

Abstract
A growing variety of technical approaches allow control over the expression of selected genes in living organisms. The ability to deliver functional exogenous genes involved in neurodegenerative diseases has opened pathological processes to experimental analysis and targeted therapeutic development in rodent and primate pre-clinical models. Biological adaptability, economic animal use, and reduced model development costs complement improved control over spatial and temporal gene expression compared with conventional transgenic models. A review of viral vector studies, typically adeno-associated virus or lentivirus, for expression of three proteins that are central to major neurodegenerative diseases, will illustrate how this approach has powered new advances and opportunities in CNS disease research.
Keywords: Parkinson’s, Alzheimer’s, Vectors, Animal models
Introduction
Neurodegenerative disease therapy is tremendously frustrating for patients and clinicians. Parkinson’s disease (PD) has a spectrum of treatment options (Poewe 2009), but drugs for Alzheimer’s disease (AD) and the more uncommon neurodegenerative diseases are limited to cholinesterase inhibitors and palliatives for acute symptoms (Rafii and Aisen 2009). The microtubule-associated protein tau, and alpha-synuclein, each precipitate in neuropathological lesions in multiple neurodegenerative diseases (Jellinger 2003; Iqbal et al. 2005). The lack of natural animal models for these diseases has severely hindered the study of disease mechanisms and the development of rational therapeutic strategies targeted at specific cellular pathology involving these proteins. Experimental disease models based on selective neurotoxins such as 6-hydroxydopamine (6-OHDA) in rats and 1-methyl-4-phenyl-1,2,3,6-tetrahy-dropyridine (MPTP) in mice and monkeys can generate extensive dopaminergic neuron loss comparable to PD, but may fail to reproduce either key neuropathological features such as Lewy bodies, or progressive motor behavior dysfunction (Gibrat et al. 2009; Krenz et al. 2009). Recent reviews covering the growing number of transgenic mouse models that express normal or mutant alpha-synuclein concur that these exhibit progressive behavioral phenotypes but relatively little neuron loss of nigral neurons or nigrostriatal projections (Martin 2007; Kahle 2008; Moore and Dawson 2008). Thus it is promising that models of PD based on localized gene transfer to generate synuclein can produce loss of dopaminergic neurons, neuronal inclusions, and progressive motor impairment in rodents and primates (Eslamboli et al. 2007; Chung et al. 2009). Although specific PD features such as resting tremor have yet to be reproduced in any animal model, gene transfer may approximate the disease sufficiently well to provide a test bed for potential new therapeutic strategies. This article highlights progress in the study of neurodegenerative diseases using viral vector somatic cell gene transfer. We focus on PD- and AD-related diseases, noting that models for Huntington’s disease have recently been reviewed elsewhere (Lundberg et al. 2008).
In contrast to neurotoxins, the somatic transgenic approach studies disease etiology for specific genes, and can be used for both wild-type alleles and disease-related mutations. The progression of vector-induced neuropathology is often accelerated compared with what occurs in transgenic mice, which may permit the development of pathology that experimental animals might not live long enough to develop otherwise. It is usually slower than the rapidly acting neurotoxins, minimizing confounds related to acute toxicity. Vector-based methods offer a number of unique advantages for disease modeling compared to transgenic mice: (1) specific targeting of disease-related brain regions, reproducing natural disease patterns; (2) expression in normal naive animals including aged subjects, avoiding developmental side effects, adaptation, compensation, or early lethality; (3) unilateral treatment for side-to-side comparisons, internal genetic control, and within-subjects experimental designs; (4) gene co-expression and simplified combinatorics; (5) dissociation of central and peripheral expression effects; and (6) rapid and (relative to the expense of purchasing or breeding transgenic mice) economical data. These advantages have been utilized for unique experiments addressing neurodegenerative disease processes and therapeutics, establishing that the approach has a distinct capacity to identify molecular targets for therapeutics development (e.g. Gorbatyuk et al. 2008; Chung et al. 2009).
Gene transfer models have limitations as well (Kirik and Björklund 2003; Kirik et al 2003). Even with advanced delivery methods that can transduce large brain volumes (Szerlip et al. 2007), transgene expression is likely to vary in relation to distance from vector delivery sites and proximal architectural features. Localized transduction may also limit phenotypic reproduction of distributed disease (Chesselet 2008). Transgene expression may supplement endogenous expression for the same gene, or result in co-expression of mutant and wild-type proteins, conditions that may differ significantly from natural disease. Vectors that can integrate into host genomes can be tumorigenic, such as the leukemias that have been traced to retroviral vector insertional mutagenesis in humans and mice (Li et al. 2002; Hacein-Bey-Abina et al. 2008; Modlich and Baum 2009). A substantial fraction of individuals in host animal populations may have existing immunological defenses against vectors (Zaiss and Muruve 2008). Diseases such as vascular amyloidopathies require peripheral and/or developmental expression better reproduced by whole-animal transgenic approaches. Finally, most gene transfer models are based on mutations or overexpression, but idiopathic AD and PD may have complex changes in expression of multiple genes that cannot presently be reproduced by gene delivery.
Vector Technology
The history of efficient brain gene transfer begins with herpes virus vectors (Geller and Breakefield 1988; Federoff et al. 1992; During et al. 1994) and later adenovirus vectors (Le Gal La Salle et al. 1993; Horellou et al. 1994; Castel-Barthe et al. 1996; Bilang-Bleuel et al. 1997; Choi-Lundberg et al. 1997). Efficiency and longevity of gene expression improved significantly with the subsequent advent of adeno-associated virus (AAV) (Kaplitt et al. 1994; McCown et al. 1996) (Fig. 1) and lentiviral vectors (Naldini et al. 1996; Blömer et al. 1997). As evidence accumulated that AAV vectors were safe and could mediate persistent expression, the efficiency of gene transfer continued to rise with the use of novel engineered promoters and the addition of new AAV serotype vectors (Davidson et al. 2000; Burger et al. 2004; Klein et al. 2006a, 2008a). Early studies with AAV and lentivirus focused on reporter gene expression and gene therapy in rodents (Kaplitt et al. 1994; Naldini et al. 1996; Blömer et al. 1997; Mandel et al. 1997; Klein et al. 1998; Lo et al. 1999) before gene transfer was successfully applied to modeling neurodegenerative disease pathology. A breakthrough paper validated the application of AAV somatic gene transfer system to this task (Senut et al. 2000). An AAV vector for polyglutamine repeats produced by this group induced a degenerative disease state in rats mimicking Huntington’s disease with respect to intraneuronal inclusions and neuronal degeneration in the striatum, cardinal neuropathology, and sequelae of the actual disease.
Fig. 1.
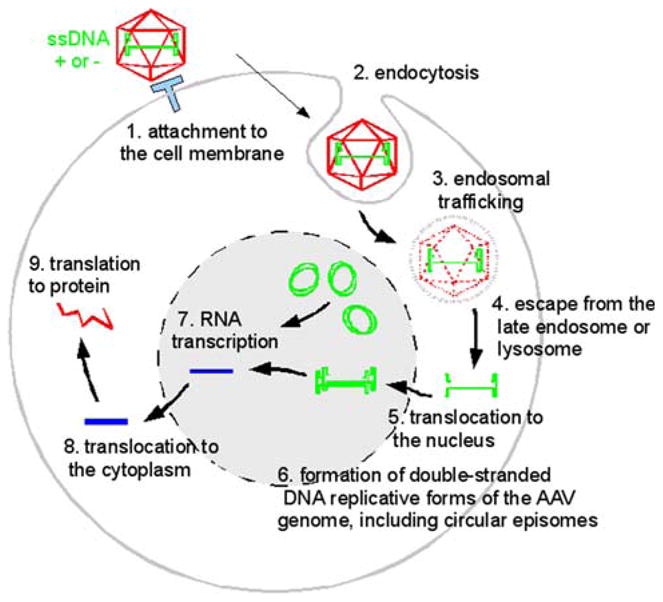
AAV is a icosahedral capsid containing either a (+) or (−)-sense single-stranded DNA. Binding to cell-surface receptors (1) triggers endocytosis (2) and passage through acidic endosomes (3) where capsid proteins are hydrolyzed. Genomic DNA escapes (4) and enters the nucleus (5) where synthesis of the complementary strand (6) permits transcription of mRNA (7) that enters the cytoplasm (8) for translation (9)
An Alpha-Synuclein Proteinopathy Model of Parkinson’s Disease
The report on the first genetic link to PD, a mutation in alpha-synuclein (Polymeropoulos et al. 1996), launched a vigorous effort to find additional genetic links and to make gene-based animal models. Because it was also known that alpha-synuclein was the protein largely comprising the Lewy body neuropathological lesions in PD (Spillantini et al. 1997), the situation was reminiscent of familial AD mutations in the gene for the neuropathological substrate protein, amyloid precursor protein (APP) (Citron et al. 1992). The first alpha-synuclein transgenic mouse line (Masliah et al. 2000) paved the way for many subsequent alpha-synuclein mouse models, as well as alternative genetic models in flies (Feany and Bender 2000), worms (Lakso et al. 2003), yeast (Outeiro and Lindquist 2003), and viral vector models in rats and monkeys (Furler et al. 2001; Kirik et al. 2002; Klein et al. 2002b, 2005; Lo Bianco et al. 2004a, b, 2008; Yamada et al. 2005; Hayashita-Kinoh et al. 2006; Yasuda et al. 2007). Even as the first genetic causes of PD were still being mapped (Polymeropoulos et al. 1996), efficient viral vectors were becoming available to mimic PD in animals (Kaplitt et al. 1994; McCown et al. 1996; Naldini et al. 1996; Blömer et al. 1997; Mandel et al. 1997; Peel et al. 1997). The use of serotypes other than AAV2 improved transduction efficiency as mentioned, as did novel engineered promoters (Klein et al. 1998, 2002a; Xu et al. 2001). Improvements were also largely due to developments in AAV purification, i.e., replacing live adenovirus with a trans-complementing plasmid system (Xiao et al. 1998; Grimm et al. 1998) and replacing CsCl with iodixanol (Zolotukhin et al. 1999). Modifications in the green fluorescent protein (GFP; Zolotukhin et al. 1996) reporter gene substantially enhanced detection of AAV-mediated expression (Peel et al. 1997; Klein et al. 1998).
