

Abstract
ROR-alpha is an orphan nuclear receptor, the inactivation of which cell-autonomously blocks differentiation of cerebellar Purkinje cells with a secondary loss of granule neurons. As part of our ENU mutagenesis screen we isolated the recessive tmgc26 mouse mutant, characterized by early onset progressive ataxia, cerebellar degeneration and juvenile lethality. Detailed analysis of the tmgc26−/− cerebella revealed Purkinje cell and granule cell abnormalities, and defects in molecular layer interneurons and radial glia. Chimera studies suggested a cell-autonomous effect of the tmgc26 mutation in Purkinje cells and molecular layer interneurons, and a non-cell autonomous effect in granule cells. The mutation was mapped to a 13Mb interval on chromosome 9, a region that contains the ROR-alpha gene. Sequencing of genomic DNA revealed a T-to-A transition in exon 5 of the ROR-alpha gene, resulting in a nonsense mutation C257X and severe truncation of the ROR-alpha protein. Together, our data identify new roles for ROR-alpha in molecular layer interneurons and radial glia development and introduce tmgc26 as a novel ROR-alpha allele suitable for further delineating molecular mechanisms of ROR-alpha action.
Keywords: mouse, ENU mutants, cerebellum, development, ROR-alpha
Introduction
During the last decade, analysis of mouse ENU mutants has significantly contributed to our understanding of mechanisms of vertebrate development (Justice, 2000; Acevedo-Arozena et al., 2008). In the central nervous system, this approach has been especially successful for identifying molecular and genetic mechanisms that drive cerebellar development. Since the cerebellum is a major center of coordination, mutants with cerebellar abnormalities are easily identifiable based on their ataxic phenotype (Chizhikov and Millen, 2003). Additionally, the adult mouse cerebellum contains only a few cell types, including granule cells, Purkinje cells, molecular layer interneurons and radial glia cells, all with distinct morphologies and each located in a discrete lamina (Goldowitz and Hamre, 1998; Maricich and Herrup, 1999; Carletti and Rossi, 2008; Schilling et al., 2008). Therefore, the cellular basis of ataxia in these mutants is relatively easy to identify.
Although the adult mouse cerebellum is a comparatively simple structure, cerebellar development is a complex multistep process precisely regulated by multiple genes (reviewed in Wang and Zoghbi, 2001, Hevner et al., 2006; Millen and Gleeson, 2008; Leto et al., 2008). One gene critically involved in cerebellar development is the orphan nuclear receptor ROR-alpha. Most of our knowledge about the role of ROR-alpha in the central nervous system comes from analysis of the staggerer (Rorasg/sg) mutant mouse (Sidman et al., 1962; Hamilton et al., 1996; Gold at al., 2007). The adult staggerer cerebellum is smaller than that of control mice and contains disorganized and immature Purkinje cells. In addition, during development a significant portion of both granule and Purkinje cells die in the staggerer cerebellum (reviewed in Gold et al., 2007). Chimera studies demonstrated that the primary cerebellar defect in staggerer is intrinsic to Purkinje cells and that the granule cell phenotype is secondary to loss of Purkinje cells (Herrup, 1983).
Here we describe the tmgc26 cerebellar mutant generated during our ENU mutagenesis screen (Goldowitz et al., 2004). Using a positional cloning strategy, we identified the tmgc26 mutation as a new allele of ROR-alpha and described Purkinje cell, granule cell, molecular layer interneuron and radial glia abnormalities in the cerebellum of this mutant. In addition, we generated and analyzed tmgc26 chimeras to dissect cell-autonomous and non-cell autonomous effects of this mutation in the developing mouse cerebellum.
Materials and Methods
Mice
tmgc26 mice were generated during our ENU mutagenesis screen (Goldowitz et al., 2004). This autosomal recessive mutation was detected in the progeny of non-inbred D7R75M males given one dose of 125 mg/kg ENU. Staggerer mice (Rorasg) were on a B6C3Fe background and were obtained from Jackson Laboratory (Bar Harbor, ME, USA). For production of chimeras, Rosa26 (Soriano, 1999) and C57BL6 or more outbred ICR mice were used. Rosa26 and C57BL6 mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and ICR mice were obtained from Charles River Laboratories (Wilmington, MA, USA). All mouse procedures followed the policies of the University of Chicago and the NIH Guidelines on Care and Use of Laboratory Animals and were in accordance with the applicable portions of the Animal Welfare Act.
Histology and antibody staining
For histological analysis, cerebella were fixed in 4% PFAin PBS for 12–24 hours, then in 10% formalin for 12 hours, sunk in 30%sucrose in PBS, and embedded in gelatin (10% gelatin, 30% sucrosein PBS). The gelatin blocks were fixed in sucrose formalinsolution (30% sucrose, 10% formalin in PBS) at 4°C for 1–2 days. Then blocks were frozen on dry ice, seriallysectioned at 20 μm on a freezing microtome and sections were stained with cresyl violet.
For antibody staining, mice were deeply anesthetized with Euthasol (40 mg/kg body weight, Delmarva Laboratories Inc., Midlothian, VA, USA) and then perfused transcardially with cold 4% PFA. The cerebella were removed and fixed in cold 4% PFA overnight, washed in PBS, sunk in 30% sucrose and frozen in OCT. Cerebella were serially sectioned sagittally at 12 μm with a cryostat, then mounted on slides and processed for immunohistochemistry essentially as described previously (Chizhikov et al., 2006). Briefly, slides were dried at room temperature for 20 min., then washed in PBS and incubated in blocking solution (PBS containing 1% normal goat serum (Sigma-Aldrich, St Louis, MO, USA) and 0.1% Triton X-100) for 1 hour at room temperature. Next, they were incubated at 4°C overnight with primary antibodies diluted in blocking solution. The following primary antibodies were used: rabbit anti-BLBP (1:300, Chemicon International, Inc., Temecula, CA, USA), rabbit anti-Pax2 (1:200, Zymed, San Francisco, CA, USA), rabbit anti-Calbindin (1:500, Chemicon International, Inc., Temecula, CA, USA), rabbit anti-GFAP (1:1000, Dako, Glostrup, Denmark) and mouse anti-Parvalbumin (1:1000, Swant, Swant, Bellinzona, Switzerland). More detailes about antibodies production and specificity tests are given in Suppl. Table 1. Following incubation with primary antibodies, slides were washed in PBS and incubated with species appropriate fluorescent-dye conjugated secondary antibodies (1:200, Jackson ImmunoResearch, West Grove, PA, USA). In some cases, sections were counterstained with DAPI (Sigma-Aldrich, St Louis, MO, USA) to visualize tissue.
