


Abstract
The generation of reactive oxygen species in the cell provokes, among other lesions, the formation of 8-oxo-7,8-dihydroguanine (8-oxoG) in DNA. Due to mispairing with adenine during replication, 8-oxoG is highly mutagenic. To minimise the mutagenic potential of this oxidised purine, human cells have a specific 8-oxoG DNA glycosylase/AP lyase (hOGG1) that initiates the base excision repair (BER) of 8-oxoG. We show here that in vitro this first enzyme of the BER pathway is relatively inefficient because of a high affinity for the product of the reaction it catalyses (half-life of the complex is >2 h), leading to a lack of hOGG1 turnover. However, the glycosylase activity of hOGG1 is stimulated by the major human AP endonuclease, HAP1 (APE1), the enzyme that performs the subsequent step in BER, as well as by a catalytically inactive mutant (HAP1-D210N). In the presence of HAP1, the AP sites generated by the hOGG1 DNA glycosylase can be occupied by the endonuclease, avoiding the re-association of hOGG1. Moreover, the glycosylase has a higher affinity for a non-cleaved AP site than for the cleaved DNA product generated by HAP1. This would shift the equilibrium towards the free glycosylase, making it available to initiate new catalytic cycles. In contrast, HAP1 does not affect the AP lyase activity of hOGG1. This stimulation of only the hOGG1 glycosylase reaction accentuates the uncoupling of its glycosylase and AP lyase activities. These data indicate that, in the presence of HAP1, the BER of 8-oxoG residues can be highly efficient by bypassing the AP lyase activity of hOGG1 and thus excluding a potentially rate limiting step.
INTRODUCTION
As a consequence of the leakage of reactive oxygen species (ROS) from mitochondria or the exposure of cells to exogenous sources of oxidative stress, potentially toxic and mutagenic lesions are produced on the DNA. A prevalent form of DNA modification appearing under oxidative stress conditions is 8-oxo-7,8-dihydroguanine (8-oxoG), a highly mutagenic lesion that can mispair with adenine (1). As is the case for most oxidised bases, the preferred repair mechanism for 8-oxoG residues is the base excision repair (BER) pathway. This pathway is initiated by the recognition of the modified base by a specific DNA glycosylase. In Escherichia coli, two DNA glycosylases prevent mutagenesis by 8-oxoG: the Fpg protein, which excises 8-oxoG from damaged DNA, and the MutY protein, which excises the adenine residues incorporated by DNA polymerases opposite 8-oxoG (2). Inactivation of both the fpg (mutM) and mutY (micA) genes of E.coli results in a strong G·C→T·A mutator phenotype (3–6). In Saccharomyces cerevisiae, a DNA glycosylase encoded by the OGG1 gene, and named yOgg1, catalyses the removal of 8-oxoG from damaged DNA (7,8). As a consequence, Ogg1-deficient strains of S.cerevisiae exhibit a G·C→T·A mutator phenotype (9). In human cells, the hOGG1 protein carries out the initial excision of mutagenic 8-oxoG residues while oxidised pyrimidines are removed by hNTH1. Recently it has been shown that ogg1 –/– mice accumulate 8-oxoG in their DNA and display a mutator phenotype in non-proliferative tissues (10,11). hOGG1 and hNTH1 share the property of being bi-functional enzymes. After removing the oxidised base through their DNA glycosylase activity, they can act as AP lyases, cleaving the DNA phosphodiester backbone at the 3′ side of the abasic site. The product resulting from the processing of the oxidised bases by the DNA glycosylase and AP lyase activities is channelled into the short patch repair pathway (12–14). The nicked DNA intermediate generated by this catalytic activity is potentially cytotoxic (15). The in vitro reconstitution of the short patch excision repair of oxidised pyrimidines initiated by the hNTH1 DNA glycosylase/AP lyase has provided the basis for a BER model (16). This model proposes that the removal of the 3′-α,β-unsaturated aldehyde resulting from the AP lyase activity of bi-functional glycosylases is carried out by the major AP endonuclease, HAP1 (APE1), leaving a 1 nt gap with a 3′-OH terminus. In the presence of XRCC1, repair is completed by the insertion of a single nucleotide by DNA polymerase β, and subsequent ligation of the resulting nick is mediated by the XRCC1/Lig III complex (14).
In vitro studies have shown that glycosylases bind tightly to abasic sites on DNA (17–21), thus provoking a reduction in enzyme turnover. The overall DNA repair process would then be slowed unless other factors were recruited to either stimulate enzyme turnover or eliminate the reaction product of this initial step in the BER pathway. In agreement with this hypothesis, it has been demonstrated that the activities of two mono-functional glycosylases, uracil DNA glycosylase (UDG) and thymidine DNA glycosylase (TDG), are enhanced by the human AP endonuclease, HAP1 (18,20). In this work, we present evidence that hOGG1 glycosylase activity is stimulated by HAP1, thereby bypassing the AP lyase reaction of hOGG1 that would otherwise be a rate limiting step in the repair of 8-oxoG residues.
