | human | 809 | 1 | 227 | |||
| Asr2 | mouse | 2901 | 20 | AA562274, AA929503, AW488339 | 875 | 97.8 | 88.7 |
| human | 2896 | 20 | AA317461, AW572565 | 876 | |||
| Trip-6 | mouse | 1754 | 9 | 480 | 86.8 | 83.4 | |
| human | 1735 | 9 | 476 | ||||
| Cip1 | mouse | 3302 | 14 | observedb | 914 | 93.5 | 85.3 |
| human | 3310 | 14 | AW675796 | 914 | |||
| Ephb4 | mouse | 4271 | 17 | AI049017 | 987 | 91.9 | 90.5 |
| human | 3945 | 17 | 987 | ||||
| Zan | mouse | 16125 | 88 | 5374 | 61.2 | 71.1 | |
| human | 8852 | 48 | 2724 | ||||
| Epo | mouse | 1385 | 5 | 192 | 78.2 | 79.6 | |
| human | 1367 | 5 | 193 | ||||
| Rpp20 | mouse | 784 | 2 | 140 | 94.3 | 87.5 | |
| human | 893 | 2 | AA405580, AA779813, AA324940 | 140 | |||
| Perq1 | mouse | 3033 | 24 | 1011 | 89.1 | 87.8 | |
| human | 3312 | 24 | 1004 | ||||
| Gnb2 | mouse | 1860 | 10 | 340 | 99.7 | 92.6 | |
| human | 1661 | 10 | 340 | ||||
| Baf53b | mouse | 1472 | 14 | 426 | 83.7 | 84.1 | |
| human | 1495 | 14 | 429 | ||||
| Tfr2 | mouse | 5′ partial | 1–6 | AA537969 | |||
| human | 5′ partial | 1–6 | |||||
aDetermined using EST, cDNA and comparative genomic data.
bEvidence for alternative splice variants seen on CLONTECH multiple tissue cDNA panels.
Figure 3.
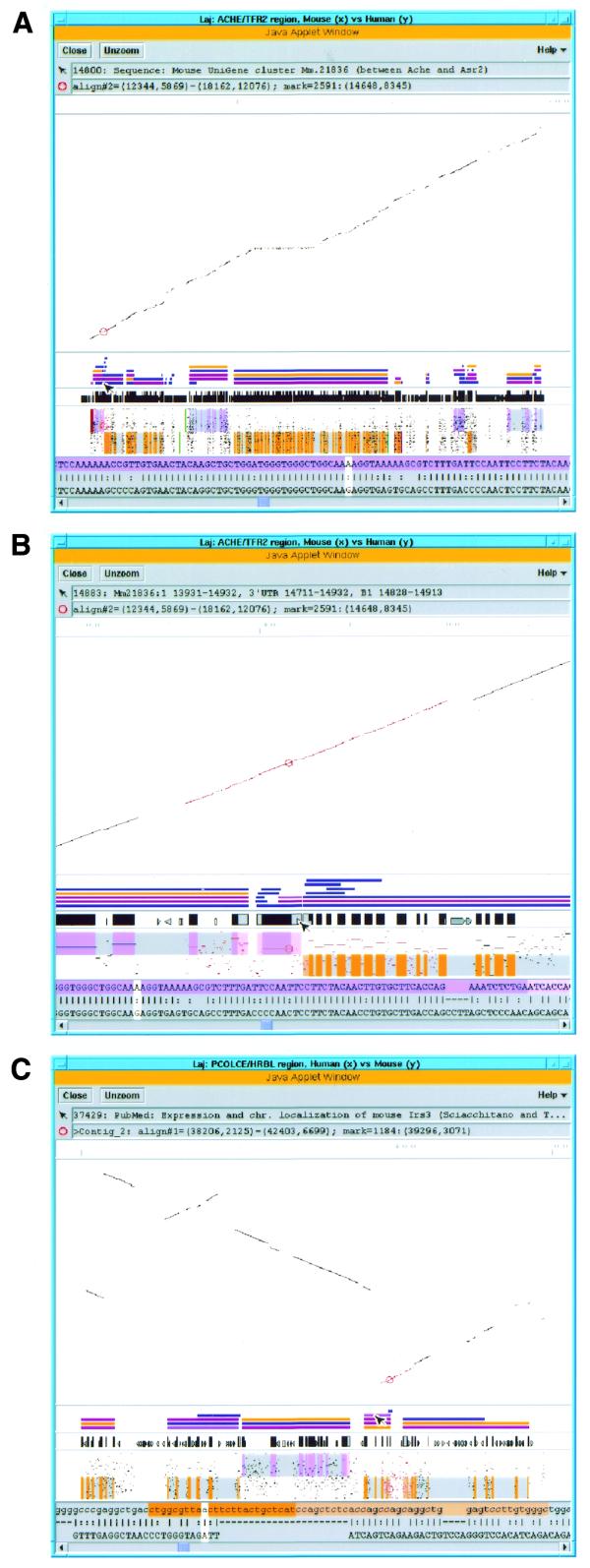
Using Laj to view complete and partial comparative genomic sequences from human chromosome 7q22 (y-axis) and the orthologous region in the mouse (x-axis). (A) View of the entire ACHE/TFR2 genomic region. The overall scale bar, dot-plot, relevant annotations, PIP (labeled as in Fig. 2B) and local alignment text are all displayed in the same window. The first message box below the menu bar displays information about the object the mouse is pointing at, in this case a hyperlink to the UniGene cluster Mm.21836. A local alignment in the ACHE/ASR2 region has been selected by clicking on it, which has caused the local alignment to be highlighted in red and the clicked position to be marked with a red circle. Information about these is provided in the second message box. The scrollable panel at the bottom of the window displays the selected local alignment in a nucleotide-level text format, with the white cursor corresponding to the marked position. (B) ‘Zooming in’ on the region selected in (A). The marked position lies in a gene flanked by ACHE and ASR2 that contains UniGene cluster Mm.21836, and the mouse pointer is currently resting on a B1 element found in the 3′ UTR of this gene. The purple and orange shading on the upper and lower halves of the PIP represent the locations of exons on the + and – strands, respectively, while the pink and light orange areas represent UTRs. Similar shading of the exons and UTRs appears in the top row (mouse sequence) of the text panel. Hyperlinked annotations include UniGene cluster Mm.21836 (blue bar above mouse pointer) flanked by Ache and Asr2 UniGene clusters, cDNAs, alternatively spliced ESTs (all blue bars), LocusLinks (pink bars) and a GeneCard for ACHE (orange bar). (C) Display of unordered partial mouse data (y-axis) aligned to complete human sequence (x-axis). Eight mouse contigs from BAC 139n8 orthologous to the PCOLCE/HRBL region on human clone AF053356 (11) are displayed, separated by horizontal gray lines. An alignment (highlighted in red) that contains the 5′ end of the IRS3L gene has been selected, and the mouse pointer is currently resting on a hyperlink (purple bar) to the PubMed entry for a paper about the mouse Irs3 gene (54). The orange-shaded text highlights the beginning of the IRS3L ORF and UTR. (See http://web.uvic.ca/~bioweb/laj.html for a display of the ordered contig data.)
Defining the boundaries of Ache and a potential novel gene