BrdU analysis
Proliferating cells in the postnatal EGL were labeled with the thymidine analog 5-bromo-2-deoxyuridine (BrdU). Mice were given a single intraperitoneal injection of BrdU (100 mg/kg body weight, Zymed, San Francisco, CA, USA) 1 hour prior to euthanasia. Cerebella were fixed, sectioned and stained with mouse anti-BrdU antibody (1:5, Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA, USA, see Suppl. Table 1 for more details regarding this antibody) exactly as described above for antibody staining. The only difference was that for BrdU analysis, slides were incubated in 2N HCl for 20 min. at room temperature and rinsed in 0.1M Borate buffer (pH 8.5) prior to incubation in blocking solution.
Generation of tmgc26<->Rosa26 chimeras
Experimental mouse chimeras were generated as described previously (Goldowitz and Mullen, 1982; Goldowitz, 1989). In brief, tmgc26 mutant embryos were produced by breeding tmgc26+/− mice, yielding a mix of homozygous mutant, heterozygous, and wild-type embryos. The wild type component of the chimera was from homozygous Rosa26 mice crossed to C57BL6 or ICR mice, resulting in obligate heterozygote Rosa26 embryos. Four- to eight-cell embryos from the tmgc26+/− mating were collected and each embryo was put in juxtaposition with a wild-type Rosa26 embryo so that they could fuse following an overnight culture. Successfully fused chimeric blastocyst embryos were then transplanted into the uterine horn of pseudo-pregnant host ICR females. Litters of experimental chimeras were born and the pups (total n=23 individuals) were monitored for gross behavioral assessment and coat color chimerism. The animals were allowed to develop until either P19 or P21 and were prepared for histology at that time.
Immunohistochemical and Beta-galactosidase staining of tmgc26<->Rosa26 chimeras
Chimeras were perfused and their cerebella were embedded in OCT as described above for antibody staining. Brains were then sectioned in the sagittal plane at 14 μm. Simultaneous visualization of β-gal activity and antibody staining were performed in a multi step method. First, sections were incubated with primary antisera in blocking solution (0.1M PBS containing 5% normal serum, 2% BSA, and 3% Triton X-100) overnight at room temperature. Following washes, a short postfixation with 4% PFA for 10 min, and subsequent washes, the tissue was stained for β-gal activity. Detection of β-gal in tissue sections was accomplished using 5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) as the chromogenic substrate. Sections were incubated in reaction buffer (0.1M PBS containing 1mg/ml X-Gal, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 0.02% Nonidet P-40, 0.01% sodium deoxycholate, and 3% Triton X-100) at 37°C for 4hrs-overnight. Slides were then washed in PBST (0.1M PBS with 0.3% Triton X-100) and the tissue was blocked again with blocking buffer. Immunoreactivity was visualized using the Vectastain Elite ABC Kit (Vector Labs, Burlingame, CA, USA) and the appropriate secondary antibody, following the manufacturer’s protocol with DAB as chromogen. Primary antisera used for these analyses included: rabbit anti-Calbindin (1:500, Chemicon International, Inc., Temecula, CA, USA), mouse anti-Parvalbumin (1:1000, Swant, Swant, Bellinzona, Switzerland) and rabbit anti-Pax6 (1:1000, Covance, Berkeley, CA, USA) (additional details regarding these antibodies are presented in Suppl. Table 1).
Identification of the tmgc26 mutation
tmgc26+/− animals were crossed to MOLF/Ei mice (Jackson Laboratory, Bar Harbor, ME, USA). F1 heterozygotes were intercrossed and produced 26 phenotypically affected F2 mice. Genomic DNA was extracted from tails/livers using the PUREGENE Purification Kit (Gentra Systems). SNP typing was performed as per the standard protocol by the Genetic Resources Core Facility at Johns Hopkins University using the Illumina Medium Density Linkage Panel, which consists of 1,449 SNPs (Valdar et al., 2006). Linkage analysis was performed across each chromosome using Allegro 2.0 (Gudbjartsson et al., 2005). Parametric LOD scores were calculated assuming a fully penetrant recessive mode of inheritance and a disease allele frequency of 1/10,000.
DNA sequencing
PCR was performed according to standard protocols. Primers and conditions used in the current study are available upon request. Bidirectional sequencing of Sephadex G-50 (Amersham Biosciences, Buckinghamshire, UK) purified PCR products was performed with the ABI BigDye Terminator Sequencing Kit (Applied Biosystems, Foster City, CA) and an automated capillary array sequencer (ABI 3100, Applied Biosystems).
Data analysis
Histological preparations were examined under a Zeiss Axioplan 2 microscope. Images were taken using a Hamamatsu C4742-95 digital camera and Axiovision Rel 4.6 (Zeiss) Software. Adobe Photoshop 7.0 was used to assemble the final plates. Sections from at least 3 wild type and mutant mice were analyzed at each developmental stage. BrdU positive cells in the EGL of wild type and tmgc26−/− mice were counted and analyzed as previously described (Faust, 2004; Pogoriler et al., 2006). BrdU-positive cells were counted on midsagittal sections in the same location in the anterior cerebellum (which corresponds to pink box in Fig. 3). For each animal, the number of BrdU-positive cells per 0.1 mm of EGL was calculated in three non-adjacent sections spaced 50–100 μm apart. Mean values obtained from multiple sections from each animal were averaged and used for statistical analysis. All quantitative data are expressed as the mean + SD. Statistical analysis wasperformed using the two tailed Student’s t test (Excel, Microsoft) or Fisher’s exact test (InStat 3.0, GraphPad Software, La Jolla, CA, USA), as required. In all instances, P<0.05 was considered to be statistically significant.
Figure 3. Proliferation of granule cell progenitors in the EGL of tmgc26−/− mutants.