MATERIALS AND METHODS
Oligonucleotide substrates and enzymes
34mer oligodeoxyribonucleotides containing a single 8-oxoG residue (a kind gift of Dr Jean Cadet, CEA-Grenoble) or a tetrahydrofuranyl residue (F) (Eurogentec) were labelled at the 5′-end using [γ-32P]ATP (3000 Ci/mmol; Amersham) and T4 polynucleotide kinase (New England Biolabs) (22). The same 34mer containing either 8-oxoG or a uracil residue (U) was labelled at the 3′-end using [α-32P]ddATP (5000 Ci/mmol; Amersham Pharmacia) and a terminal transferase (New England Biolabs), producing a 35mer oligonucleotide. In both cases, the 32P-labelled strands were hybridised with a complementary sequence, containing a cytosine (C) opposite the lesion, yielding the duplexes 8-oxoG:C, F:C and U:C, respectively. The duplex DNA containing a 5′ side incised AP site was prepared as follows: a reaction mixture (20 µl) containing 1 pmol of the 32P-labelled [F:C] duplex was incubated at 37°C in reaction buffer (25 mM Tris–HCl pH 8.0, 75 mM NaCl, 1 mM MgCl2) with an excess of endonuclease IV (endo IV) protein. Following incision of >97% of the AP sites, endo IV was heat-inactivated at 90°C for 15 min. The [3H]Me-FapyG-poly(dG–dC):poly(dG–dC) used was prepared as previously described (23). The recombinant human OGG1, GST–OGG1, HAP1 and mutant derivative HAP1-D210N enzymes were purified as described (24–26). Endo IV and Fpg enzymes were from our laboratory stocks. UDG was from New England Biolabs.
Enzymatic assays
In a standard reaction (16 µl final volume), 50 fmol of the 8-oxoG:C-labelled duplex was incubated in reaction buffer [25 mM Tris–HCl pH 8.0, 75 mM NaCl, 1 mM MgCl2, 0.4 mg/ml bovine serum albumin (BSA)] with various amounts of hOGG1 and HAP1. Reactions were carried out at 37°C for 30 min. To measure the total amount of generated AP sites, an aliquot of each reaction mixture (8 µl) was mixed with 4 µl of a solution containing 30 mM EDTA and 0.6 N NaOH. The mixtures were then heated for 30 min at 90°C and mixed with 4.5 µl of formamide dye, followed by heating for 5 min at 95°C before loading onto the gels. Alternatively, when measuring DNA strand nicking, reactions were stopped by the addition of NaBH4 (50 mM final concentration) and then mixed with 6 µl of formamide dye and heated for 5 min at 95°C. The products of the reactions were resolved by denaturing 20% PAGE (19:1 acrylamide:bisacrylamide) (22). Gels were scanned and band intensities quantified using a Storm PhosphorImager (Molecular Dynamics).
The assay mixture for Me-FapyG DNA glycosylase activity (100 µl) was composed of 25 mM Tris–HCl pH 7.6, 1 mM MgCl2, 0.4 mg/ml BSA, [3H]Me-FapyG-poly(dG–dC):poly(dG–dC), and purified hOGG1 and HAP1 proteins. The reactions were carried out at 37°C for 90 min. [3H]Me-FapyG residues, represented by ethanol-soluble radioactivity, were separated and quantified in a scintillation counter (23).
Electrophoretic mobility shift assay (EMSA)
For the equilibrium analysis, a reaction mixture (10 µl) containing 25 mM Tris–HCl pH 8.0, 75 mM NaCl, 1 mM MgCl2, 0.4 mg/ml BSA, 10 fmol of the 34mer 5′-32P-labelled F:C duplex and 215 fmol of GST–OGG1 fusion protein was incubated for 30 min at 37°C to allow GST–OGG1–DNA complex formation. Reactions were then mixed with 3 µl of reaction buffer solution containing or not containing 250 fmol of HAP1 and incubated at 37°C for the times indicated. The mixtures were loaded onto a pre-running non-denaturing 8% polyacrylamide gel (37.5:1 acrylamide:bisacrylamide). The electrophoresis buffer contained 6 mM Tris–HCl pH 7.8, 5 mM sodium acetate and 0.1 mM EDTA (27). The gel was subjected to a constant voltage of 8 V/cm applied for 1 h at 4°C. Following gel electrophoresis, the gel was dried on 3MM Whatman paper and scanned using a Storm PhosphorImager (Molecular Dynamics).
For the determination of the dissociation constants, the hOGG1–DNA product complex was preformed as described above and at time 0, a 100-fold excess of unlabelled 8-oxoG:C oligonucleotide was added to sequester the dissociated protein. After different times (10 s to 1 h) at 37°C the mixture was loaded on a pre-running non-denaturing polyacrylamide gel. When analysing the effect of HAP1 on the dissociation constants, 2.5 pmol of the endonuclease were added at time 0. The results were fitted to an exponential curve ([hOGG1–DNA]t = [hOGG1–DNA]t = 0 e–k t) and the dissociation constant (koff) was derived from it.