A recent search for genes that are expressed during mouse development revealed the expression of a novel cDNA fragment, D5Ertd655e, in a 2-day-old mouse embryo (27). D5Ertd655e is part of a mouse UniGene cluster (Mm.21836) that is homologous to clusters observed in human and rat (Hs.157779 and Rn.13571, respectively). This 3′ cDNA sequence is identical to the 3′ end of our predicted gene lying between Ache and Asr2 and contains the polyadenylation signal previously attributed to an alternative form of Ache (28). We searched the data banks and found no mRNA or EST sequence that contained Ache exon 6 and the second (alternative) polyadenylation site. We did find additional murine ESTs that overlap with the D5Ertd655e, providing evidence that the entire predicted open reading frame (ORF) is transcribed in mouse and rat (see Table 1 and http://web.uvic.ca/~bioweb/laj.html for further details).
Initially it was assumed that two major mRNA species of mouse Ache (2.5 and 3.7 kb) seen in northern blot experiments differed only in the 3′ UTR region and that the larger mRNA species was due to the use of a second polyadenylation site (28). The only published northern blot evidence supporting alternate polyadenylation usage comes from an experiment utilizing a 1.5 kb probe that begins 200 bp 3′ of the first polyadenylation signal and extends through the second polyadenylation signal (28). This probe would also contain the last two exons of the ubiquitously expressed Asr2, which would complicate the interpretation of these northern blots. Although the existence of alternate polyadenylation usage in Ache is still cited (29–31), the most abundant Ache mRNA species are believed to range from 2 to 2.4 kb (29). While it is still possible that this novel ORF is part of an uncharacterized variant of acetylcholinesterase, or that the two genes are co-transcribed, there is currently no sequence data supporting this. Taken together, our preliminary evidence suggests that a new gene separate from Ache exists between Ache and Asr2 (Fig. 2B).
GNB2, CGG repeats and a recombined erythroleukemic cell line
A deletion on mouse chromosome 5 resulting in a rearrangement of the 5′ UTR of Gnb2 with part of exon 1 of Epo causes the abnormal erythropoietin gene expression found in murine erythroleukemic cell line IW32 (see GenBank: MUSERPB) (32). We find that this deletion removes a 45 kb region containing Rpp20, Perq1 and Gnb2 (Fig. 2B). The region extending 2.5 kb from the deletion breakpoint on the Gnb2 side (positions 251000–253567 on Fig. 2B) is 79.4% similar between human and mouse and is very GC-rich in both species (68.6 and 59.8%, respectively). In the 5′ UTR part of this same 2.5 kb region, 10 consecutive CGG repeats were identified by screening a brain cDNA library for CGG repeats in a search for neurological disease candidate genes (33). Eleven consecutive CGG repeats are seen in AF053356 (11), suggesting that this region may be polymorphic. Recent findings demonstrate a strong correlation between chromosome deletion breakpoints and CGG repeats in patients with Jacobsen (11q-) syndrome (34,35), which raises interesting questions about a human breakpoint analogous to the rearrangement event characterized in the mouse erythroleukemic cell line IW32.
Extending the mouse and human physical maps
Identifying mouse orthologs for human genes found on human chromosome 7 and then probing mouse BAC libraries with these orthologs has proven highly successful in providing sequence-ready maps for mouse (8). We also find that in difficult-to-map areas of chromosome 7 not represented by contiguous YAC sequence, human orthologs of mouse genes identified in orthologous regions can help to orient and define human physical maps. With PCR primers designed from human orthologs for Hrbl, Thg-1, Ze03f02, D5Wsu46e and Cyp3a13, we ordered seven BACs previously identified by probing with the end sequence of NH0452F23 (AZ254552; see Fig. 1 for a representative pair). Further PCR and hybridization experiments join the EPO contig (AF053356) to a nearly completed sequence contig that extends all the way up to AZGP1 and CYP3A4 (Fig. 1). On the ACHE side of our 365 kb contig, BAC end sequence from clone NH0126L15 (AC011895) identifies several clones that were previously mapped near MUC3. Specifically, end sequence from clone NH1001A20 lines up with clone CIT-HSP-306C13 whose other end aligns to the locus designated MUC3 on Figure 1, which contains two genes, MUC3A and MUC3B (36).