Using human alpha-synuclein cDNA received from Matthew Farrer in 1999, we noticed a consistent neuron morphological phenotype caused by alpha-synuclein at 3 weeks after AAV2 vector injections into the rat substantia nigra (SN). We did not fully appreciate the mimicry of PD at the time, but eventually concluded that the classic neuropathology in PD, Lewy neurites, was reproduced by the axon morphology changes we observed in rats (Klein et al. 2002b). Synuclein-transduced nigrostriatal axons were dystrophic, with both swollen and shrunken neuronal processes and large axonal spheroids, closely resembling Lewy neurites in alpha-synucleinopathy diseases (Spillantini et al. 1997, 1998b). However, we were puzzled by the lack of dopaminergic cell loss or behavioral effects when we examined expression intervals between 1 and 6 months. Insight was provided by the study by Furler et al. (2001), who demonstrated that SN dopamine neurons can tolerate high levels of alpha-synuclein overexpression without degenerating in rat SN transduced with an efficient AAV2 vector. We did achieve alpha-synuclein-induced loss of dopamine neurons in addition to the Lewy-like neuropathology when we used a long interval of 1 year, and a relatively high vector dose (3 × 1010 vector genomes). The cell loss was not sufficient to drive the behavioral pathology, amphetamine-stimulated rotation that occurs after extensive unilateral loss of SN dopamine neurons (Klein et al. 2002b). The absence of substantial motor symptoms until dopaminergic loss is severe is consistent with the clinical properties of actual PD (Thomas and Beal 2007).
A more extensive study by Kirik et al. (2002) showed a progressive AAV2 alpha-synuclein induced dopaminergic degeneration and dopamine loss, as well as dynamic time-dependent changes in the Lewy-like neuropathology and the nigrostriatal innervation. Still the degree of cell loss caused by alpha-synuclein did not result in a clear behavioral phenotype, although clever use of a drug to deplete endogenous dopamine levels uncovered an additive effect with alpha-synuclein gene transfer (Kirik et al. 2002). This team of researchers produced an exciting story of alpha-synuclein proteinopathy in rats, and in monkeys, by demonstrating that a postural bias developed progressively, consistent with the unilateral alpha-synuclein neuropathology and degeneration of the nigrostriatal pathway (Kirik et al. 2003). Innovative studies with lentivirus (Lo Bianco et al. 2002) and AAV (Yamada et al. 2004; St Martin et al. 2007) have helped elucidate the pathogenic mechanism of alpha-synuclein in the rodent and primate brain, leading to methods of protecting against alpha-synuclein induced neurodegeneration (Lo Bianco et al. 2004b, 2008; Yamada et al. 2005; Yasuda et al. 2007; Hayashita-Kinoh et al. 2006). Our studies with alpha-synuclein provided a unique example of alpha-synuclein filaments viewed by electron microscopy in the SN in an animal model, confirming the formation of fibrillar lesions (Fig. 2; Klein et al. 2005). We attempted to improve expression levels of alpha-synuclein by way of the promoter system used (Klein et al. 2005), and by using efficient AAV serotypes 9 and 10 (Klein et al. 2008b), to induce dopaminergic deficits commensurate with behavioral effects. However, our conclusion is that the rat nigrostriatal pathway can tolerate high levels of alpha-synuclein overexpression, as these efforts did elevate expression but did not induce significant changes in either cell loss or behavior. There seems to be a large fraction of the rat SN that is resistant to alpha-synuclein overexpression, and failure to develop motor behavior phenotype in numerous studies may be due to preservation of these neurons (Klein et al. 2002b; Kirik et al. 2002; Lo Bianco et al. 2002; Yamada et al. 2004). Differential phosphorylation of synuclein has been proposed to mediate such vulnerability, and gene transfer of phosphorylation-resistant synuclein created by site-directed mutagenesis was reported to exacerbate toxicity and dopaminergic neuron loss in rat SN (Gorbatyuk et al. 2008). However, similar comparisons of AAV vectors for wild-type and phosphorylation-mutant synuclein yielded conflicting results that will require further study (McFarland et al. 2009).
Fig. 2.
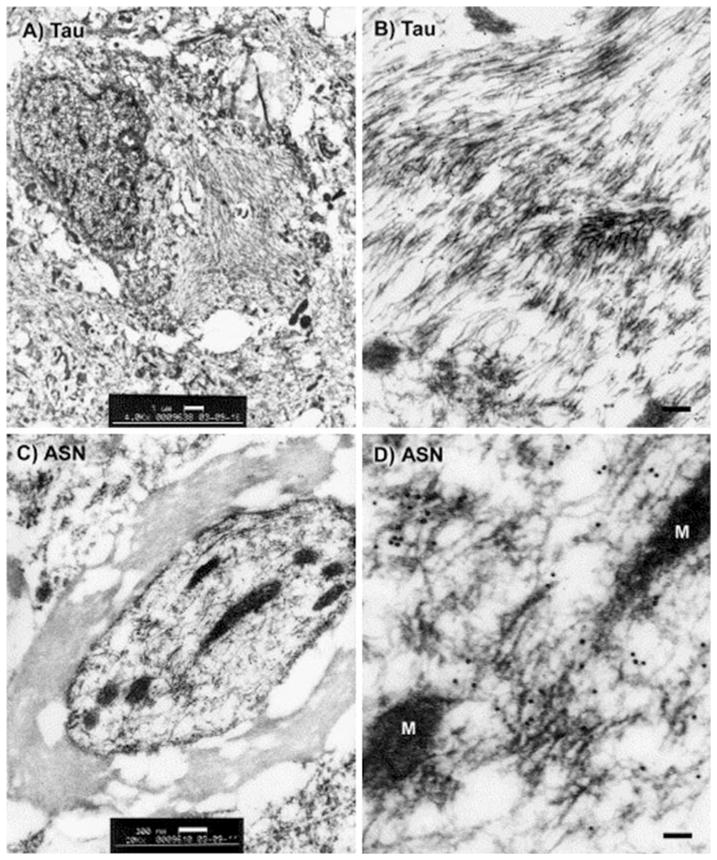
Electron microscopy for tau or alpha-synuclein in the rat, 4 months after gene transfer with either tau (a, b) or alpha-synuclein (ASN; c, d) AAV vectors. Tau filaments in the basal forebrain (a) or substantia nigra (b), labeled with 10 nm gold particles and an antibody specific for hyperphosphorylated tau, found only in cases of tau vector injections. c, d ASN filaments in a myelinated axon in the midbrain labeled with 10 nm gold particles and a human-specific ASN antibody, found only in cases of ASN vector injections. M, mitochondria. Reprinted from Klein et al. (2004, 2005) with permission
Tau Gene Transfer Models of Tauopathies
Neurofibrillary tangles are one of the classic neuropathological lesions in AD but occur in a number of other neurodegenerative diseases as well as normal aging (Ballatore et al. 2007; Iqbal et al. 2005). Tangles are mainly composed of the microtubule-associated protein tau and ubiquitin. The traditional view held that pathogenesis of tauopathy diseases involved tau hyperphosphorylation, dissociation from microtubules, self-polymerization into fibrils, and subsequent microtubule dysfunction, defective axonal transport, and neurodegeneration responsible for dementia (see Dickson 1999 for review). However, a consensus is emerging that tangles might not be the most pernicious forms of tau, cannot explain cognitive decline, and may in fact represent adaptive cellular responses (Congdon and Duff 2008; Castellani et al. 2008, etc.). Instead, “prefibrillary” tau, still poorly characterized, may be responsible for synaptic dysfunction and neurodegeneration underlying behavioral deficits (Brunden et al. 2008). This may identify new therapeutic targets and strategies.