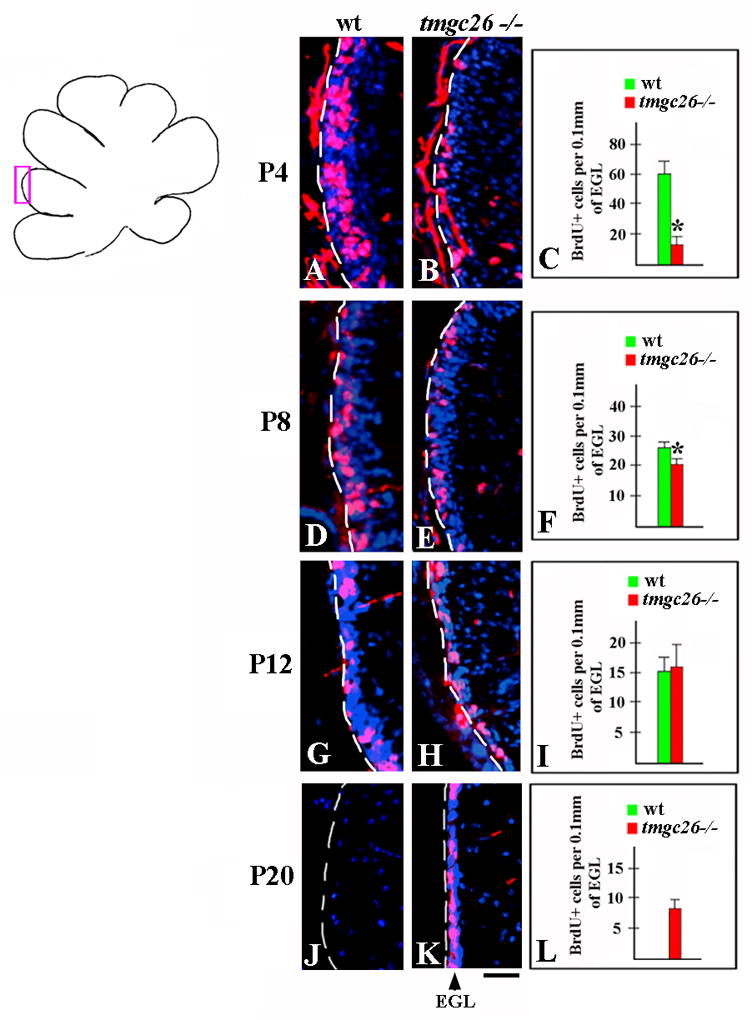
Sagittal sections from wild type (A, D, G, J) and tmgc26 −/− mutant mice (B, E, H, K) at the indicated stages co-stained with DAPI (blue) and anti-BrdU antibody (red). All panels correspond to the pink box in the adjacent diagram. The red staining outside the EGL is nonspecific staining of the pia. (C, F, I, L) Quantification of numbers of BrdU+ cells per 0.1mm of EGL in wild type and tmgc26 −/− mutant mice. Reduced proliferation of granule cell progenitors in tmgc26 mutants was observed at P4 and P8 but not at P12. * P<0.05, two tailed Student’s t test. At P20, there was no EGL in wild type mice (J). In contrast, EGL containing numerous proliferating granule cell progenitors was still detected in tmgc26−/− mice at that time (K and L). Scale bar: 15 μm
Results
tmgc26 is a new ENU mutation that affects the cerebellum
The tmgc26 mutation was identified during our ENU mutagenesis screen (Goldowitz et al., 2004). tmgc26−/− mice demonstrated severe early onset progressive ataxia and hindlimb paralysis. Many homozygous mutants died starting at postnatal day 12 (P12) with rare mutants surviving past weaning. Analysis of tmgc26−/− mutants that survived beyond the 3rd postnatal week revealed a diminutive cerebellum with rudimentary foliation (Fig. 1A–D) and a reduced number of granule cells (Fig. 1E, F). In contrast to adult wild type mice, where Purkinje cells form a monolayer, in tmgc26 mutants many Purkinje cells were detected in ectopic positions within the internal granule cell layer (IGL) (Fig. 1E, F). Molecular layer abnormalities were also evident in tmgc26 mutant mice. In wild type P21 cerebellum, the molecular layer is populated by numerous interneurons, including basket and stellate cells (Carletti and Rossi, 2008). The molecular layer of tmgc26 homozygous mice was very thin and contained very few cells (Fig. 1G and H). In addition, the vast majority of cells found in tmgc26 mutant molecular layer expressed a granule cell marker Pax6 (Friedel et al., 2007), suggesting that they were ectopic granule cells rather than molecular layer interneurons (Fig. 1I and J). Taken together, these data suggest loss or displacement of basket and stellate cells, in tmgc26 mutant cerebellum. Heterozygous mice had no obvious behavioral or morphological phenotypes.
Figure 1. Cerebellar abnormalities in tmgc26−/− mutants.

Dorsal whole-mount views (A, B) and cresyl violet stained sagittal sections of the cerebellar vermis (C–H) of wild type (A, C, E, G) and tmgc26−/− mutant mice (B, D, F, H) at P21. The cerebellum of a tmgc26−/− mouse is small and shows reduced foliation.
(E, F) The panels correspond to the pink box in the adjacent diagram. In contrast to wild type mice, in tmgc26−/− mutants, Purkinje cells (PC) are mixed with granule cells within the internal granule cell layer (IGL, indicated by bracket).
(G, H) Higher magnification of the region corresponding to the pink boxes in panels E and F. The molecular layer (ML) is thin in tmgc26−/− mutants. Numerous molecular layer interneurons (arrowheads) were present in the wild type molecular layer (G) but very few cells were found in the the tmgc26−/− mutant molecular layer (H).
(I, J) Sagittal sections from P21 wild type (I) and tmgc26 −/− mutant mice (J) co-stained with DAPI (blue) and Pax6 antibody (red). In the wild type molecular layer, numerous Pax6-negative interneurons were detected (arrows in panel I). Rare cells detected in the tmgc26−/− molecular layer expressed the granule cell marker Pax6 (arrowheads in panel J) suggesting that they were mislocated granule cells. Scale bar: A, B, 2 mm; C, D, 630 μm; E, F, 120 μm; G–J, 35 μm.
To better understand how the tmgc26 mutation affects cerebellar development, we performed detailed analysis of major cerebellar cellular populations, including Purkinje cells, granule cells, molecular layer interneurons and radial glia, in adult and developing tmgc26−/− mutants.
The tmgc26 mutation affects development of Purkinje cells
We compared the development of Purkinje cells in wild type and tmgc26−/− mutants using an anti-Calbindin antibody, which specifically labels Purkinje cells in the cerebellum. During early postnatal development, Purkinje cells begin forming a monolayer below the EGL and undergo a period of major dendritic outgrowth (Goldowitz and Hamre, 1998). At P8, wild type Purkinje cells appeared in a monolayer with clearly identifiable axons extending toward the cerebellar surface (Fig. 2A). In contrast, in tmgc26−/− mutant cerebella, Purkinje cells were multilayered and small compared to wild type Purkinje cells (Fig. 2B). By this developmental stage, tmgc26 −/− Purkinje cells had begun dendrite formation, although their dendrites were smaller and less complex than that in wild type littermates (Fig. 2A, B). At P21, mutant Purkinje cells remained multilayered. They were still small and their dendrites were virtually unrecognizable (Fig. 2C, D), suggesting dendrite degeneration.
Figure 2. Purkinje cell abnormalities in tmgc26−/− mutants.