Using Laj to display comparative genomic information
Laj gives the reader an immediately accessible interactive supplement to the text, providing considerably more power for viewing and interpreting data than can be provided on a printed page. It can easily be set up as a Web-based applet, allowing scientists to curate their sequence data and display the information to the public from their own Web sites. Furthermore, this electronic supplement can be updated as research progresses. The applet can be viewed from any computer with Internet access and a Web browser that supports Java 1.2 (e.g., a PC running Windows 95/98/NT or Linux, or a Sparc workstation running Solaris), thus enabling a wide audience to readily examine the information. Laj can display a dot-plot, a PIP and a nucleotide-level local alignment simultaneously. It also supports a variety of annotations, including locations of exons and repeat elements, color underlays that can be used to represent GENSCAN predictions or any other regions of the annotator’s choice and hyperlinks to relevant Web resources such as sequences from GenBank (37), UniGene clusters (22), GeneCards (25), LocusLinks (26) and PubMed references. Note that when provided with exon locations in the first sequence, Laj displays them in two ways: as tall black boxes in the diagram above the PIP, and as shaded regions in the top row of the text alignments (purple for the forward strand, orange for the reverse strand). This serves as a visual link between the graphical and text representations of the alignments and facilitates the visualization of intron–exon junctions.
Laj is available for download at http://bio.cse.psu.edu/ and is well documented, so interested researchers can (i) use it as a stand-alone tool to display and annotate their sequences, or (ii) set up their comparative sequence analyses for display on the Internet. Once Laj is installed, the user need only supply the data and annotations in specific formats. PipMaker, along with some small programs available from the PipMaker site, can rapidly transform input files obtained from various other bioinformatic sources (e.g., GENSCAN and RepeatMasker2) into a format suitable for Laj, thus increasing Laj’s utility as a comparative workbench. For more information about Laj, including online documentation, a software tour describing the program itself and additional examples of annotated genomic regions, please see http://bio.cse.psu.edu/.
DISCUSSION
Comparative sequence analysis is a useful supplement to traditional gene-finding methods for completing the catalog of all mammalian genes. We describe 640 kb of mouse genomic sequence orthologous to a gene-dense, GC-rich isochore in the EPO region on human chromosome 7q22. Our direct comparison of the contiguous portion of this sequence from ACHE to ZAN allowed us to characterize a novel cation-chloride cotransporter-interacting protein CIP1, provide evidence of the possibility of a gene flanked by ACHE and ASR2, and characterize the genomic structure of ASR2, TRIP6, EPHB4 and ZAN. In addition, our comparative and physical mapping data have determined the orientation of the AF053356 contig on the physical map of chromosome 7q22. Even in regions where high quality sequence analysis has been done in one species, as was the case for the ZAN/TFR2 region (11), we find that a dual-species sequence comparison incorporating EST data, cDNA data and automated gene prediction can: (i) identify genomic regions that have been misassembled or have recombined; (ii) extend cDNA sequences with higher confidence; and (iii) correct cDNA sequences that have frameshift or recombination errors. Human–mouse comparisons are also valuable for predicting the locations of regulatory elements. In addition to identifying putative promoter regions for genes whose regulation is unknown, we were able to verify that functionally characterized regulatory elements for ACHE and EPO stand out in large genomic comparisons.
Isochores are generally described as regions of DNA >300 kb that are homogenous in their nucleotide composition. Regions of high G-C content (>52% G-C) are classified as H3 isochores and are believed to comprise ∼3% of the genome, yet harbor 28% of the genes (reviewed in 38 and 39). The human genomic region we examined (ACHE to TFR2) has an average G-C content of 53.1% and is therefore a clear example of an H3 isochore. The orthologous mouse genomic DNA has a G-C content of 50%, which is to be expected given that the mouse genome has a lower average G-C content (40). An abundance of CpG islands in this region (Fig. 2B) and the presence of DNase hypersensitive sites near EPO (41) suggest that this segment is one of the sub-regions of 7q22 that replicates at the onset of S-phase (42). The high gene density typically associated with an H3 isochore (43) is not quite met in this region, which can be explained in part by the length of the zonadhesin gene and the abundance of repeat elements. Overall, the intergenic distances between the human and mouse genes in the ACHE/TFR2 region are quite similar, except for a 10.2 kb expansion seen in the human CIP1/EPHB4 region orthologous to the mouse intergenic region (positions 52210–57931, Fig. 2B). A 7.4 kb region of this expansion consists of sequence from what appears to be an insertion of a single Harlequin retroviral element.
The density of repeats found in this region of 7q22 (37.1% Alu and 50.2% total repetitive elements; Table 3) is higher than the average seen for H3 isochores (∼20% Alu and 35% total repetitive elements) and the average seen in the genome overall (44). In mouse, we find 36.3% of the sequence consists of repeats, of which 22.8% are SINEs. The trend of having fewer overall repeats in mouse compared with humans is consistent with most human–mouse genomic comparisons to date: 40 to 33% (45), 59 to 42% (9) and 32 to 14% (15); see Mallon et al. (46) for a summary of eight additional comparisons. An exception is the TCR-β V region, which shows 30% overall repeat content in human compared to 44% in mouse (47,48).