Normal and abnormal tau function have been studied in a wide variety of organisms including invertebrates (Lee et al. 2005). A number of transgenic mouse models for tau overexpression emerged in the 1990s with aberrant somatodendritic localization of tau, which is normally found in axons, although full-blown tangles were elusive (see Lee et al. 2005 for review). Ground-breaking discoveries of familial tau mutations in cases of frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17; Hutton et al. 1998; Poorkaj et al. 1998; Spillantini et al. 1998a, for example) led to a tau transgenic mouse model overexpressing the pathogenic P301L tau mutation, with mature neurofibrillary tangles, pronounced motor deficits, and neurodegeneration (Lewis et al. 2000). We received the P301L tau from Jada Lewis and Michael Hutton in 1999 and began expressing it in the basal forebrain of rats with an AAV2 vector (Klein et al. 2004). Early experiments clearly demonstrated that the model achieved aberrant somatodendritic tau expression as in tauopathies. Markers for neurofibrillary pathology, including various antibodies for phospho-tau epitopes, antibodies for pathological tau conformations, and Gallyas silver staining indicated tau hyperphosphorylation, aggregation, and the formation of outright tangles (Klein et al. 2004). Electron microscopy confirmed the first example of mature neurofibrillary tangles in rats, induced by P301L tau gene transfer into the medial septum of the basal forebrain or the SN (Fig. 2; Klein et al. 2004, 2005). The tau filaments and tangles were viewed by electron microscopy at an interval of 4 months, but cells positive for Gallyas silver staining, another marker for tangles, appeared as early as 3 weeks after gene transfer, demonstrating that tangles can form quite rapidly in vivo. This rapid time course is similar to what was observed in the elegant cellular model for tauopathy developed by Hall and colleagues, who transduced lamprey hindbrain anterior bulb neurons in situ to express human tau constructs. Progressive neurofibrillary pathology in this model reproduces hyperphosphorylation, filament formation, dendritic tau accumulation, and neuron death observed in human disease (Hall and Yao 2005). Because tangles can form in such a short interval, tangle-bearing neurodegenerative diseases might involve a sudden change in tau function disrupting critical cellular activities, rather than a slow process taking decades. This would not be inconsistent with a slowly progressive age-related neurodegeneration, but raises the possibility that individual tangles may develop relatively abruptly compared with the evolution of dementia. The model conclusively proves that neurofibrillary pathology does not require non-neuronal, developmental, or peripheral expression of mutant tau, and still affords superior regional reproduction of AD tangle localization compared to existing transgenic models.
Taking advantage of the AAV system to combine transgenes, we expressed the tau vector in amyloid-bearing mice, in order to comprehensively mimic both the plaques and tangles in AD. We achieved efficient gene transfer of the reporter gene GFP in double transgenic APP/presenilin mice, and were able to show dense neuritic tau pathology embedded within and surrounding plaques in the hippocampus (Klein et al. 2004). A more recent study with a lentivirus vector for mutant tau resulted in tangle formation in amyloid transgenic mice after 13 months, but not in non-transgenic mice (Osinde et al. 2008). This will be a useful model to explore mechanisms linking tau and beta amyloid pathology interact, for example the aggravation of tauopathy associated with stimulation of glycogen synthase kinase 3-beta activity in mice transgenic for mutant APP (Terwel et al. 2008).
Next, we targeted tau expression to the rat SN, as we did earlier with alpha-synuclein (Klein et al. 2002b), for two reasons. The first reason was disease relevance, as there is nigral tau pathology in AD, FTDP-17, corticobasal degeneration, and progressive supranuclear palsy, as well as nigral degeneration in the latter three diseases (Wakabayashi et al. 1994; Mirra et al. 1999; Di Maria et al. 2000; Poorkaj et al. 2002; Schneider et al. 2002). Second, SN anatomy is favorable to quantitative analysis. In rats, SN consists of a discrete population of about 10,000 dopamine neurons. The combination of relatively low interanimal variation and highly reproducible stereological methods translates into an ability to resolve small effects (15%). Further, the nigrostriatal pathway has no contralateral projections, thereby facilitating within-animal designs. In other words, unilateral treatments afford a valid internal control from the untreated, separate side. The nigrostriatal pathway, therefore, provides an index of neurodegeneration in vivo that is relatively easy to quantify in order to test for specific neurotoxic effects. Compared to AAV2 GFP gene transfer, which did not alter numbers of dopamine neurons relative to untreated samples, an equal vector dose of AAV2 P301L tau vector caused a progressive loss of dopamine neurons from 28% at 3 weeks to a 56% loss by 4 months. Whether the rapid neurofibrillary tangle formation underlies the neuronal death is unclear. We know that tangles form as soon as 3 weeks (Klein et al. 2004), a time point with significant cell loss. However, only a small fraction of the cells positive for markers of human and phospho-tau were also positive for markers of tangles, such as globose morphology, Gallyas silver staining, and a neurofibrillary tangle-specific antibody (Klein et al. 2004, 2005). If the number of neurons bearing tangles is not sufficient to account for the degree of cell loss, other mechanisms are more likely to mediate neuronal death. These might include truncated tau abrogation of mitochondrial function (Quintanilla et al. 2009), stimulation of inflammatory responses (Zilka et al. 2009), or pre-tangle tau disruption of microtubule function (Iqbal et al. 2009; LaPointe et al. 2009).
Compared to an equal dose of an AAV2 alpha-synuclein vector, there was clearly a more damaging outcome as a result of tau gene transfer. We were always able to detect alpha-synuclein immunoreactivity in SN pars compacta neurons despite some degree of dopaminergic cell loss in that area (Klein et al. 2002b, 2005). In stark contrast, tau vector injections always produced clear evidence of transgene expression, above and below the pars compacta, with the adjacent pars compacta devoid of tau-immunoreactive neurons. Because the adjacent sections stained for tyrosine hydroxylase or a general neuronal marker showed the same overlapping blank areas, we concluded that the pars compacta dopamine neurons are highly sensitive to tau overexpression, which obliterates staining for tau or neuronal markers in that area (Klein et al. 2005, 2008b). Another difference between the tau and alpha-synuclein outcomes was expression in the striatum. Whereas the alpha-synuclein was greatly enriched in the terminal area of the nigrostriatal pathway as indicated by western blot samples of SN and striatum, in the case of tau, we were unable to detect the recombinant human tau in the striatum, suggesting either axonal death or lack of anterograde axonal transport (Klein et al. 2005). The third major difference between tau and alpha-synuclein in the rat/vector model is the functional readout of behavior. Amphetamine-stimulated rotational behavior, a measure of inter-hemispheric imbalance in dopamine levels, is readily observed in tau vector-injected rats, but not alpha-synuclein vector-injected rats (Klein et al. 2005, 2008b). Distinct specific disease states are induced by either tau or alpha-synuclein in terms of toxic potency, effects on axonal function, and behavior. The toxic potential of alpha-synuclein models early pre-symptomatic disease, whereas tau has potential for modeling late stage disease with behavioral deficits in conjunction with severe neuronal loss.
Newly isolated serotypes of recombinant AAV have become available and mediate increased spread and efficiency of gene transfer in brain (Burger et al. 2004; Klein et al. 2006b, 2008b, for example). We tested some of them functionally in our tau model and found differences in expression levels and degree of dopamine neuron loss (Klein et al. 2006a, 2008b). An AAV9 vector for human P301L tau produced enhanced transgene expression, and greater loss of striatal dopamine content, than identically packaged AAV2 or AAV8 vectors (Klein et al. 2008b) (Fig. 3). Robust tau expression at 1 week preceded loss of dopamine at 2 weeks and behavioral phenotype at 3–4 weeks. With the efficient AAV9 vector, tau gene transfer caused a progressive disease with roughly 90% loss of SN dopamine neurons, the first example of a nearly complete dopaminergic lesion using a vector approach and unprecedented outside of neurotoxin models.
Fig. 3.