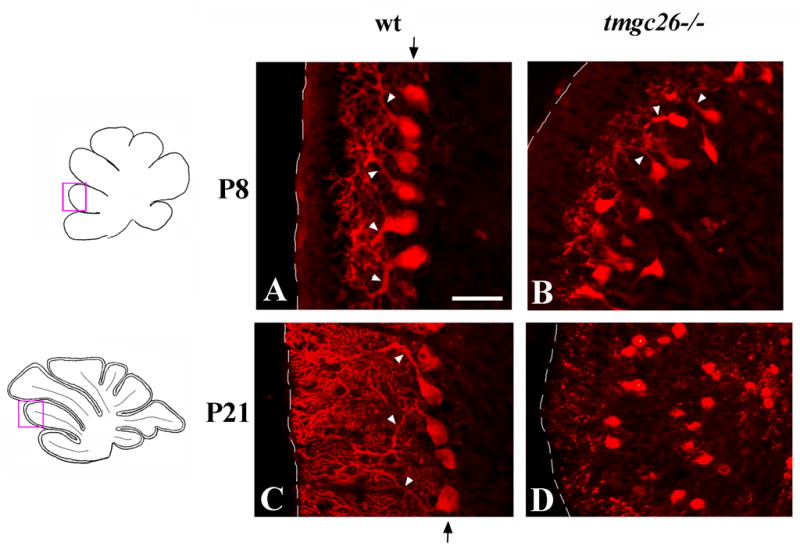
Sagittal sections from wild type (A, C) and tmgc26−/− mutant mice (B, D) at the indicated stages stained with anti-Calbindin antibody. All panels correspond to the pink boxes in the adjacent diagrams. Arrowheads point to dendrites of Purkinje cells. At P8, dendrites of tmgc26 −/− mutant Purkinje cells are small and less complex than those of wild type Purkinje cells (B). At P21, dendrites of tmgc26−/− mutant Purkinje cells are unrecognizable. Arrow (A, C) points to the Purkinje cell layer in wild type mice. In tmgc26−/− mutants Purkinje cells do not form a monolayer (B, D). Scale bar: A, B, 35 μm; C, D, 40 μm.
The tmgc26 mutation affects proliferation of granule cells during early postnatal development
Since the granule cell population was dramatically reduced in tmgc26−/− mutant cerebella, we investigated proliferation of granule cell progenitors in this mutant. During cerebellar development, granule cell progenitors form the external granule cell layer (EGL) where they proliferate extensively and then differentiate and migrate into the IGL, their final destination in the adult cerebellum (reviewed in Chizhikov and Millen, 2003). At P4, we observed a dramatic reduction in the proliferation of granule cell progenitors in tmgc26−/−mutants compared to wild type littermates. There was a 76% decrease in number of BrdU+ cells in the EGL in tmgc26−/− mutants compared to wild type littermates (60.5+7.5 BrdU+ cells per 0.1mm of EGL in wild type mice (n=4 mice) versus 14.7+3.1 BrdU+ cells per 0.1mm of EGL in tmgc26−/− mutants (n=3 mice), two tailed Student’s t test P=0.0003) (Fig. 3A–C). At P8 the difference was less obvious (a 26% decrease in tmgc26−/− mutants, 25.8±2.5 BrdU+ cells per 0.1mm of EGL in wild type mice (n=4 mice) versus 19.3+2.5 BrdU+ cells per 0.1mm of EGL in tmgc26−/− mutants (n=4 mice), two tailed Student’s t test P=0.01) (Fig. 3D–F). At P12, there was no significant difference between wild type and tmgc26−/− mice (15.8+2.2 BrdU+ cells per 0.1mm of EGL in wild type mice (n=4 mice) versus 16.7+3.8 BrdU+ cells per 0.1mm of EGL in tmgc26−/− mutants (n=4 mice), two tailed Student’s t test P=0.67) (Fig. 3G–I). In wild type mice, the EGL disappeared by P16, with all granule cell progenitors differentiated into granule neurons and migrated into the IGL. In the cerebellum of tmgc26−/− mice the EGL, however, the EGL persisted much longer, until at least P20 (Fig. 3J-L). Interestingly, even at that late stage, numerous BrdU+ cells were detected in the tmgc26−/− EGL (Fig. 3K), indicating that it still contains granule cell progenitors rather than differentiated granule neurons.
Molecular layer interneuron abnormalities in tmgc26−/− mice
Next we studied development of molecular layer interneurons in tmgc26−/− mice. During development, molecular layer interneuron precursors arise in the embryonic cerebellar ventricular zone. These cells continue to divide while they enter the overlying parenchyma and migrate through the white matter toward their final destinations in the molecular layer (Reviewed in Carletti and Rossi, 2008). Both early postmitotic molecular layer interneurons and their precursors specifically express Pax2 (Maricich and Herrup, 1999; Weisheit et al., 2006). By P14, these cells downregulate Pax2 but can be recognized at this and later developmental stages based on their Parvalbumin expression (Maricich and Herrup, 1999; Collin et al, 2005).
At P5, we detected no significant abnormalities in the molecular layer interneuron lineage in tmgc26−/− cerebella. Indeed, numerous Pax2+ precursors of molecular layer interneurons were detected in the white matter of tmgc26−/− cerebella (Fig. 4A, C, arrow). In addition, similar to wild type mice, Pax2+ cells were also detected within the molecular layer of tmgc26−/− mice (Fig. 4B, D). At P21, however, no Parvalbumin staining was detected in the molecular layer or elswhere in tmgc26−/− cerebella (Fig. 4E, F). Taken together, these data suggest that in tmgc26−/− mice molecular layer interneurons are normally generated and populate the molecular layer. However, loss of Parvalbumin staining and the lack of cell bodies in the molecular layer of the adult mutant mice (Fig. 1G, H) suggest that the tmgc26 mutation precludes survival of these cells.
Figure 4. Molecular layer abnormalities in tmgc26−/− mutants.

Sagittal sections from wild type (A, B, E) and tmgc26 −/− mutant mice (C, D, F) at the indicated stages co-stained with DAPI (blue) and Pax2 antibody (red) (A–D) or with Parvalbumin (green) and Calbindin (red) antibodies (E, F). All panels correspond to the pink boxes in the adjacent diagrams.
(A, C) Arrows point to Pax2+ precursors of molecular layer interneurons in the white matter of wild type (A) and tmgc26 −/− mutant mice (B). Panels B and D correspond to boxed regions in panels A and C, respectively. Arrowheads point to Pax2+ interneurons detected in the molecular layer of both wild type (B) and tmgc26 −/− mutant mice (D) at P5. Scale bar: A, C, 160 μm; B, D, 80 μm; E, F, 40 μm.
(E–F) Parvalbumin + Calbindin co-labeling was used to better discriminate molecular layer interneurons from Purkinje cells (as described by Collin et al., 2005). Purkinje cells coexpress Parvalbumin and Calbindin and appear yellow (arrows). Molecular layer interneurons express Parvalbumin but not Calbindin and appear green (arrowheads). In wild type P21 cerebella, numerous Parvalbumin-positive cells are present in the molecular layer. In tmgc26 −/− mutants no Parvalbumin-positive cells were detected in the molecular layer or elswhere in the cerebellum at P21. PC-Purkinje cells, ML- molecular layer, IGL- internal granule cell layer, PC+IGL – Purkinje cells mixed with granule cells within the internal granule cell layer in tmgc26 −/− mutants.