Table 3. Distribution of repetitive elements.
| Mouse (288 854 bp) | Human (258 784 bp) | ||||
| |
|
Number of elements |
Percentage of sequence |
Number of elements |
Percentage of sequence |
| SINEs | 537 | 22.79 | 383 | 37.79 | |
| Alu | – | – | 367 | 37.09 | |
| MIR | 3 | 0.13 | 16 | 0.70 | |
| B1 | 228 | 8.79 | – | – | |
| B2-B4 | 229 | 12.02 | – | – | |
| ID | 77 | 1.84 | – | – | |
| LINEs | 29 | 2.86 | 41 | 5.30 | |
| LINE1 | 18 | 2.34 | 11 | 2.09 | |
| LINE2 | 8 | 0.25 | 28 | 2.47 | |
| LTR elements | 40 | 3.15 | 10 | 4.12 | |
| MaLR | 27 | 2.27 | 1 | 0.01 | |
| Retroviruses | 5 | 0.24 | 7 | 1.57 | |
| MER4_group | 0 | 0.00 | 1 | 0.03 | |
| DNA elements | 9 | 0.51 | 11 | 1.13 | |
| MER1 type | 6 | 0.38 | 8 | 0.88 | |
| MER2 type | 3 | 0.14 | 0 | 0.00 | |
| Mariners | 0 | 0.00 | 1 | 0.03 | |
| Unclassified repeats | 7 | 0.54 | 0 | 0.00 | |
| Small RNAs | 6 | 0.17 | 1 | 0.11 | |
| Satellites | 24 | 1.25 | 0 | 0.00 | |
| Simple repeats | 183 | 4.05 | 61 | 1.19 | |
| Low complexity regions | 47 | 1.14 | 23 | 0.67 | |
| Total repeats | 36.34 | 50.17 | |||
A direct comparison of the distribution of mouse and human repetitive elements over the mouse region contained in BAC423o3 and BAC558e17 and the orthologous region in human. The comparison is made for the ACHE/TFR2 region spanning the first to last local alignments in Figure 2B (see PIP positions 6059–294912).
Several EST matches were observed in non-repetitive regions that have not been assigned to a gene (see http://web.uvic.ca/~bioweb/laj.html for details). Some of these reside close to the 5′ UTR of the Cip1, Rpp20 and Gnb2 genes, and the 5′ and 3′ end of Perq1; they likely represent parts of these genes. For example, the conserved portion of the region 5′ of the Cip1 gene (55–60 kb) corresponds to a single 5′ EST (BB592601) sequence containing one canonical splice site and no obvious ORFs. The two non-coding regions at 206–220 and 253–272 kb are relatively long for this gene-dense region, contain small conserved segments and yet appear to lack genes. The intergenic region corresponding to 253–272 kb consists of 56% repeat elements in mouse and 76% in human. The conserved segments here do not correspond to any EST sequences and thus may contain regulatory elements for GNB2 and BAF53B. The non-coding region found at 206–220 kb consists of 59 and 56% repeat elements in mouse and human, respectively. The conserved regions here do not correspond to any ESTs and are close to a region suggested to play a role in EPO regulation (Table 1) (49). In addition to the deficiency of ESTs and obvious ORFs, the abundance and spacing of repeat elements helps substantiate the lack of genes in these regions.
Male reproductive proteins are known to show a high level of divergence (50). Several sequence differences were noted between the mouse zonadhesin cDNA predicted from our genomic cDNA and the previously reported complete mouse cDNA sequence (MMU97068) (24). Furthermore, a comparison ignoring the duplicated partial D3 domains in the mouse shows that human and mouse zonadhesin genes share 61% amino acid identity and 71% nucleotide identity over the coding region. This is considerably lower than the 85% average coding DNA identity seen for other genes in this region (Table 2) and the 85% genome-wide average coding identity previously reported (51). The rapid evolution of the zonadhesin gene, the observed recombination between Gnb2 and Epo and the relatively high repeat content observed in this GC-rich isochore suggest that this is an unstable and dynamic region. Paradoxically, the conserved gene order between human and mouse underscore this region’s chromosomal stability during evolution. It is becoming clear that the genome cannot be treated as a uniform landscape, even within GC-rich isochores.
While any comparative genomic publication should provide readers with enough information to recreate, annotate and inspect the data on their own, this can be quite tedious and time-consuming, especially for long, gene-rich segments. The dot-plot is a universally accepted way of comparing and displaying large genomic regions. PIPs are being used as an additional means of presentation in a growing number of publications because of their utility in highlighting conserved regions and their informative, relatively compact output for short comparisons. However, while a dot-plot can display comparisons of virtually any length on a single page by compressing the scale, PIPs require a more-or-less fixed resolution to be legible (e.g., one page per 160 kb), which can cause large comparisons to exceed journal page limits. Laj effectively addresses this dilemma by displaying both a dot-plot and a PIP on a single page, regardless of the region’s size, but allowing the reader to ‘zoom in’ to see finer detail as needed (Fig. 3A and B).