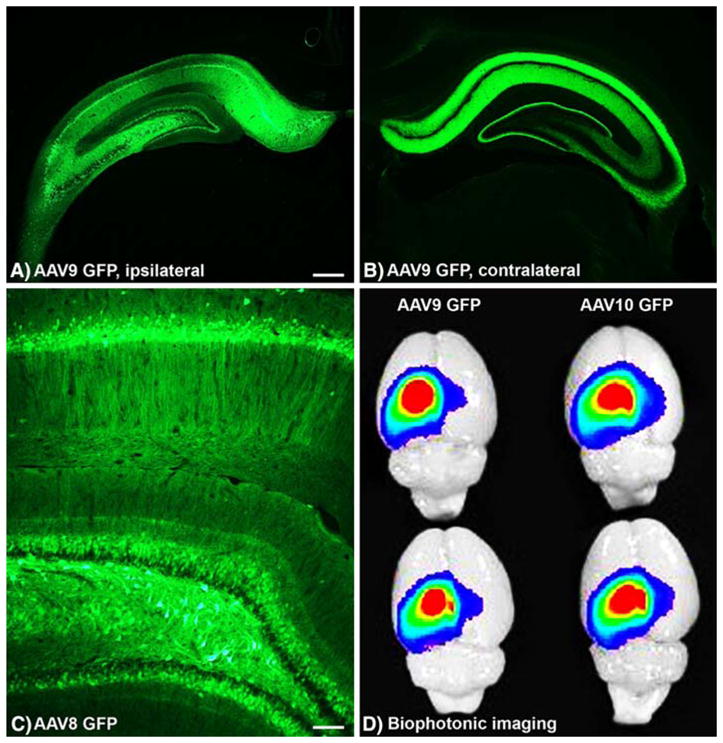
Widespread expression of recombinant green fluorescent protein (GFP) in the rat hippocampus via adeno-associated virus (AAV) vector gene transfer. a, b GFP expressed from AAV9 on the ipsilateral, injected side and the contralateral, uninjected side. Contralateral GFP is largely due to projections from neurons on the injected side. The scarcity of GFP+ somata contralateral to injection argues against transduction subsequent to retrograde transport of vector. c GFP from AAV8 vector. Native GFP fluorescence in a–c, i.e., no immunofluorescent enhancement. d Hippocampal GFP visualized in whole AAV-GFP-injected brains (2 per vector group). Spread and intensity of gene transfer efficiency can be rapidly quantified with the technique. Bars: a, b = 540 μm; c = 104 μm. Reprinted from Klein et al. (2008) with permission
Using lower doses of an AAV9 wild-type tau vector for a partial lesioning effect, we investigated whether aged tissue is more susceptible to neurodegenerative disease by comparing the experimental disease state in young and aged rats (Klein et al. 2009). We compared early and late time points (2 and 8 weeks), and 2 vector doses, after first ascertaining with an AAV9 GFP vector that gene transfer efficiency was equivalent for the two ages. Then, with the tau vector, we observed significant effects of age on the degree of cell loss at both intervals. The most outstanding age difference was observed for the short expression duration and low dose combination, suggesting greater susceptibility to the onset of early stage disease and low level tau expression. Significant rotational behavior occurred only in the aged rats at the high vector dose. Aging effects on microglial activation were more pronounced. We observed a clear microgliosis associated specifically with tau, but not GFP gene transfer, and the effect was stronger in aged samples at both intervals and doses. Because the maximal microgliosis was seen after short expression, but neuronal loss increased after this interval, microgliosis may play a causative role in neuronal death. The study demonstrated that wild-type tau is sufficient for inducing dopaminergic neuron loss (Klein et al. 2009), confirming our earlier comparison of wild-type and P301L tau (Klein et al. 2005), while revealing that microglia function may be a critical early step in tau pathogenesis. DNA microarray data after tau gene transfer also implicate the importance of cytokines and inflammation at an early point in the disease state (Klein et al. unpublished data).
The profound neuronal loss that can be generated by vector-induced tauopathy may help resolve the contested issue of whether tangles are toxic or protective. It is possible, for example, that the severity of neuronal loss compared with actual disease and transgenic rodents is related to a rapid rate of accumulation of aggregated tau. Fast accumulation would be more likely to overwhelm physical or biochemical processes (e.g., intracellular transport) necessary for sustained survival (De Vos et al. 2008; Spires-Jones et al. 2009). In slowly progressing disease, a gradual accumulation of aggregated tau over decades may permit adaptation to intracellular volume displacement. This could maintain intracellular transport sufficient for survival in neurons with slowly growing tangles, while allowing tangles to exert neuroprotective effects such as those hypothesized for oxidative stress (Castellani et al. 2008) or microtubule function (Spires-Jones et al. 2009). Our histological data do make it clear that the number of ‘ghost’ tangles observed is far too low for degradation-resistant tangles to be an obligatory route to neuron death. It is possible that intracellular tangles are protecting the neurons that contain them from cell death mechanisms that claim the large proportion of neurons without tangles. The use of different gene variants, promoters, serotypes, and multiplicity-of-infection parameters will allow controlled use of the gene transfer approach to analyze how disease phenotypes such as tau aggregation, impaired cytoplasmic transport, oxidative stress, and cell-cycle re-entry interact to bring about cell death.
Gene Transfer Modulation of Parkin Function Related to Dopaminergic Neurodegeneration
Mutations in parkin, an enzyme in the protein ubiquitination cascade, cause an inherited form of PD, similar to alpha-synuclein (Shimura et al. 2001). Parkin has been proposed to ubiquitinate alpha-synuclein under some conditions, so loss-of-function mutations in an enzyme that targets alpha-synuclein for degradation could underlie alpha-synuclein/ubiquitin neuropathology in PD (Shimura et al. 2001). In a vector model to cause parkin dysfunction, Dong et al. (2003) overexpressed a known parkin enzyme substrate called CDCrel-1 in the SN of rats and found a robust lesioning effect on the nigrostriatal system. The model has also been used by Jung et al. (2008) to study the effects of co-expression with heat shock proteins (Hsp), with Hsp70 protecting against CDCrel-1. Another parkin substrate called Pael-R, when expressed with an adenovirus vector, produces similar effects on the rat nigrostriatal system of parkin knockout, but not wild-type mice (Kitao et al. 2007). Although blocking parkin function results in dopaminergic neurodegeneration, turning up parkin levels has protective effects, as observed in a number of model systems and for a variety of toxic insults (discussed in Klein et al. 2006b). Parkin co-expression has protected against alpha-synuclein-induced neurodegeneration using either lentivirus (Lo Bianco et al. 2004b) or AAV in rats (Yamada et al. 2005). Similar effects were observed in monkeys (Yasuda et al. 2007). The action of parkin as a general neuroprotective factor is underscored by improved behavioral outcome after 6-OHDA lesions (Manfredsson et al. 2007), or neuronal preservation after low level tau protein expression (Klein et al. 2006b) in AAV/rat experiments. Both parkin and Hsp70 may act to eliminate unwanted and misfolded proteins. In fact, Hsp70 has been shown to provide neuroprotection against alpha-synuclein in a Drosophila model (Auluck et al. 2002), against MPTP lesions in AAV-transduced mice (Dong et al. 2005), and against CDCrel-1 (Jung et al. 2008), making Hsp70 a candidate factor worth pursuing. Lo Bianco et al. (2008) recently described the yeast-derived factor Hsp104 which protected against alpha-synuclein with lentivirus co-expression in rats. The high throughput screening systems of yeast (Liang et al. 2008) and C. elegans (Hamamichi et al. 2008) have uncovered a host of potential protectants against alpha-synuclein toxicity. While parkin and Hsp70 are protective against more than one type of toxic overexpression, hits described in the nematode study were specific for alpha-synuclein versus polyglutamine repeats (Hamamichi et al. 2008). These factors could help target protective function to a specific type of lesion and limit interference with unrelated cell functions. The proteins such as parkin, Hsp70, and the ones uncovered by the screening studies that protect against alpha-synuclein, are by and large associated with protein trafficking and protein degradation. Investigation of these protective factors will lead to clues on alpha-synuclein’s neurotoxic mechanisms and an enzyme therapeutic target.
Gene Transfer Models of Beta Amyloidopathy
Despite the centrality of beta amyloid in Alzheimer’s research (Chiang et al. 2008), remarkably few papers have described gene transfer models of brain beta amyloidosis. Although early vectors were conducive to using somatic gene transfer to look at serum amyloidosis (Kindy et al. 1998), transgenic mice expressing human disease mutations have been the predominant model for studying central amyloid (Rockenstein et al. 2007, Duyckaerts et al. 2008). This was partly because techniques for making whole-animal transgenics outpaced advances in the development of effective gene transfer vectors for brain (Pearson and Choi 1993; Duff et al. 1994; Borchelt et al. 1996). However, a remarkably consistent failure to observe plaques when APP constructs were expressed only in brain was perceived as a deficiency of these models, before the importance of soluble beta amyloid was appreciated. Once plaque-like deposits could be reproduced transgenic mice rapidly became the models of choice. It can now be argued that models demonstrating brain beta amyloid generation without plaque formation provide unique opportunities to focus experimental attention on the toxicity of soluble forms (Bayer et al. 2008).
Gene transfer vectors did find early and extensive application for making in vitro models (Marotta et al. 1989), permitting cell culture studies of transfected cells over-expressing wild-type and mutant APP, secretases, and other gene products that influence APP processing (Askanas et al. 1996; Luo et al. 1999). These allowed many important advances with respect to understanding cellular processing and regulation of APP, and effects of mutations on cellular physiology (Fukuchi et al. 1992; De Strooper et al. 1995; Lynn et al. 1995; Uetsuki et al. 1999; Eckert et al. 2001; Magrané et al. 2005, etc.).