Radial glia abnormalities in tmgc26−/− mice
In addition to neurons, the mouse cerebellum also contains Golgi epithelial cells and their radial glial fibers, which are critical for proper neuronal migration (Hatten 1999). In wild type mice, by P4, the cell bodies of these glial cells align with Purkinje cells and extend long radial fibersto the pial surface of the cerebellum (Fig. 5A). In tmgc26−/− mutant cerebella, the radial glia were abnormally distributed inthe cerebellar cortex, although many still extendedradial fibers to the pial surface (Fig. 5B). However, by P21, radial glia in tmgc26 mice correctly formed a monolayer (Fig. 5C, D), suggesting that in tmgc26 mutants, radial glia development is delayed but not completely disrupted.
Figure 5. Radial glia defects in tmgc26−/− mutants.

Sagittal sections of cerebellar vermis of wild type (A, C) and tmgc26 −/− mutant mice (B, D) at the indicated stages stained with anti-BLBP (A, B) and anti-GFAP (C, D) antibodies. All panels correspond to the pink boxes in the adjacent diagrams. White arrowheads point to radial glia fibers extending to the cerebellar surface (asterisk). (A, B) Arrow points to radial glia aligned into a monolayer in a P4 wild type cerebellum but not in a tmgc26−/− mutant cerebellum. Scale bar: A, B, 30 μm; C, D, 60 μm.
tmgc26 chimeras reveal direct and indirect influences of the tmgc26−/− mutation
To identify the primary cellular target(s) of the tmgc26 mutation we used an experimental mouse chimera approach. Embryos from tmgc26+/− matings were fused with wild-type Rosa26 embryos (which carry the lacZ transgene under a strong ubiquitous promoter). A total of 23 chimeras were generated with varying cellular contributions from the tmgc26 and wild type Rosa26 strains. Of these animals, 14 showed no noticeable behavioral phenotype and revealed no cerebellar abnormalities, presumably because they received tmgc26+/− or tmgc26+/+ cells (highlighted green in Suppl. Table 2). The remaining 10 animals were ataxic and/or had a smaller cerebellum at the gross macroscopic level, indicating that they contained tmgc26−/− cells. Three of these were 100% tmgc26 (Suppl. Table 2, blue). The remaining seven contained significant cellular contributions from both tmgc26 and Rosa26 embryos (Suppl. Table 2, black). These were considered true experimental chimeras and used for further analysis.
Similar to tmgc26−/− cerebella, the cerebella of our experimental chimeras were small, had rudimentary foliation and a persistent EGL at P19–21. The degree of abnormality, however, varied from chimera to chimera. Interestingly, there was a correlation between the cerebellar size in our experimental chimeras and the presence of wild type Purkinje cells: the larger the cerebellum, the greater number of wild type Purkinje cells it contained. Notably, granule cells could be of either genotype (data not shown). At higher magnification it was clear that wild type Purkinje cells were large and normally placed with extensive dendritic arborization (Fig. 6A, B, open arrowheads), while mutant Purkinje cells were small with a small atrophic dendrite and were often in ectopic positions within the IGL (Fig. 6A, B, black arrowheads). These data argue that the tmgc26 mutation cell-autonomously affects Purkinje cells.
Figure 6. tmgc26<->Rosa26 chimeras suggest cell autonomous effects of the mutation in Purkinje cells and molecular layer interneurons.

(A) Calbindin and β-gal double staining of the cerebellum of chimera X2006 at P19. (B) Higher magnification of the region boxed in panel “A”. Open arrowheads point to β-gal-positive (wild type) Purkinje cells. Black arrowheads point to mutant Purkinje cells. Only wild type Purkinje cells have a normal placement, orientation and size, whereas calbindin-positive β-gal-negative (mutant) cells are small, misoriented and many are displaced in the IGL. (C) Parvalbumin and β-gal double staining of the cerebellum of chimera X2009 at P21. (D) Higher magnification of the region boxed in panel “C”. Virtually all the parvalbumin positive molecular layer interneurons are wild type (β-gal-positive, black arrowheads). The only Parvalbumin-positive/β-gal- negative cells are the displaced Purkinje cells (arrows). (E) Pax6 and β-gal double staining of the cerebellum of chimera X1995 at P19. Both wild type (βgal-positive, black arrowheads) and mutant (βgal-negative, open arrowheads) granule cells were found within both the EGL and IGL. Scale bar: A, 160 μm; B, 70 μm; C, 320 μm; D, 50 μm; E, 40 μm.
We also examined the interneuron population using anti-Parvalbumin immunocytochemistry. Interestingly, in our experimental chimeras virtually all Parvalbumin-positive cells in the molecular layer were wild type (Fig. 6C and D). This is particularly interesting as many of our experimental chimeras were mostly composed of mutant cells (Suppl. Table 2). Therefore, similar to Purkinje cells, the tmgc26 mutation likely affects development of molecular layer interneurons cell-autonomously.
In contrast to Purkinje cells and molecular layer interneurons, we did not see any evidence of cell-autonomous effects of the tmgc26 mutation on development of granule cells. Both wild type and mutant Pax6-positive granule cells were present in both EGL and IGL in all our experimental chimeras (Fig. 6E) suggesting that the longer persistence of the EGL in tmgc26−/− mutants is caused by non cell-autonomous mechanisms.
tmgc26 is a new allele of ROR-alpha
Since the tmgc26 phenotype did not precisely match any previously described mouse mutant phenotype, we decided to define the tmgc26 mutation molecularly using positional cloning. tmgc26+/− animals were crossed to MOLF mice and F1 heterozygotes were intercrossed. Genome-wide linkage analysis using 26 affected F2 progeny of the F1 intercross revealed maximum evidence for linkage (LOD = 16.35) within a 13 Mb region on distal chromosome 9 between the flanking SNPs rs3714012 and rs6257131. According to the UCSC Mouse Genome Browser (May 2007), this region contains 155 known genes, including ROR-alpha, which is disrupted in the staggerer mutant mouse. Since staggerer homozygous mice have obvious cerebellar defects including Purkinje cell and granule cell abnormalities (Gold et al., 2007), we sequenced ROR-alpha in tmgc26−/− mutants. We found a T-to-A transition at nucleotide 347 of exon 5 of the ROR-alpha gene that was predicted to result in a nonsense mutation (C257X) leading to severe truncation of the ROR-alpha protein (Fig. 7).
Figure 7. ROR-alpha mutations in tmgc26 and staggerer mice.
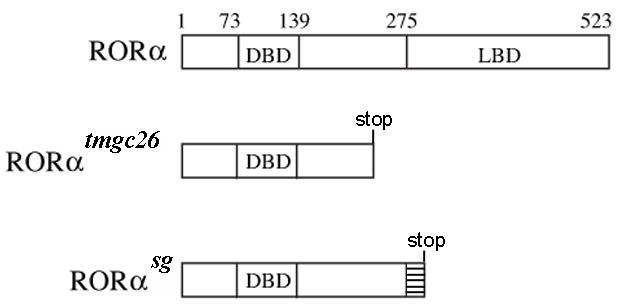
Schematic diagram of the wild type mouse ROR-alpha protein and ROR-alpha proteins predicted to be produced by tmgc26 and staggerer mutations. DBD- DNA binding domain, LB-ligand binding domain. Aberrant amino acid sequence in staggerer ROR-alpha protein is represented by a hatched box.