Many large blocks of contiguous human sequence are becoming available, and they can be used as templates to compare and order partial sequence data from other mammalian genomes. To illustrate Laj’s usefulness for this purpose, Figure 3C uses Laj to display our unordered partial sequence data from BAC 139n8, orthologous to the EPO contig (see http://web.uvic.ca/~bioweb/laj.html for the ordered data). Based on the similarity in gene content, order and organization observed between human and mouse in this region of 7q22, it is likely that this region is also conserved in other mammals that diverged <80 million years ago. This a priori knowledge of gene order and structure will help significantly in ordering partial sequence data from other species, and demonstrates the need for tools that can display such information. The importance of considering evolutionary history before making assumptions when ordering partial sequences from more divergent species is seen in the comparison of human and Fugu rubripes genomic sequence in the PCOLCE region, which reveals conservation in the PCOLCE gene itself but no conserved linkage to the surrounding genes (52).
Java applets like Laj and Alfresco (18) can deliver easy access to interactive, informative displays of comparative genomic information. While these programs share some common features, their formats and areas of emphasis are quite different. Alfresco’s main goal is to serve as a graphical front end for various external analysis programs, while Laj is primarily intended to display results from PipMaker, and thus places more emphasis specifically on sequence alignments. [For a direct comparison of these applets in action on the BTK locus, see http://www.sanger.ac.uk/Software/Alfresco (MMU58105–HSU78027) and http://bio.cse.psu.edu/ (BTK)]. Tools like these will be ideal for researchers who want quick access to, and an interactive display of, the comparative genomic data that is becoming available for many disease regions at an accelerating rate.
Complete human and mouse genomic sequences will soon be freely available, prompting many groups to engage in comparative sequence analysis of their region of interest. The resulting publications will contain only the most novel findings; most of the hard-won observations will not appear in print. An electronic supplement to such a paper can present these additional observations, such as links to related network resources, as well as provide details that facilitate verification of the published conclusions. Moreover, the supplement can be corrected and updated after the journal publication appears. Centralized organism-wide archives of annotated genomic alignments present another potential use for tools like Laj. Such repositories already exist for enteric bacteria (53; http://globin.cse.psu.edu/enterix/) and a Caenorhabditis elegans to Caenorhabditis briggsae genomic alignment (17; http://www.cse.ucsc.edu/~kent/intronerator).
DDBJ/EMBL/GenBank accession nos: AF312032–AF312033
References
- 1.Johnson E.J., Scherer,S.W., Osborne,L., Tsui,L.C., Oscier,D., Mould,S. and Cotter,F.E. (1996) Molecular definition of a narrow interval at 7q22.1 associated with myelodysplasia. Blood, 87, 3579–3586. [PubMed] [Google Scholar]
- 2.Le Beau M.M., Espinosa,R.,III, Davis,E.M., Eisenbart,J.D., Larson,R.A. and Green,E.D. (1996) Cytogenetic and molecular delineation of a region of chromosome 7 commonly deleted in malignant myeloid diseases. Blood, 88, 1930–1935. [PubMed] [Google Scholar]
- 3.Fischer K., Frohling,S., Scherer,S.W., McAllister Brown,J., Scholl,C., Stilgenbauer,S., Tsui,L.C., Lichter,P. and Dohner,H. (1997) Molecular cytogenetic delineation of deletions and translocations involving chromosome band 7q22 in myeloid leukemias. Blood, 89, 2036–2041. [PubMed] [Google Scholar]
- 4.Fischer K., Brown,J., Scherer,S.W., Schramm,P., Stewart,J., Fugazza,G., Pascheberg,U., Peter,W., Tsui,L.C., Lichter,P. and Dohner,H. (1998) Delineation of genomic regions in chromosome band 7q22 commonly deleted in myeloid leukemias. Recent Results Cancer Res., 144, 46–52. [DOI] [PubMed] [Google Scholar]
- 5.Liang H., Fairman,J., Claxton,D.F., Nowell,P.C., Green,E.D. and Nagarajan,L. (1998) Molecular anatomy of chromosome 7q deletions in myeloid neoplasms: evidence for multiple critical loci. Proc. Natl Acad. Sci. USA, 9, 3781–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekelund J., Lichtermann,D., Hovatta,I., Ellonen,P., Suvisaari,J., Terwilliger,J.D., Juvonen,H., Varilo,T., Arajarvi,R., Kokko-Sahin,M.L., Lonnqvist,J. and Peltonen,L. (2000) Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum. Mol. Genet., 9, 1049–1057. [DOI] [PubMed] [Google Scholar]