The earliest gene transfer models for CNS amyloidopathy used a two-step approach, transfecting neuron-like transformed cells in culture with human APP constructs and subsequently transplanting those into rodent brains (Neve et al. 1992; Fukuchi et al. 1994; Bayer et al. 1996). These early experiments achieved long-term expression, but tended to show what now would be regarded as severe neuropathology relative to APP transgenic mice or human AD, with marked atrophy, neuronal loss, tauopathy, and extracellular beta amyloid deposits even with wild-type APP sequences. More troubling was the fact that plaque-like features were rarely if ever observed. Results with early whole-animal transgenic mice expressing APP C-terminal peptides (probably in glia selectively) (Sandhu et al. 1991), or made with yeast artificial chromosomes containing human APP sequences (Lamb et al. 1997), were similar although relatively sparse thioflavin-positive amyloid beta deposits were observed in mice transgenic for the Swedish double mutation APP with survivals of 14 months or more and 4–6 transgene copies per cell (Kulnane and Lamb 2001).
Attempts to use adenovirus gene delivery vectors to express human APP in rat brain did demonstrate rapid protein expression in hippocampus, but also severe toxicity within the first week that precluded use as a chronic disease model (Masumura et al. 2000). Lentiviral vectors proved better for long-term APP or presenilin 1 brain expression, but still did not show plaques even after 6 months (Shaughnessy et al. 2005; Hong et al. 2006). We made AAV2 vectors for the gene encoding the Swedish double mutation (K595N/M596L) of APP, under control of a chicken beta actin/cytomegalovirus promoter/enhancer (Gong et al. 2006). Robust neuronal expression of human protein was observed 6 and 12 months after rat hippocampal injections, but no plaque-like deposits were observed by thioflavin S or immunolabeling. Gliosis and cell death were also absent. Irregular granular deposits were observed in dentate gyrus, reminiscent of those observed in transplant studies (Fukuchi et al. 1994) but it could not be ascertained whether these amyloid beta1-42-immunoreactive structures were intra- or extracellular. After 12 month expression, these animals were impaired on water maze acquisition and retention. New AAV vectors designed to selectively produce amyloid beta 1-40, or 1-42 also produced behavioral deficits in rats after 3 month expression durations (Lawlor et al. 2007), in conjunction with elevated soluble but not insoluble beta amyloid. However, although neither plaques nor pathology were evident, the amyloid 1-42 vector rats exhibited some amorphous deposits that did not bind thioflavin. The characteristics of what may be intra-neuronal deposits in several of these models warrant further study in light of data implicating such structures in toxicity (Ohyagi 2008).
Gene transfer vectors are increasingly being used not to merely reproduce disease pathology but to modify it. One example of this is the use of intracranial lentivirus vectors to investigate how differential expression of ApoE isozymes influences beta amyloid accumulation (Dodart et al. 2005). Many of these efforts involve peripheral transduction to generate vaccine epitopes (Frazer et al. 2008; Mouri et al. 2007; Zou et al. 2008), but some focus on brain transduction to mediate enzymatic amyloid reduction (e.g. Hong et al. 2006; Carty et al. 2008; Iwata et al. 2004; Spencer et al. 2008), inhibit gene expression (Singer et al. 2005), or deliver modified amyloid antibodies (Fukuchi et al. 2006; Levites et al. 2006). A persuasive case can be made that intracranial antibody gene therapy for Alzheimer’s will be safer and more effective than peripheral strategies, because the vascular disease highly prevalent in Alzheimer’s patients will interfere with functional action across the blood–brain barrier (Vasilevko and Cribbs 2006). This emphasizes the need to determine how age-and disease-related phenomena affect somatic gene transfer. Beta amyloid in particular is likely to have complex interactions with viral (Wojtowicz et al. 2002) and non-viral gene transfer (Ajmani et al. 2001) that will need to be understood to control gene delivery in brains with abnormal amyloid loads.
Summary and Conclusions
Brain gene transfer has developed into an adaptable toolkit for the neuroscientist, and more and more experiments on the mechanisms of neurodegenerative diseases employ this approach. The ability to target brain regions and to study specific gene products in a rapid, flexible, and cost-effective manner are the most important advantages of the methods, which enable unique opportunities for discovery not possible with other paradigms. The ability to use rat models allows access to a wealth of behavioral, electrophysiological, and biochemical data that are far more extensive than for mice (e.g., hippocampal synaptic physiology). Partial lesions such as those induced by alpha-synuclein gene transfer reflect an early pre-symptomatic disease state, ideal for studying factors that could modulate the subsequent clinical outcome. Tau gene transfer is effective for a broader range of lesioning, which we demonstrated by our studies of vector dose–response, different AAV variants, and in aged versus young subjects. Differences between beta amyloid deposition between transgenic and gene transfer approaches may illuminate the conditions necessary for plaque formation, and isolate effects of soluble and insoluble forms.
The literature on gene vector models of neurodegenerative diseases in mammals is minuscule relative to the thousands of papers studying neurodegeneration with neurotoxins and transgenic mice. Although the vector models of alpha-synuclein, tau, and amyloid have been overshadowed by the transgenic mouse approach, the vector model may be leading for the recently described hallmark TDP-43 pathology associated with amyotrophic lateral sclerosis and frontotemporal lobar degeneration with ubiquitin deposits (Neumann et al. 2006; Tatom et al. 2009). The fact that no diseases are yet associated with elevated TDP-43 expression may spur closer genetic re-examination of human cases. It also serves as a reminder that overexpression models may not reproduce necessary or sufficient components of real diseases.
Although overexpression of synuclein (Singleton et al. 2003), APP (Schupf et al. 2007), tau (Hutton et al. 1998), and polyglutamine (Heng et al. 2008) do occur in their associated neurodegenerative diseases, it remains questionable just how useful gene transfer models will be to study diseases where overexpression per se is not important. The greatest promise for vector models may lie in their utility as unique platforms for developing small-molecule therapeutics. As an alternative to transgenic animals, they afford superior experimental design, precision of interpretation, and simplicity in combining models, economy, and speed. The ability to generate disease-specific neuropathological phenomena across the range of species used in preclinical testing (particularly primates) will facilitate the translational development of innovative treatments. An appreciation for gene transfer models will undoubtedly increase significantly once they actually facilitate the clinical translation of new therapeutic agents.
Acknowledgments
B. Scott Pruett and James Zavecz critiqued the manuscript. National Institute of Neurological Disorders and Stroke R01 NS048450, National Institute on Aging P10485, and the U.S. Department of Veterans Affairs Medical Research Service supported the work.
Contributor Information
Ronald L. Klein, Department of Pharmacology, Toxicology & Neuroscience, and Gene Therapy Program, Louisiana State University Health Sciences Center, Shreveport, LA 71130, USA
David B. Wang, Department of Pharmacology, Toxicology & Neuroscience, and Gene Therapy Program, Louisiana State University Health Sciences Center, Shreveport, LA 71130, USA
Michael A. King, Email: making@ufl.edu, Department of Pharmacology & Therapeutics, University of Florida College of Medicine, Gainesville, FL 32610, USA. NF/SG VA Medical Center, Gainesville, FL 32608, USA
References
- Ajmani PS, Wang W, Tang F, King MA, Meyer EM, Hughes JA. Transgene delivery with a cationic lipid in the presence of amyloid beta (betaAP) peptide. Neurochem Res. 2001;26(3):195–202. doi: 10.1023/a:1010956231321. [DOI] [PubMed] [Google Scholar]
- Askanas V, McFerrin J, Baqué S, Alvarez RB, Sarkozi E, Engel WK. Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc Natl Acad Sci USA. 1996;93(3):1314–1319. doi: 10.1073/pnas.93.3.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295(5556):865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neuro-degeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Fossgreen A, Czech C, Beyreuther K, Wiestler OD. Plaque formation in brain transplants exposed to human beta-amyloid precursor protein 695. Acta Neuropathol. 1996;92(2):130–137. doi: 10.1007/s004010050500. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Breyhan H, Duan K, Rettig J, Wirths O. Intraneuronal beta-amyloid is a major risk factor—novel evidence from the APP/PS1KI mouse model. Neurodegener Dis. 2008;5(3–4):140–142. doi: 10.1159/000113684. [DOI] [PubMed] [Google Scholar]