To confirm that the ROR-alpha mutation caused the tmgc26 phenotype, we performed complementation analysis by mating tmgc26+/− mice with staggerer heterozygotes. Of the 24 pups born, 7 were ataxic, confirming that tmgc26 represents a new mutation of the ROR-alpha gene.
Since both tmgc26 and stagger mice were present in our colony, we directly compared their behavioral and gross cerebellar phenotypes. Although homozygous tmgc26 and staggerer mutants were indistinguishable based on their behavior or their mature cerebellar morphology (data not shown), we observed some differences. While most homozygous staggerer mice (82%, 18 out of 22) survived until weaning, many tmgc26−/− mice died beginning from P12 with only 48% (19 out of 40) surviving until P21 (P=0.01, Fisher’s exact test) (Fig. 8A). In addition, at P21, the cerebellar weight of surviving tmgc26 homozygous mutants was significantly lower than that of homozygous staggerer mutants (0.03+0.004g in homozygous staggerer mice (n=12 mice) versus 0.021+0.005g in tmgc26−/− mice (n=19 mice), two tailed Student’s t test P=0.0001) (Fig. 8B). In contrast, no difference in cerebellar weights between control tmgc26 (wild type and heterozygous littermates of tmgc26 homozygous mutants, n=17) and control staggerer mice (wild type and heterozygous littermates of staggerer homozygous mutants, n=10) was detected (0.058+0.005g versus 0.056+0.003g, two tailed Student’s t test P=0.4). Together, these data suggest a more severe manifestation of our tmgc26 mutation compared to staggerer mutation.
Figure 8. Reduced survival and cerebellar weight in tmgc26 mutants compared to staggerer mutants.
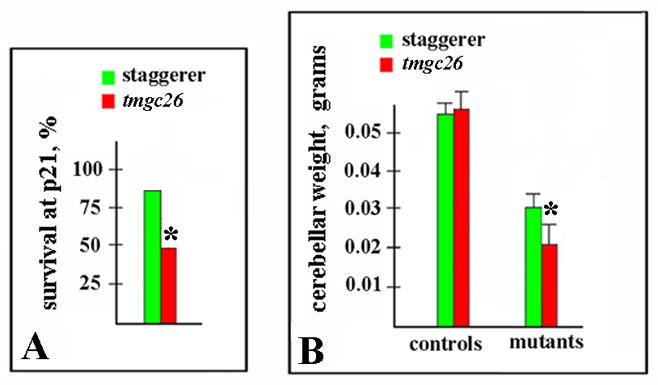
(A) A Much lower proportion of homozygous tmgc26 mutant mice survived until P21 than that of homozygous staggerer mice. (*P=0.01, Fisher’s exact test).
(B) At P21, the average cerebellar weight of tmgc26 homozygous mutants was significantly smaller than that of staggerer homozygous mutants (*P=0.0001, two tailed Student’s t test), while no difference in cerebellar weights between control tmgc26 (wild type and heterozygous littermates of tmgc26 homozygous mutants) and control staggerer mice (wild type and heterozygous littermates of staggerer homozygous mutants) was detected (P=0.4, two tailed Student’s t test).
Discussion
The orphan nuclear receptor ROR-alpha is involved in cerebellar development, regulation of circadian behavior, cholesterol and lipid metabolism, immune function, and bone formation (Gold et al., 2007). In the current study we identified tmgc26 as a new ROR-alpha allele. We documented Purkinje cell and granule cell abnormalities in the cerebella of these mice, and also identified a novel role for ROR-alpha in survival of cerebellar molecular layer interneurons and development of radial glia.
tmgc26 is a new ROR-alpha allele
To date, the majority of our knowledge regarding the role of ROR-alpha in cerebellar development and maintenance has come from analysis of staggerer mutant mice (Sidman, 1962; Hamilton et al., 1996). This mutant line arose spontaneously at the Jackson laboratory more than 40 years ago and was later found to harbor a ROR-alpha intragenic deletion that removes the sixth exon of the gene. This deletion causes a more than 10 fold reduction in ROR-alpha mRNA in the staggerer mouse, suggesting decreased mRNA stability. In addition, loss of the sixth exon causes a frameshift in the ROR-alpha mRNA that generates a premature stop codon and causes loss of the ligand binding domain (Hamilton et al., 1996) (Fig. 7). Recently it has been shown that removal of the ligand binding domain completely abolishes ROR-alpha transcriptional activity, suggesting loss of functional activity of the staggerer ROR-alpha protein (Qui et al., 2007). In addition, two independent ROR-alpha-null mouse lines, created by gene targeting, possess cerebellar phenotypes virtually indistinguishable from that of the staggerer mouse (Dussault et al. 1998; Steinmayr et al., 1998) further supporting the conclusion that staggerer is a null ROR-alpha allele.
In the current study we determined that the tmgc26 mutation is a new allele of ROR-alpha. This allele harbors a T-to-A substitution in exon 5 of the ROR-alpha gene resulting in a nonsense mutation C257X and severe truncation of the ROR-alpha protein. Although we do not know if this mutation affects ROR-alpha mRNA stability, it is predicted to truncate ROR-alpha at amino acid 257, producing a protein that is 16 amino acids shorter than the staggerer protein (which preserves 272 amino acids of ROR alpha, Hamilton et al., 1996). Similar to the truncated staggerer ROR-alpha protein, the tmgc26 ROR-alpha lacks the entire ligand binding domain and, therefore, likely represents another null allele of ROR-alpha.
Surprisingly, our comparison of tmgc26 and staggerer homozygous mice revealed some phenotypic differences between these two mutants. While homozygous staggerer mice survive until weaning, many of our tmgc26−/− mutants died as early as P12–P15. In addition, at P21, there was a statistically significant reduction of cerebellar weight of tmgc26−/− mice compared to staggerer homozygous mutants. Together, these data suggest a more severe manifestation of our tmgc26 mutation compared to staggerer mutation. One possibility is that staggerer is not a complete null allele and that the loss of 16 additional amino acids in ROR-alpha observed in our tmgc26 mutant further decreases ROR-alpha activity. However, this is unlikely since transcription activation using in vitro assays revealed no residual activity for the staggerer ROR-alpha protein (Qui et al., 2007). It is important to note, that while our tmgc26 mutation arose on a mixed noninbred background, the staggerer mice that we used for comparison, were on a B6C3Fe background. Therefore, it is possible that the more severe effect of the tmgc26 mutation is caused by modifiers present in its genetic background. Currently, the ROR-alpha interacting genetic elements are unknown and our new tmgc26 strain may be useful for their identification.