- 7.Battey J., Jordan,E., Cox,D. and Dove,W. (1999) An action plan for mouse genomics. Nature Genet., 21, 73–75. [DOI] [PubMed] [Google Scholar]
- 8.Thomas J.W., Summers,T.J., Lee-Lin,S.Q., Maduro,V.V., Idol,J.R., Mastrian,S.D., Ryan,J.F., Jamison,D.C. and Green,E.D. (2000) Comparative genome mapping in the sequence-based era: early experience with human chromosome 7. Genome Res., 10, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martindale D.W., Wilson,M.D., Wang,D., Burke,R.D., Chen,X., Duronio,V. and Koop,B.F. (2000) Comparative genomic sequence analysis of the Williams syndrome region (LIMK1-RFC2) of human chromosome 7q11.23. Mamm. Genome, 11, 890–898. [DOI] [PubMed] [Google Scholar]
- 10.Harris N.L. (1997) Genotator: a workbench for sequence annotation. Genome Res., 7, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gloeckner G., Scherer,S., Schattevoy,R., Boright,A., Weber,J., Tsui,L.C. and Rosenthal,A. (1998) Large-scale sequencing of two regions in human chromosome 7q22: analysis of 650 kb of genomic sequence around the EPO and CUTL1 loci reveals 17 genes. Genome Res., 8, 1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J. and Madden,T.L. (1997) PowerBLAST: a new network BLAST application for interactive or automated sequence analysis and annotation. Genome Res., 7, 649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuehl P.M., Weisemann,J.M., Touchman,J.W., Green,E.D. and Boguski,M.S. (1999) An effective approach for analyzing ‘prefinished’ genomic sequence data. Genome Res., 9, 189–194. [PMC free article] [PubMed] [Google Scholar]
- 14.Glusman G. and Lancet,D. (2000) GESTALT: a workbench for automatic integration and visualization of large-scale genomic sequence analyses. Bioinformatics, 16, 482–483. [DOI] [PubMed] [Google Scholar]
- 15.Lund J., Chen,F., Hua,A., Roe,B., Budarf,M., Emanuel,B.S. and Reeves,R.H. (2000) Comparative sequence analysis of 634 kb of the mouse chromosome 16 region of conserved synteny with the human velocardiofacial syndrome region on chromosome 22q11.2. Genomics, 63, 374–383. [DOI] [PubMed] [Google Scholar]
- 16.Kent W.J. and Zahler,A.M. (2000) Conservation, regulation, synteny and introns in a large-scale C. briggsae–C. elegans genomic alignment. Genome Res., 10, 1115–1125. [DOI] [PubMed] [Google Scholar]
- 17.Kent W.J. and Zahler,A.M. (2000) The Intronerator: exploring introns and alternative splicing in Caenorhabditis elegans. Nucleic Acids Res., 28, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jareborg N. and Durbin,R. (2000) Alfresco—A workbench for comparative genomic sequence analysis. Genome Res., 10, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouck J., Miller,W., Gorrell,J.H., Muzny,D. and Gibbs,R.A. (1998) Analysis of the quality and utility of random shotgun sequencing at low redundancies. Genome Res., 8, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz S., Zhang,Z., Frazer,K.A., Smit,A., Riemer,C., Bouck,J., Gibbs,R., Hardison,R. and Miller,W. (2000) PipMaker—A web server for aligning two genomic DNA sequences. Genome Res., 10, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burge C. and Karlin,S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- 22.Schuler G.D. (1997) Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J. Mol. Med., 75, 694–698. [DOI] [PubMed] [Google Scholar]
- 23.Shapira M., Tur-Kaspa,I., Bosgraaf,L., Livni,N., Grant,A.D., Grisaru,D., Korner,M., Ebstein,R.P. and Soreq,H. (2000) A transcription-activating polymorphism in the ACHE promoter associated with acute sensitivity to anti-acetylcholinesterases. Hum. Mol. Genet., 9, 1273–1281. [DOI] [PubMed] [Google Scholar]
- 24.Gao Z. and Garbers,D.L. (1998) Species diversity in the structure of zonadhesin, a sperm-specific membrane protein containing multiple cell adhesion molecule-like domains. J. Biol. Chem., 273, 3415–3421. [DOI] [PubMed] [Google Scholar]
- 25.Rebhan M., Chalifa-Caspi,V., Prilusky,J. and Lancet,D. (1998) GeneCards: a novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics, 14, 656–664. [DOI] [PubMed] [Google Scholar]
- 26.Maglott D.R., Katz,K.S., Sicotte,H. and Pruitt,K.D. (2000) NCBI’s LocusLink and RefSeq. Nucleic Acids Res., 28, 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko M.S., Kitchen,J.R., Wang,X., Threat,T.A., Hasegawa,A., Sun,T., Grahovac,M.J., Kargul,G.J., Lim,M.K., Cui,Y. et al. (2000) Large-scale cDNA analysis reveals phased gene expression patterns during preimplantation mouse development. Development, 127, 1737–1749. [DOI] [PubMed] [Google Scholar]