- Bilang-Bleuel A, Revah F, Colin P, Locquet I, Robert JJ, Mallet J, Horellou P. Intrastriatal injection of an adenoviral vector expressing glial-cell-line-derived neurotrophic factor prevents dopaminergic neuron degeneration and behavioral impairment in a rat model of Parkinson disease. Proc Natl Acad Sci USA. 1997;94(16):8818–8823. doi: 10.1073/pnas.94.16.8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blömer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol. 1997;71(9):6641–6649. doi: 10.1128/jvi.71.9.6641-6649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13(6):159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Brunden KR, Trojanowski JQ, Lee VM. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis. 2008;14(4):393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10(2):302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Carty NC, Nash K, Lee D, Mercer M, Gottschall PE, Meyers C, Muzyczka N, Gordon MN, Morgan D. Adeno-associated viral (AAV) serotype 5 vector mediated gene delivery of endothelin-converting enzyme reduces Abeta deposits in APP + PS1 transgenic mice. Mol Ther. 2008;16(9):1580–1586. doi: 10.1038/mt.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel-Barthe MN, Jazat-Poindessous F, Barneoud P, Vigne E, Revah F, Mallet J, Lamour Y. Direct intracerebral nerve growth factor gene transfer using a recombinant adenovirus, effect on basal forebrain cholinergic neurons during aging. Neurobiol Dis. 1996;3(1):76–86. doi: 10.1006/nbdi.1996.0008. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Nunomura A, Lee HG, Perry G, Smith MA. Phosphorylated tau: toxic, protective, or none of the above. J Alzheimers Dis. 2008;14(4):377–383. doi: 10.3233/jad-2008-14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF. In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson’s disease? Exp Neurol. 2008;209(1):22–27. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PK, Lam MA, Luo Y. The many faces of amyloid beta in Alzheimer’s disease. Curr Mol Med. 2008;8(6):580–584. doi: 10.2174/156652408785747951. [DOI] [PubMed] [Google Scholar]
- Choi-Lundberg DL, Lin Q, Chang YN, Chiang YL, Hay CM, Mohajeri H, Davidson BL, Bohn MC. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science. 1997;275(5301):838–841. doi: 10.1126/science.275.5301.838. [DOI] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29(11):3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360(6405):672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Congdon EE, Duff KE. Is tau aggregation toxic or protective? J Alzheimers Dis. 2008;14(4):453–457. doi: 10.3233/jad-2008-14415. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, Zabner J, Ghodsi A, Chiorini JA. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA. 2000;97(7):3428–3432. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 1995;14(20):4932–4938. doi: 10.1002/j.1460-2075.1995.tb00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Di Maria E, Tabaton M, Vigo T, Abbruzzese G, Bellone E, Donati C, Frasson E, Marchese R, Montagna P, Munoz DG, Pramstaller PP, Zanusso G, Ajmar F, Mandich P. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47:374–377. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Tau and synuclein and their role in neuropathology. Brain Pathol. 1999;9:657–661. doi: 10.1111/j.1750-3639.1999.tb00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2005;102(4):1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Ferger B, Paterna JC, Vogel D, Furler S, Osinde M, Feldon J, Büeler H. Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proc Natl Acad Sci USA. 2003;100(21):12438–12443. doi: 10.1073/pnas.2132992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Wolfer DP, Lipp HP, Büeler H. Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther. 2005;11(1):80–88. doi: 10.1016/j.ymthe.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Duff K, McGuigan A, Huxley C, Schulz F, Hardy J. Insertion of a pathogenic mutation into a yeast artificial chromosome containing the human amyloid precursor protein gene. Gene Ther. 1994;1(1):70–75. [PubMed] [Google Scholar]
- During MJ, Naegele JR, O’Malley KL, Geller AI. Long-term behavioral recovery in parkinsonian rats by an HSV vector expressing tyrosine hydroxylase. Science. 1994;266(5189):1399–1403. doi: 10.1126/science.266.5189.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert A, Steiner B, Marques C, Leutz S, Romig H, Haass C, Müller WE. Elevated vulnerability to oxidative stress-induced cell death and activation of caspase-3 by the Swedish amyloid precursor protein mutation. J Neurosci Res. 2001;64(2):183–192. doi: 10.1002/jnr.1064. [DOI] [PubMed] [Google Scholar]
- Eslamboli A, Romero-Ramos M, Burger C, Bjorklund T, Muzyczka N, Mandel RJ, Baker H, Ridley RM, Kirik D. Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain. 2007;130(3):799–815. doi: 10.1093/brain/awl382. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404(6776):394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Federoff HJ, Geschwind MD, Geller AI, Kessler JA. Expression of nerve growth factor in vivo from a defective herpes simplex virus 1 vector prevents effects of axotomy on sympathetic ganglia. Proc Natl Acad Sci USA. 1992;89(5):1636–1640. doi: 10.1073/pnas.89.5.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer ME, Hughes JE, Mastrangelo MA, Tibbens JL, Federoff HJ, Bowers WJ. Reduced pathology and improved behavioral performance in Alzheimer’s disease mice vaccinated with HSV amplicons expressing amyloid-beta and interleukin-4. Mol Ther. 2008;16(5):845–853. doi: 10.1038/mt.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi K, Kamino K, Deeb SS, Furlong CE, Sundstrom JA, Smith AC, Martin GM. Expression of a carboxy-terminal region of the beta-amyloid precursor protein in a heterogeneous culture of neuroblastoma cells: evidence for altered processing and selective neurotoxicity. Brain Res Mol Brain Res. 1992;16(1–2):37–46. doi: 10.1016/0169-328x(92)90191-d. [DOI] [PubMed] [Google Scholar]
- Fukuchi KI, Kunkel DD, Schwartzkroin PA, Kamino K, Ogburn CE, Furlong CE, Martin GM. Overexpression of a C-terminal portion of the beta-amyloid precursor protein in mouse brains by transplantation of transformed neuronal cells. Exp Neurol. 1994;127(2):253–264. doi: 10.1006/exnr.1994.1101. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Tahara K, Kim HD, Maxwell JA, Lewis TL, Accavitti-Loper MA, Kim H, Ponnazhagan S, Lalonde R. Anti-Abeta single-chain antibody delivery via adeno-associated virus for treatment of Alzheimer’s disease. Neurobiol Dis. 2006;23(3):502–511. doi: 10.1016/j.nbd.2006.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furler S, Paterna JC, Weibel M, Büeler H. Recombinant AAV vectors containing the foot and mouth disease virus 2A sequence confer efficient bicistronic gene expression in cultured cells and rat substantia nigra neurons. Gene Ther. 2001;8(11):864–873. doi: 10.1038/sj.gt.3301469. [DOI] [PubMed] [Google Scholar]
- Geller AI, Breakefield XO. A defective HSV-1 vector expresses Escherichia coli beta-galactosidase in cultured peripheral neurons. Science. 1988;241(4873):1667–1669. doi: 10.1126/science.241.4873.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat C, Saint-Pierre M, Bousquet M, Lévesque D, Rouillard C, Cicchetti F. Differences between subacute and chronic MPTP mice models: investigation of dopaminergic neuronal degeneration and alpha-synuclein inclusions. J Neurochem. 2009;109(5):1469–1482. doi: 10.1111/j.1471-4159.2009.06072.x. [DOI] [PubMed] [Google Scholar]
- Gong Y, Meyer EM, Meyers CA, Klein RL, King MA, Hughes JA. Memory-related deficits following selective hippocampal expression of Swedish mutation amyloid precursor protein in the rat. Exp Neurol. 2006;200(2):371–377. doi: 10.1016/j.expneurol.2006.02.136. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci USA. 2008;105(2):763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther. 1998;9(18):2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall GF, Yao J. Modeling tauopathy: a range of complementary approaches. Biochim Biophys Acta (BBA) (Mol Basis Disease) 2005;1739(2–3):224–239. doi: 10.1016/j.bbadis.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA. Hypothesis-based RNAi screening identifies neuro-protective genes in a Parkinson’s disease model. Proc Natl Acad Sci USA. 2008;105(2):728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashita-Kinoh H, Yamada M, Yokota T, Mizuno Y, Mochizuki H. Down-regulation of alpha-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson’s disease rat model. Biochem Biophys Res Commun. 2006;341(4):1088–1095. doi: 10.1016/j.bbrc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Heng MY, Detloff PJ, Albin RL. Rodent genetic models of Huntington disease. Neurobiol Dis. 2008;32:1–9. doi: 10.1016/j.nbd.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Hong CS, Goins WF, Goss JR, Burton EA, Glorioso JC. Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer’s disease-related amyloid-beta peptide in vivo. Gene Ther. 2006;13(14):1068–1079. doi: 10.1038/sj.gt.3302719. [DOI] [PubMed] [Google Scholar]
- Horellou P, Vigne E, Castel MN, Barnéoud P, Colin P, Perricaudet M, Delaère P, Mallet J. Direct intracerebral gene transfer of an adenoviral vector expressing tyrosine hydroxylase in a rat model of Parkinson’s disease. NeuroReport. 1994;6(1):49–5. doi: 10.1097/00001756-199412300-00014. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso AC, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739(2–3):198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Alonso AC, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118(1):53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B, Gerard NP, Gerard C, Ozawa K, Saido TC. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J Neurosci. 2004;24(4):991–998. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18(Suppl 6):S2–S12. doi: 10.1002/mds.10557. [DOI] [PubMed] [Google Scholar]