ROR alpha and the development of major classes of cerebellar cells
Previously, most ROR-alpha cerebellar studies focused on Purkinje cells and granule cells. During cerebellar development ROR-alpha is expressed in Purkinje cells and its inactivation either by the staggerer mutation or by the tmgc26 mutation causes very similar cell-autonomous Purkinje cell abnormalities (Herrup, 1983 and current study). In contrast to Purkinje cells, granule cells do not express ROR-alpha (Ino, 2004). Using the staggerer allele it has been shown that granule cell abnormalities, which begin with reduced proliferation of granule progenitors in ROR-alpha deficient mice (Gold et al., 2003; 2007), are secondary to Purkinje cell defects (Herrup, 1983). During cerebellar development Purkinje cells stimulate proliferation of granule cell progenitors by secreting mitogenic signals such as Shh (Wechsler-Reya and Scott, 1999; Wallace, 1999; Dahmane and Ruiz i Altaba, 1999). Recently, it has been shown that ROR-alpha directly activates transcription of the Shh gene in Purkinje cells (Gold et al., 2003) explaining how loss of this gene in staggerer Purkinje cells decreases granule cell proliferation.
In the current study using the tmgc26 allele, we confirmed that inactivation of ROR-alpha severely affects proliferation of granule cell progenitors, in particular at P4. Interestingly, as development proceeded, the difference in granule progenitor proliferation between tmgc26−/− mutant and wild type mice became less obvious, suggesting that our tmgc26 mutation more significantly affects proliferation of granule progenitors at early developmental stages. Furthermore, the EGL of tmgc26−/− mutants contained numerous proliferating granule cell progenitors even at P20, a time when all granule cell progenitors have already differentiated into granule neurons in wild type mice. There are several possible explanations of the longer persitance of the EGL in tmgc26−/− mutants. For example, in addition to affecting proliferation of granule cell progenitors at early postnatal stages, the tmgc26 mutation may also cause a prolonged clonal expansion of granule progenitors at later stages. Alternatively, the mutation may affect granule cell differentiation and migration into the IGL.
In addition to Purkinje and granule cell defects, we have documented abnormalities in two other cerebellar populations: molecular layer interneurons and radial glia, both of which have never been reported to require ROR-alpha for their development. Similar to Purkinje cells, molecular layer interneurons express ROR-alpha (Ino, 2004). As revealed by Pax2 staining, in the early postnatal tmgc26 mutant cerebellum, precursors of molecular layer interneurons normally populate the white matter and the developing molecular layer. By P21, however, very few cells were detected in the molecular layer in the tmgc26−/− cerebellum, with the vast majority of expressing the granule cell marker Pax6. In addition, no Parv+/Calb- cells were detected in the molecular layer or elsewhere in the cerebellum of this mutant. This suggests that the tmgc26 mutation does not significantly disrupt early development of molecular layer interneurons but affects their survival. Furthermore, in our tmgc26−/− chimeras all detectable Parvalbumin-positive molecular layer neurons were derived from the wild type embryos. These data suggest that ROR-alpha acts cell-autonomously during molecular layer interneuron development, although we cannot completely exclude the possibility that cell-extrinsic events also affect development of tmgc26−/− molecular layer interneurons in our chimeras. We also acknowledge that in the current study we collected our chimeras at P19–21 and did not study them during earlier developmental stages. We could not, therefore, directly analyze the generation, migration and degeneration of tmgc26−/− molecular layer interneurons or their precursors in these chimeras. It is possible that in our chimeras, tmgc26−/− cells do not contribute to the molecular layer interneuron lineage or that if generated, tmgc26−/− molecular layer interneuron precursors do not properly migrate in the presence of wild type cells. Based on the results of our analysis of tmgc26−/− mutant cerebella, the lack of tmgc26−/− molecular layer interneurons in our chimeras is most likely caused by death of these cells after their arrival in the molecular layer. In the original staggerer mutant, molecular layer interneurons were reported to be normal (Landis and Sidman, 1978) and the requirement of ROR-alpha for development of these cells has not been reported for other ROR-alpha mutants (Gold et al., 2007). Previous studies of staggerer chimeras were unable to address development of molecular layer interneurons since they could not distinguish between wild type and staggerer cells in the molecular layer as an appropriate marker of cell genotype was not available at that time (Herrup and Mullen, 1979).
Previous fate mapping studies revealed that both Purkinje cells and molecular layer interneurons derive from Ptf1a+ progenitors in the embryonic cerebellar ventricular zone and require Ptf1a function for proper development (Hoshino et al., 2005; Pasqual et al., 2007; reviewed in Carletti and Rossi, 2008). Results of our current study strongly suggest that these two populations also rely on Ror-alpha for their normal survival and maintenance in the mature cerebellar cortex.
In contrast to molecular layer interneurons, radial glia do not express ROR-alpha. Thus, the radial glia defects in our tmgc26 mutant mice must be secondary to Purkinje cell or granule cell abnormalities. Interestingly, it has been shown that radial glia cells receive Shh signals from Purkinje cells during development (Corrales et al., 2004, 2006; Chizhikov et al., 2007). The role of this signaling in radial glia development remains unknown. Therefore, ROR-alpha-deficient mice may be a useful model to study radial glia – Purkinje cell interactions in more detail.
Supplementary Material
Acknowledgments
We thank Richard Cushing, Meifen Lu, and Noble Del Mar for their expert technical assistance and Marissa Blank for valuable comments on this manuscript. This work was supported by NIH R01 MG61915 and NIH RO1 HD52472 grants to Dan Goldowitz and by NIH RO1 NS050386 grant to Kathleen Millen.