- 28.Li Y., Camp,S., Rachinsky,T.L., Getman,D. and Taylor,P. (1991) Gene structure of mammalian acetylcholinesterase. Alternative exons dictate tissue-specific expression. J. Biol. Chem., 266, 23083–23090. [PubMed] [Google Scholar]
- 29.Atanasova E., Chiappa,S., Wieben,E. and Brimijoin,S. (1999) Novel messenger RNA and alternative promoter for murine acetylcholinesterase. J. Biol. Chem., 274, 21078–21084. [DOI] [PubMed] [Google Scholar]
- 30.Edwalds-Gilbert G., Veraldi,K.L. and Milcarek,C. (1997) Alternative poly(A) site selection in complex transcription units: means to an end? Nucleic Acids Res., 25, 2547–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo Z.D., Camp,S., Mutero,A. and Taylor,P. (1998) Splicing of 5′ introns dictates alternative splice selection of acetylcholinesterase pre-mRNA and specific expression during myogenesis. J. Biol. Chem., 273, 28486–28495. [DOI] [PubMed] [Google Scholar]
- 32.Chretien S., Duprez,V., Maouche,L., Gisselbrecht,S., Mayeux,P. and Lacombe,C. (1997) Abnormal erythropoietin (Epo) gene expression in the murine erythroleukemia IW32 cells results from a rearrangement between the G-protein β2 subunit gene and the Epo gene. Oncogene, 15, 1995–1999. [DOI] [PubMed] [Google Scholar]
- 33.Kleiderlein J.J., Nisson,P.E., Jessee,J., Li,W.B., Becker,K.G., Derby,M.L., Ross,C.A. and Margolis,R.L. (1998) CCG repeats in cDNAs from human brain. Hum. Genet., 103, 666–673. [DOI] [PubMed] [Google Scholar]
- 34.Tunnacliffe A., Jones,C., Le Paslier,D., Todd,R., Cherif,D., Birdsall,M., Devenish,L., Yousry,C., Cotter,F.E. and James,M.R. (1999) Localization of Jacobsen syndrome breakpoints on a 40-Mb physical map of distal chromosome 11q. Genome Res., 9, 44–52. [PMC free article] [PubMed] [Google Scholar]
- 35.Jones C., Mullenbach,R., Grossfeld,P., Auer,R., Favier,R., Chien,K., James,M., Tunnacliffe,A. and Cotter,F. (2000) Co-localisation of CCG repeats and chromosome deletion breakpoints in Jacobsen syndrome: evidence for a common mechanism of chromosome breakage. Hum. Mol. Genet., 9, 1201–1208. [DOI] [PubMed] [Google Scholar]
- 36.Pratt W.S., Crawley,S., Hicks,J., Ho,J., Nash,M., Kim,Y.S., Gum,J.R. and Swallow,D.M. (2000) Multiple transcripts of MUC3: evidence for two genes, MUC3A and MUC3B. Biochem. Biophys. Res. Commun., 275, 916–923. [DOI] [PubMed] [Google Scholar]
- 37.Benson D.A., Karsch-Mizrachi,I., Lipman,D.J., Ostell,J., Rapp,B.A. and Wheeler,D.L. (2000) GenBank. Nucleic Acids Res., 28, 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernardi G. (1995) The human genome: organization and evolutionary history. Annu. Rev. Genet., 29, 445–476. [DOI] [PubMed] [Google Scholar]
- 39.Gardiner K. (1996) Base composition and gene distribution: critical patterns in mammalian genome organization. Trends Genet., 12, 519–524. [DOI] [PubMed] [Google Scholar]
- 40.Robinson M., Gautier,C. and Mouchiroud,D. (1997) Evolution of isochores in rodents. Mol. Biol. Evol., 14, 823–828. [DOI] [PubMed] [Google Scholar]
- 41.Beru N., McDonald,J. and Goldwasser,E. (1989) Activation of the erythropoietin gene due to the proximity of an expressed gene. DNA, 8, 253–259. [DOI] [PubMed] [Google Scholar]
- 42.Federico C., Saccone,S. and Bernardi,G. (1998) The gene-richest bands of human chromosomes replicate at the onset of the S-phase. Cytogenet. Cell Genet., 80, 83–88. [DOI] [PubMed] [Google Scholar]
- 43.Gardiner K. (1995) Human genome organization. Curr. Opin. Genet. Dev., 5, 315–322. [DOI] [PubMed] [Google Scholar]
- 44.Duret L., Mouchiroud,D. and Gautier,C. (1995) Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-rich isochores. J. Mol. Evol., 40, 308–317. [DOI] [PubMed] [Google Scholar]
- 45.Ellsworth R.E., Jamison,D.C., Touchman,J.W., Chissoe,S.L., Braden Maduro,V.V., Bouffard,G.G., Dietrich,N.L., Beckstrom-Sternberg,S.M., Iyer,L.M., Weintraub,L.A. et al. (2000) Comparative genomic sequence analysis of the human and mouse cystic fibrosis transmembrane conductance regulator genes. Proc. Natl Acad. Sci. USA, 97, 1172–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mallon A., Platzer,M., Bate,R., Gloeckner,G., Botcherby,M.R., Nordsiek,G., Strivens,M.A., Kioschis,P., Dangel,A., Cunningham,D., et al. (2000) Comparative genome sequence analysis of the Bpa/Str region in mouse and man. Genome Res., 10, 758–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowen L., Koop,B.F. and Hood,L. (1996) The complete 685-kilobase DNA sequence of the human β T cell receptor locus. Science, 272, 1755–1762. [DOI] [PubMed] [Google Scholar]
- 48.Brown G., Martindale,D.W., Wilson,M.D. and Koop,B.F. (2000) In Sankoff,D. and Nadeau,J.H. (eds), Comparative Genomics. Kluwer Academic Press, Dordrecht, The Netherlands, Vol. 1, pp. 59–69.