- Jung AE, Fitzsimons HL, Bland RJ, During MK, Young D. HSP70 and constitutively active HSF1 mediate protection against CDCrel-1-mediated toxicity. Mol Ther. 2008;16(6):1048–1055. doi: 10.1038/mt.2008.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ. alpha-Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):87–95. doi: 10.1007/s00401-007-0302-x. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O’Malley KL, During MJ. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8(2):148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- Kindy MS, King AR, Yu J, Gerardot C, Whitley J, de Beer FC. Adenoviral expression of murine serum amyloid A proteins to study amyloid fibrillogenesis. Biochem J. 1998;332(Pt 3):721–728. doi: 10.1042/bj3320721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Björklund A. Modeling CNS neurodegeneration by overexpression of disease-causing proteins using viral vectors. Trends Neurosci. 2003;26(7):386–392. doi: 10.1016/S0166-2236(03)00164-4. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22(7):2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson’s disease. Proc Natl Acad Sci USA. 2003;100(5):2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao Y, Imai Y, Ozawa K, Kataoka A, Ikeda T, Soda M, Nakimawa K, Kiyama H, Stern DM, Hori O, Wakamatsu K, Ito S, Itohara S, Takahashi R, Ogawa S. Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Hum Mol Genet. 2007;16(1):50–60. doi: 10.1093/hmg/ddl439. [DOI] [PubMed] [Google Scholar]
- Klein RL, Meyer EM, Peel AL, Zolotukhin S, Meyers C, Muzyczka N, King MA. Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp Neurol. 1998;150:183–194. doi: 10.1006/exnr.1997.6736. [DOI] [PubMed] [Google Scholar]
- Klein RL, Hamby ME, Hirko AC, Gong Y, Wang S, Hughes J, King MA, Meyer EM. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp Neurol. 2002a;176:66–74. doi: 10.1006/exnr.2002.7942. [DOI] [PubMed] [Google Scholar]
- Klein RL, King MA, Hamby ME, Meyer EM. Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum Gene Ther. 2002b;13:605–612. doi: 10.1089/10430340252837206. [DOI] [PubMed] [Google Scholar]
- Klein RL, Lin WL, Dickson DW, Lewis J, Hutton M, Duff K, Meyer EM, King MA. Rapid neurofibrillary tangle formation after localized gene transfer of mutated tau. Am J Pathol. 2004;164:347–353. doi: 10.1016/S0002-9440(10)63124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Lin WL, Dickson DW. Tau gene transfer, but not alpha-synuclein, induces both progressive dopamine neuron degeneration and rotational behavior in the rat. Neurobiol Dis. 2005;20:64–73. doi: 10.1016/j.nbd.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Henderson KM, Petrucelli L. Parkin is protective for substantia nigra dopamine neurons in a tau gene transfer neurodegeneration model. Neurosci Lett. 2006a;401:130–135. doi: 10.1016/j.neulet.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RW. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006b;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Diaczynsky CG, Salvatore MF. Tau expression levels from various adeno-associated virus vector serotypes produce graded neurodegenerative disease states. Eur J NeuroSci. 2008a;27:1615–1625. doi: 10.1111/j.1460-9568.2008.06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP. AAV 8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol Ther. 2008b;16:89–96. doi: 10.1038/sj.mt.6300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Diaczynsky CG, Wang DB. Pronounced microgliosis and neurodegeneration in aged rats after tau gene transfer. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2008.12.002. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz A, Falkenburger BH, Gerhardt E, Drinkut A, Schulz JB. Aggregate formation and toxicity by wild-type and R621C synphilin-1 in the nigrostriatal system of mice using adenoviral vectors. J Neurochem. 2009;108(1):139–146. doi: 10.1111/j.1471-4159.2008.05755.x. [DOI] [PubMed] [Google Scholar]
- Kulnane LS, Lamb BT. Neuropathological characterization of mutant amyloid precursor protein yeast artificial chromosome transgenic mice. Neurobiol Dis. 2001;8(6):982–992. doi: 10.1006/nbdi.2001.0446. [DOI] [PubMed] [Google Scholar]
- Lakso M, Vartiainen S, Moilanen AM, Sirviö J, Thomas JH, Nass R, Blakely RD, Wong G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem. 2003;86(1):165–172. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- Lamb BT, Call LM, Slunt HH, Bardel KA, Lawler AM, Eckman CB, Younkin SG, Holtz G, Wagner SL, Price DL, Sisodia SS, Gearhart JD. Altered metabolism of familial Alzheimer’s disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum Mol Genet. 1997;6(9):1535–1541. doi: 10.1093/hmg/6.9.1535. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87(2):440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor PA, Bland RJ, Das P, Price RW, Holloway V, Smithson L, Dicker BL, During MJ, Young D, Golde TE. Novel rat Alzheimer’s disease models based on AAV-mediated gene transfer to selectively increase hippocampal Abeta levels. Mol Neurodegener. 2007;2:11. doi: 10.1186/1750-1326-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gal La Salle G, Robert JJ, Berrard S, Ridoux V, Stratford-Perricaudet LD, Perricaudet M, Mallet J. An adenovirus vector for gene transfer into neurons and glia in the brain. Science. 1993;259(5097):988–990. doi: 10.1126/science.8382374. [DOI] [PubMed] [Google Scholar]
- Lee VM, Kenyon TK, Trojanowski JQ. Transgenic animal models of tauopathies. Biochim Biophys Acta. 2005;1739:251–259. doi: 10.1016/j.bbadis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Levites Y, Jansen K, Smithson LA, Dakin R, Holloway VM, Das P, Golde TE. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. 2006;26(46):11923–11928. doi: 10.1523/JNEUROSCI.2795-06.2006. Erratum in J Neurosci (2006) 26(49), preceding 12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25(4):402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- Liang J, Clark-Dixon C, Wang S, Flower TR, Williams-Hart T, Zweig R, Robinson LC, Tatchell K, Witt SN. Novel suppressors of alpha-synuclein toxicity identified using yeast. Hum Mol Genet. 2008;17(23):3784–3795. doi: 10.1093/hmg/ddn276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci USA. 2002;99(16):10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Déglon N, Pralong W, Aebischer P. Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson’s disease. Neurobiol Dis. 2004a;17(2):283–289. doi: 10.1016/j.nbd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci USA. 2004b;101(50):17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Shorter J, Régulier E, Lashuel H, Iwatsubo T, Lindquist S, Aebischer P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest. 2008;118(9):3087–3097. doi: 10.1172/JCI35781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WD, Qu G, Sferra TJ, Clark R, Chen R, Johnson PR. Adeno-associated virus-mediated gene transfer to the brain: duration and modulation of expression. Hum Gene Ther. 1999;10(2):201–213. doi: 10.1089/10430349950018995. [DOI] [PubMed] [Google Scholar]
- Lundberg C, Björklund T, Carlsson T, Jakobsson J, Hantraye P, Déglon N, Kirik D. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr Gene Ther. 2008;8(6):461–473. doi: 10.2174/156652308786847996. [DOI] [PubMed] [Google Scholar]
- Luo JJ, Wallace W, Riccioni T, Ingram DK, Roth GS, Kusiak JW. Death of PC12 cells and hippocampal neurons induced by adenoviral-mediated FAD human amyloid precursor protein gene expression. J Neurosci Res. 1999;55(5):629–642. doi: 10.1002/(SICI)1097-4547(19990301)55:5<629::AID-JNR10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Lynn BD, Marotta CA, Nagy JI. Propagation of intercellular calcium waves in PC12 cells overexpressing a carboxy-terminal fragment of amyloid precursor protein. Neurosci Lett. 1995;199(1):21–24. doi: 10.1016/0304-3940(95)12028-3. [DOI] [PubMed] [Google Scholar]
- Magrané J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci. 2005;25(47):10960–10969. doi: 10.1523/JNEUROSCI.1723-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel RJ, Spratt SK, Snyder RO, Leff SE. Midbrain injection of recombinant adeno-associated virus encoding rat glial cell line-derived neurotrophic factor protects nigral neurons in a progressive 6-hydroxydopamine-induced degeneration model of Parkinson’s disease in rats. Proc Natl Acad Sci USA. 1997;94(25):14083–14088. doi: 10.1073/pnas.94.25.14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredsson FP, Burger C, Sullivan LF, Muzyczka N, Lewin AS, Mandel RJ. rAAV-mediated nigral human parkin over-expression partially ameliorates motor deficits via enhanced dopamine neurotransmission in a rat model of Parkinson’s disease. Exp Neurol. 2007;207(2):289–301. doi: 10.1016/j.expneurol.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Marotta CA, Chou WG, Majocha RE, Watkins R, LaBonne C, Zain SB. Overexpression of amyloid precursor protein A4 (beta-amyloid) immunoreactivity in genetically transformed cells: implications for a cellular model of Alzheimer amyloidosis. Proc Natl Acad Sci USA. 1989;86(1):337–341. doi: 10.1073/pnas.86.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. Transgenic mice with human mutant genes causing Parkinson’s disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration. Rev Neurosci. 2007;18(2):115–136. doi: 10.1515/revneuro.2007.18.2.115. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287(5456):1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Masumura M, Hata R, Nishimura I, Uetsuki T, Sawada T, Yoshikawa K. Caspase-3 activation and inflammatory responses in rat hippocampus inoculated with a recombinant adenovirus expressing the Alzheimer amyloid precursor protein. Brain Res Mol Brain Res. 2000;80(2):219–227. doi: 10.1016/s0169-328x(00)00163-7. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Xiao X, Li J, Breese GR, Samulski RJ. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 1996;713(1–2):99–107. doi: 10.1016/0006-8993(95)01488-8. [DOI] [PubMed] [Google Scholar]
- McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT, McLean PJ. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol. 2009;68(5):515–524. doi: 10.1097/NEN.0b013e3181a24b53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Murrell JR, Gearing M, Spillantini MG, Goedert M, Crowther RA, Levey AI, Jones R, Green J, Shoffner JM, Wainer BH, Schmidt ML, Trojanowski JQ, Ghetti B. Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol. 1999;58:335–345. doi: 10.1097/00005072-199904000-00004. [DOI] [PubMed] [Google Scholar]
- Modlich U, Baum C. Preventing and exploiting the oncogenic potential of integrating gene vectors. J Clin Invest. 2009;19(4):755–758. doi: 10.1172/JCI38831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, Dawson TM. Value of genetic models in understanding the cause and mechanisms of Parkinson’s disease. Curr Neurol Neurosci Rep. 2008;8(4):288–296. doi: 10.1007/s11910-008-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Hara H, Mizoguchi H, Tabira T, Nabeshima T. Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J. 2007;21(9):2135–2148. doi: 10.1096/fj.06-7685com. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996;93(21):11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neve RL, Kammesheidt A, Hohmann CF. Brain transplants of cells expressing the carboxyl-terminal fragment of the Alzheimer amyloid protein precursor cause specific neuropathology in vivo. Proc Natl Acad Sci USA. 1992;89(8):3448–3452. doi: 10.1073/pnas.89.8.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyagi Y. Intracellular amyloid beta-protein as a therapeutic target for treating Alzheimer’s disease. Curr Alzheimer Res. 2008;5(6):555–561. doi: 10.2174/156720508786898514. [DOI] [PubMed] [Google Scholar]
- Osinde M, Clavaguera F, May-Nass R, Tolnay M, Dev KK. Lentivirus Tau (P301S) expression in adult amyloid precursor protein (APP)-transgenic mice leads to tangle formation. Neuropathol Appl Neurobiol. 2008;34(5):523–531. doi: 10.1111/j.1365-2990.2008.00936.x. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302(5651):1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BE, Choi TK. Expression of the human beta-amyloid precursor protein gene from a yeast artificial chromosome in transgenic mice. Proc Natl Acad Sci USA. 1993;90(22):10578–10582. doi: 10.1073/pnas.90.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ. Efficient transduction of green fluorescent protein in spinal cord neurons using adeno-associated virus vectors containing cell type-specific promoters. Gene Ther. 1997;4(1):16–24. doi: 10.1038/sj.gt.3300358. [DOI] [PubMed] [Google Scholar]
- Poewe W. Treatments for Parkinson disease—past achievements and current clinical needs. Neurology. 2009;72(7 Suppl):S65–S73. doi: 10.1212/WNL.0b013e31819908ce. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT, Schaffer AA, Lazzarini AM, Nussbaum RL, Duvoisin RC. Mapping of a gene for Parkinson’s disease to chromosome 4q21–q23. Science. 1996;274(5290):1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VM, Schellenberg GD. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression results in mitochondrial dysfunction in cortical neurons. Implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009 Apr 23; doi: 10.1074/jbc.M808908200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii MS, Aisen PS. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Crews L, Masliah E. Transgenic animal models of neurodegenerative diseases and their application to treatment development. Adv Drug Deliv Rev. 2007;59(11):1093–1102. doi: 10.1016/j.addr.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Sandhu FA, Salim M, Zain SB. Expression of the human beta-amyloid protein of Alzheimer’s disease specifically in the brains of transgenic mice. J Biol Chem. 1991;266(32):21331–21334. [PubMed] [Google Scholar]
- Schneider JA, Bienias JL, Gilley DW, Kvarnberg DE, Mufson EJ, Bennett DA. Improved detection of substantia nigra pathology in Alzheimer’s disease. J Histochem Cytochem. 2002;50:99–106. doi: 10.1177/002215540205000111. [DOI] [PubMed] [Google Scholar]
- Schupf N, Patel B, Pang D, Zigman WB, Silverman W, Mehta PD, Mayeux R. Elevated plasma beta-amyloid peptide Abeta(42) levels, incident dementia, and mortality in Down syndrome. Arch Neurol. 2007;64:1007–1013. doi: 10.1001/archneur.64.7.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senut MC, Suhr ST, Kaspar B, Gage FH. Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J Neurosci. 2000;20(1):219–229. doi: 10.1523/JNEUROSCI.20-01-00219.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy L, Thomas MB, Wakefield J, Chamblin B, Nair A, Koentgen F, Ramabhadran R. Lentiviral vector-based models of amyloid pathology: from cells to animals. Curr Alzheimer Res. 2005;2(2):239–247. doi: 10.2174/1567205053585963. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001;293(5528):263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8(10):1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Spencer B, Marr RA, Rockenstein E, Crews L, Adame A, Potkar R, Patrick C, Gage FH, Verma IM, Masliah E. Long-term neprilysin gene transfer is associated with reduced levels of intracellular Abeta and behavioral improvement in APP transgenic mice. BMC Neurosci. 2008;9:109. doi: 10.1186/1471-2202-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol. 1998a;153(5):1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Masegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998b;95(11):6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32(3):150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem. 2007;100(6):1449–1457. doi: 10.1111/j.1471-4159.2006.04310.x. [DOI] [PubMed] [Google Scholar]
- Szerlip NJ, Walbridge S, Yang L, Morrison PF, Degen JW, Jarrell ST, Kouri J, Kerr PB, Kotin R, Oldfield EH, Lonser RR. Real-time imaging of convection-enhanced delivery of viruses and virus-sized particles. J Neurosurg. 2007;107(3):560–567. doi: 10.3171/JNS-07/09/0560. [DOI] [PubMed] [Google Scholar]
- Tatom JB, Wang DB, Dayton RD, Hutton ML, Dickson DW, Klein RL. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig’s disease in rats via TDP-43 overexpression. Mol Ther. 2009;17(4):607–613. doi: 10.1038/mt.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172(3):786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Spec2):R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- Uetsuki T, Takemoto K, Nishimura I, Okamoto M, Niinobe M, Momoi T, Miura M, Yoshikawa K. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci. 1999;19(16):6955–6964. doi: 10.1523/JNEUROSCI.19-16-06955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilevko V, Cribbs DH. Novel approaches for immunotherapeutic intervention in Alzheimer’s disease. Neurochem Int. 2006;49(2):113–126. doi: 10.1016/j.neuint.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Oyanagi K, Makifuchi T, Ikuta F, Homma A, Homma Y, Horikawa Y, Tokiguchi S. Corticobasal degeneration: etiopathological significance of the cytoskeletal alterations. Acta Neuropathol. 1994;87:545–553. doi: 10.1007/BF00293314. [DOI] [PubMed] [Google Scholar]
- Wojtowicz WM, Farzan M, Joyal JL, Carter K, Babcock GJ, Israel DI, Sodroski J, Mirzabekov T. Stimulation of enveloped virus infection by beta-amyloid fibrils. J Biol Chem. 2002 Sep 20;277(38):35019–35024. doi: 10.1074/jbc.M203518200. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72(3):2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Janson CG, Mastakov M, Lawlor P, Young D, Mouravlev A, Fitzsimons H, Choi KL, Ma H, Dragunow M, Leone P, Chen Q, Dicker B, During MJ. Quantitative comparison of expression with adeno-associated virus (AAV-2) brain-specific gene cassettes. Gene Ther. 2001;8(17):1323–1332. doi: 10.1038/sj.gt.3301529. [DOI] [PubMed] [Google Scholar]
- Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem. 2004;91(2):451–461. doi: 10.1111/j.1471-4159.2004.02728.x. [DOI] [PubMed] [Google Scholar]
- Yamada M, Mizuno Y, Mochizuki H. Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson’s disease. Hum Gene Ther. 2005;16(2):262–270. doi: 10.1089/hum.2005.16.262. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Miyachi S, Kitagawa R, Wada K, Nihira T, Ren YR, Hirai Y, Ageyama N, Terao K, Shimada T, Takada M, Mizuno Y, Mochizuki H. Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience. 2007;144(2):743–753. doi: 10.1016/j.neuroscience.2006.09.052. [DOI] [PubMed] [Google Scholar]
- Zaiss AK, Muruve DA. Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 2008;15(11):808–816. doi: 10.1038/gt.2008.54. [DOI] [PubMed] [Google Scholar]
- Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, Novak M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J Neuroimmunol. 2009;209(1–2):16–25. doi: 10.1016/j.jneuroim.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Hauswirth WW, Guy J, Muzyczka N. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J Virol. 1996;70(7):4646–4654. doi: 10.1128/jvi.70.7.4646-4654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6(6):973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- Zou J, Yao Z, Zhang G, Wang H, Xu J, Yew DT, Forster EL. Vaccination of Alzheimer’s model mice with adenovirus vector containing quadrivalent foldable Abeta(1–15) reduces Abeta burden and behavioral impairment without Abeta-specific T cell response. J Neurol Sci. 2008;272(1–2):87–98. doi: 10.1016/j.jns.2008.05.003. [DOI] [PubMed] [Google Scholar]