References
- 1.Acevedo-Arozena A, Wells S, Potter P, Kelly M, Cox RD, Brown SD. ENU mutagenesis, a way forward to understand gene function. Annu Rev Genomics Hum Genet. 2008;9:49–69. doi: 10.1146/annurev.genom.9.081307.164224. [DOI] [PubMed] [Google Scholar]
- 2.Chizhikov V, Millen KJ. Development and malformations of the cerebellum in mice. Mol Genet Metab. 2003;80:54–65. doi: 10.1016/j.ymgme.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Chizhikov VV, Lindgren AG, Currle DS, Rose MF, Monuki ES, Millen KJ. The roof plate regulates cerebellar cell-type specification and proliferation. Development. 2006;133:2793–804. doi: 10.1242/dev.02441. [DOI] [PubMed] [Google Scholar]
- 4.Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci. 2007;27:9780–9. doi: 10.1523/JNEUROSCI.5586-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carletti B, Rossi F. Neurogenesis in the cerebellum. Neuroscientist. 2008;14:91–100. doi: 10.1177/1073858407304629. [DOI] [PubMed] [Google Scholar]
- 6.Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, Marty A, Llano I. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131:5581–90. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- 8.Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–21. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 9.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 10.Dussault I, Fawcett D, Matthyssen A, Bader JA, Giguére V. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70:147–53. doi: 10.1016/s0925-4773(97)00187-1. [DOI] [PubMed] [Google Scholar]
- 11.Faust PL. Abnormal cerebellar histogenesis in PEX2 Zellweger mice reflects multiple neuronal defects induced by peroxisome deficiency. J Comp Neurol. 2003;461:394–413. doi: 10.1002/cne.10699. [DOI] [PubMed] [Google Scholar]
- 12.Friedel RH, Kerjan G, Rayburn H, Schüller U, Sotelo C, Tessier-Lavigne M, Chédotal A. Plexin-B2 controls the development of cerebellar granule cells. J Neurosci. 2007;27:3921–32. doi: 10.1523/JNEUROSCI.4710-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gold DA, Gent PM, Hamilton BA. ROR alpha in genetic control of cerebellum development: 50 staggering years. Brain Res. 2007;1140:19–25. doi: 10.1016/j.brainres.2005.11.080. [DOI] [PubMed] [Google Scholar]
- 14.Gold DA, Baek SH, Schork NJ, Rose DW, Larsen DD, Sachs BD, Rosenfeld MG, Hamilton BA. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40:1119–31. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldowitz D, Mullen RJ. Granule cell as a site of gene action in the weaver mouse cerebellum: evidence from heterozygous mutant chimeras. J Neurosci. 1982;2:1474–85. doi: 10.1523/JNEUROSCI.02-10-01474.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldowitz D. The weaver granuloprival phenotype is due to intrinsic action of the mutant locus in granule cells: evidence from homozygous weaver chimeras. Neuron. 1989;2:1565–75. doi: 10.1016/0896-6273(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 17.Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–82. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 18.Goldowitz D, Frankel WN, Takahashi JS, Holtz-Vitaterna M, Bult C, Kibbe WA, Snoddy J, Li Y, Pretel S, Yates J, Swanson DJ. Large-scale mutagenesis of the mouse to understand the genetic bases of nervous system structure and function. Brain Res Mol Brain Res. 2004;132:105–15. doi: 10.1016/j.molbrainres.2004.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A. Allegro version 2. Nat Genet. 2005;37:1015–6. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature. 1996;379:736–9. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 21.Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–39. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- 22.Herrup K, Mullen RJ. Staggerer chimeras: intrinsic nature of Purkinje cell defects and implications for normal cerebellar development. Brain Res. 1979;178:443–57. doi: 10.1016/0006-8993(79)90705-4. [DOI] [PubMed] [Google Scholar]
- 23.Herrup K. Role of staggerer gene in determining cell number in cerebellar cortex. I. Granule cell death is an indirect consequence of staggerer gene action. Brain Res. 1983;313:267–74. doi: 10.1016/0165-3806(83)90225-0. [DOI] [PubMed] [Google Scholar]
- 24.Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–33. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Hoshino M, Nakamura S, Mori K, Kawauchi T, Terao M, Nishimura YV, Fukuda A, Fuse T, Matsuo N, Sone M, Watanabe M, Bito H, Terashima T, Wright CV, Kawaguchi Y, Nakao K, Nabeshima Y. Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron. 2005;47:201–13. doi: 10.1016/j.neuron.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Ino H. Immunohistochemical characterization of the orphan nuclear receptor ROR alpha in the mouse nervous system. J Histochem Cytochem. 2004;52:311–23. doi: 10.1177/002215540405200302. [DOI] [PubMed] [Google Scholar]
- 27.Justice MJ. Capitalizing on large-scale mouse mutagenesis screens. Nat Rev Genet. 2000;1:109–15. doi: 10.1038/35038549. [DOI] [PubMed] [Google Scholar]
- 28.Landis DM, Sidman RL. Electron microscopic analysis of postnatal histogenesis in the cerebellar cortex of staggerer mutant mice. J Comp Neurol. 1978;179:831–63. doi: 10.1002/cne.901790408. [DOI] [PubMed] [Google Scholar]
- 29.Leto K, Bartolini A, Rossi F. Development of cerebellar GABAergic interneurons: origin and shaping of the “minibrain” local connections. Cerebellum. 2008;7:523–9. doi: 10.1007/s12311-008-0079-z. [DOI] [PubMed] [Google Scholar]
- 30.Maricich SM, Herrup K. Pax-2 expression defines a subset of GABAergic interneurons and their precursors in the developing murine cerebellum. J Neurobiol. 1999;41:281–94. doi: 10.1002/(sici)1097-4695(19991105)41:2<281::aid-neu10>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 31.Millen KJ, Gleeson JG. Cerebellar development and disease. Curr Opin Neurobiol. 2008;18:12–9. doi: 10.1016/j.conb.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascual M, Abasolo I, Mingorance-Le Meur A, Martínez A, Del Rio JA, Wright CV, Real FX, Soriano E. Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc Natl Acad Sci U S A. 2007;104:5193–5198. doi: 10.1073/pnas.0605699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development. 2006;133:3929–37. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu CH, Shimokawa N, Iwasaki T, Parhar IS, Koibuchi N. Alteration of cerebellar neurotropin messenger ribonucleic acids and the lack of thyroid hormone receptor augmentation by staggerer-type retinoic acid receptor-related orphan receptor-alpha mutation. Endocrinology. 2007;148:1745–53. doi: 10.1210/en.2006-1131. [DOI] [PubMed] [Google Scholar]
- 35.Schilling K, Oberdick J, Rossi F, Baader SL. Besides Purkinje cells and granule neurons: an appraisal of the cell biology of the interneurons of the cerebellar cortex. Histochem Cell Biol. 2008;130:601–15. doi: 10.1007/s00418-008-0483-y. [DOI] [PubMed] [Google Scholar]
- 36.Sidman RL, Lane PW, Dickie MM. Staggerer, a new mutation in the mouse affecting the cerebellum. Science. 1962;137:610–2. doi: 10.1126/science.137.3530.610. [DOI] [PubMed] [Google Scholar]
- 37.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 38.Steinmayr M, André E, Conquet F, Rondi-Reig L, Delhaye-Bouchaud N, Auclair N, Daniel H, Crépel F, Mariani J, Sotelo C, Becker-André M. staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc Natl Acad Sci U S A. 1998;31:3960–5. doi: 10.1073/pnas.95.7.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valdar W, Solberg LC, Gauguier D, Burnett S, Klenerman P, Cookson WO, Taylor MS, Rawlins JN, Mott R, Flint J. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet. 2006;38:879–87. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- 40.Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–8. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 41.Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–91. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- 42.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 43.Weisheit G, Gliem M, Endl E, Pfeffer PL, Busslinger M, Schilling K. Postnatal development of the murine cerebellar cortex: formation and early dispersal of basket, stellate and Golgi neurons. Eur J Neurosci. 2006;24:466–78. doi: 10.1111/j.1460-9568.2006.04915.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.