- 49.Ebert B.L. and Bunn,H.F. (1999) Regulation of the erythropoietin gene. Blood, 94, 1864–1877. [PubMed] [Google Scholar]
- 50.Wyckoff G.J., Wang,W. and Wu,C.I. (2000) Rapid evolution of male reproductive genes in the descent of man. Nature, 403, 304–309. [DOI] [PubMed] [Google Scholar]
- 51.Makalowski W. and Boguski,M.S. (1998) Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc. Natl Acad. Sci. USA, 95, 9407–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riboldi Tunnicliffe G., Gloeckner,G., Elgar,G.S., Brenner,S. and Rosenthal,A. (2000) Comparative analysis of the PCOLCE region in Fugu rubripes using a new automated annotation tool. Mamm. Genome, 11, 213–219. [DOI] [PubMed] [Google Scholar]
- 53.Florea L., Riemer,C., Schwartz,S., Zhang,Z., Stojanovic,N., Miller,W. and McClelland,M. (2000) Web-based visualization tools for bacterial genome alignments. Nucleic Acids Res., 28, 3486–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sciacchitano S. and Taylor,S.I. (1997) Cloning, tissue expression and chromosomal localization of the mouse IRS-3 gene. Endocrinology, 138, 4931–4940. [DOI] [PubMed] [Google Scholar]
- 55.Getman D.K., Mutero,A., Inoue,K. and Taylor,P. (1995) Transcription factor repression and activation of the human acetylcholinesterase gene. J. Biol. Chem., 270, 23511–23519. [DOI] [PubMed] [Google Scholar]
- 56.Rossman T.G. and Wang,Z. (1999) Expression cloning for arsenite-resistance resulted in isolation of tumor-suppressor fau cDNA: possible involvement of the ubiquitin system in arsenic carcinogenesis. Carcinogenesis, 20, 311–316. [DOI] [PubMed] [Google Scholar]
- 57.Caron L., Rousseau,F., Gagnon,E. and Isenring,P. (2000) Cloning and functional characterization of a cation-Cl-cotransporter interacting protein. J. Biol. Chem., 275, 32027–32036. [DOI] [PubMed] [Google Scholar]
- 58.Nakai K. and Horton,P. (1999) PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci., 24, 34–36. [DOI] [PubMed] [Google Scholar]
- 59.Yi J. and Beckerle,M.C. (1998) The human TRIP6 gene encodes a LIM domain protein and maps to chromosome 7q22, a region associated with tumorigenesis. Genomics, 49, 314–316. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y., Dooher,J.E., Koedood Zhao,M. and Gilmore,T.D. (1999) Characterization of mouse Trip6: a putative intracellular signaling protein. Gene, 234, 403–409. [DOI] [PubMed] [Google Scholar]
- 61.Bennett B.D., Wang,Z., Kuang,W.J., Wang,A., Groopman,J.E., Goeddel,D.V. and Scadden,D.T. (1994) Cloning and characterization of HTK, a novel transmembrane tyrosine kinase of the EPH subfamily. J. Biol. Chem., 269, 14211–14218. [PubMed] [Google Scholar]
- 62.Bennett B.D., Zeigler,F.C., Gu,Q., Fendly,B., Goddard,A.D., Gillett,N. and Matthews,W. (1995) Molecular cloning of a ligand for the EPH-related receptor protein–tyrosine kinase Htk. Proc. Natl Acad. Sci. USA, 92, 1866–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerety S.S., Wang,H.U., Chen,Z.F. and Anderson,D.J. (1999) Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell, 4, 403–414. [DOI] [PubMed] [Google Scholar]
- 64.Helbling P.M., Saulnier,D.M. and Brandli,A.W. (2000) The receptor tyrosine kinase EphB4 and ephrin-B ligands restrict angiogenic growth of embryonic veins in Xenopus laevis. Development, 127, 269–278. [DOI] [PubMed] [Google Scholar]
- 65.Andres A.C., Reid,H.H., Zurcher,G., Blaschke,R.J., Albrecht,D. and Ziemiecki,A. (1994) Expression of two novel eph-related receptor protein tyrosine kinases in mammary gland development and carcinogenesis. Oncogene, 9, 1461–1467. [PubMed] [Google Scholar]
- 66.Ciossek T., Lerch,M.M. and Ullrich,A. (1995) Cloning, characterization and differential expression of MDK2 and MDK5, two novel receptor tyrosine kinases of the eck/eph family. Oncogene, 11, 2085–2095. [PubMed] [Google Scholar]
- 67.Ashworth A., Malik,A.N., Walkley,N.A., Kubota,H. and Willison,K.R. (1994) The Tcp-1-related gene, Cctg, maps to mouse chromosome 3. Mamm. Genome, 5, 509–510. [DOI] [PubMed] [Google Scholar]
- 68.Hardy D.M. and Garbers,D.L. (1995) A sperm membrane protein that binds in a species-specific manner to the egg extracellular matrix is homologous to von Willebrand factor. J. Biol. Chem., 270, 26025–26028. [DOI] [PubMed] [Google Scholar]
- 69.Gao Z., Harumi,T. and Garbers,D.L. (1997) Chromosome localization of the mouse zonadhesin gene and the human zonadhesin gene (ZAN). Genomics, 41, 119–122. [DOI] [PubMed] [Google Scholar]
- 70.Lacombe C. and Mayeux,P. (1999) The molecular biology of erythropoietin. Nephrol. Dial. Transplant., 14, 22–28. [DOI] [PubMed] [Google Scholar]
- 71.Blanchard K.L., Acquaviva,A.M., Galson,D.L. and Bunn,H.F. (1992) Hypoxic induction of the human erythropoietin gene: cooperation between the promoter and enhancer, each of which contains steroid receptor response elements. Mol. Cell. Biol., 12, 5373–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eder P.S., Kekuda,R., Stolc,V. and Altman,S. (1997) Characterization of two scleroderma autoimmune antigens that copurify with human ribonuclease P. Proc. Natl Acad. Sci. USA, 94, 1101–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freund C., Dotsch,V., Nishizawa,K., Reinherz,E.L. and Wagner,G. (1999) The GYF domain is a novel structural fold that is involved in lymphoid signaling through proline-rich sequences. Nature Struct. Biol., 6, 656–660. [DOI] [PubMed] [Google Scholar]
- 74.Offermanns S. and Simon,M.I. (1998) Genetic analysis of mammalian G-protein signalling. Oncogene, 17, 1375–1381. [DOI] [PubMed] [Google Scholar]
- 75.Zhao K., Wang,W., Rando,O.J., Xue,Y., Swiderek,K., Kuo,A. and Crabtree,G.R. (1998) Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell, 95, 625–636. [DOI] [PubMed] [Google Scholar]
- 76.Kawabata H., Yang,R., Hirama,T., Vuong,P.T., Kawano,S., Gombart,A.F. and Koeffler,H.P. (1999) Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem., 274, 20826–20832. [DOI] [PubMed] [Google Scholar]