For the DNA binding assay, reaction mixtures (10 µl) were prepared containing 25 mM Tris–HCl pH 8.0, 75 mM NaCl, 1 mM MgCl2, 0.4 mg/ml BSA, 200 fmol of G:C duplex competitor oligonucleotide, 10 fmol of the 34mer 5′-32P-labelled F:C or endo IV-treated F:C duplexes and increasing amounts of hOGG1 protein, as indicated in the figure legends. After incubation for 30 min at 37°C, samples were analysed as described above.
RESULTS
Stimulation of the hOGG1 8-oxoG DNA glycosylase activity by HAP1
Kinetics studies on the enzymatic activities of hOGG1 have shown that this enzyme is very inefficient. The kcat values reported previously (24,28) suggest that, in vitro, the purified protein takes >20 min to perform a single repair event. It has been postulated that additional factors might be required by this and other DNA glycosylases for adequate BER to occur in the cell (16). One likely candidate for the activation of the first step of 8-oxoG repair is HAP1, which carries out the subsequent reaction (14). To test if HAP1 stimulated hOGG1 activity, the two initial steps of the 8-oxoG repair pathway were reconstituted in vitro using an 8-oxoG-containing oligonucleotide as substrate. In order to analyse the effect of HAP1 on the first catalytic reaction performed by hOGG1, the excision of the 8-oxoG residue, incubations including the two enzymes and the 8-oxoG-containing oligonucleotide were stopped by treating the reaction mixtures with 0.2 N NaOH. This treatment cleaves the oligonucleotides carrying an AP site. Therefore, in this assay, the fraction of cleaved oligonucleotide reflects the level of 8-oxoG DNA glycosylase activity of hOGG1. In the absence of NaOH treatment only the products of the AP lyase activity are revealed. Figure 1 shows that the presence of HAP1 increased the 8-oxoG DNA glycosylase activity of hOGG1. This stimulation was dependent on the concentration of HAP1 (Fig. 1A, lanes 4–9). Increasing amounts of HAP1 induced up to a 6-fold stimulation of the glycosylase reaction performed by hOGG1 (Fig. 1B). HAP1 alone was not able to cleave the substrate, eliminating the possibility of contamination of the HAP1 preparation with an 8-oxoG DNA glycosylase (Fig. 1A, lane 2).
Figure 1.
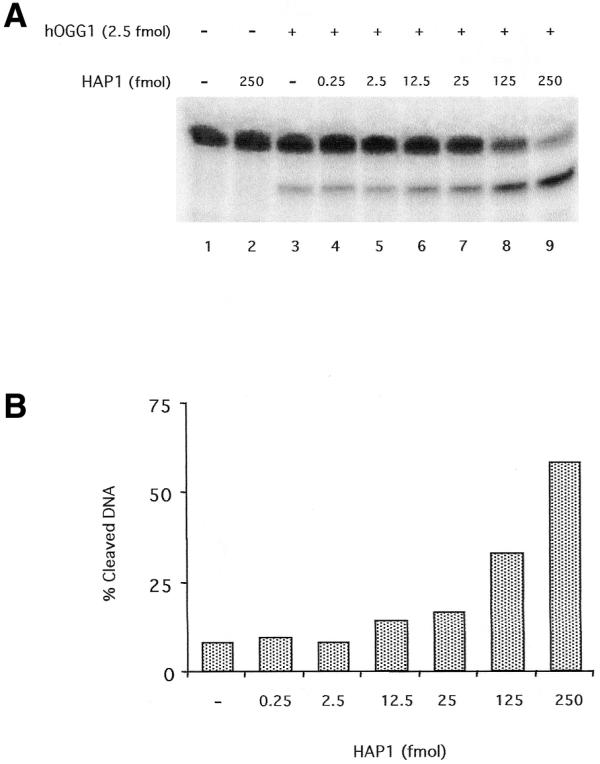
Effect of the AP endonuclease HAP1 upon the 8-oxoG DNA glycosylase activity of hOGG1. (A) hOGG1 glycosylase activity on a 34mer [8-oxoG:C] duplex 32P-labelled at the 5′-end on the lesion-containing strand. A limiting amount (2.5 fmol) of hOGG1 was incubated under standard assay conditions with the double-stranded oligonucleotide substrate (50 fmol) and increasing amounts of HAP1. Reactions were stopped by treatment with 10 mM EDTA and 0.2 N NaOH and heating for 30 min at 90°C as described in Materials and Methods. The products of the reactions were separated by denaturing 20% PAGE. (B) Quantification of the results from the gel displayed in (A).
To determine whether the catalytic activity of HAP1 is necessary to stimulate the glycosylase activity of hOGG1, a mutant form of the AP endonuclease was used in the 8-oxoG DNA glycosylase assay described above. Mutant HAP1-D210N protein, although catalytically inactive, retains the capacity to bind AP sites (26). Figure 2 shows that this inactive mutant is able to stimulate the hOGG1 DNA glycosylase activity to the same extent as the wild-type HAP1 in the range of concentrations tested. In agreement with these results, HAP1 was also found to stimulate 8-oxoG excision from an oligonucleotide substrate as determined by high pressure liquid chromatography followed by electrochemical detection (22) (data not shown).
Figure 2.
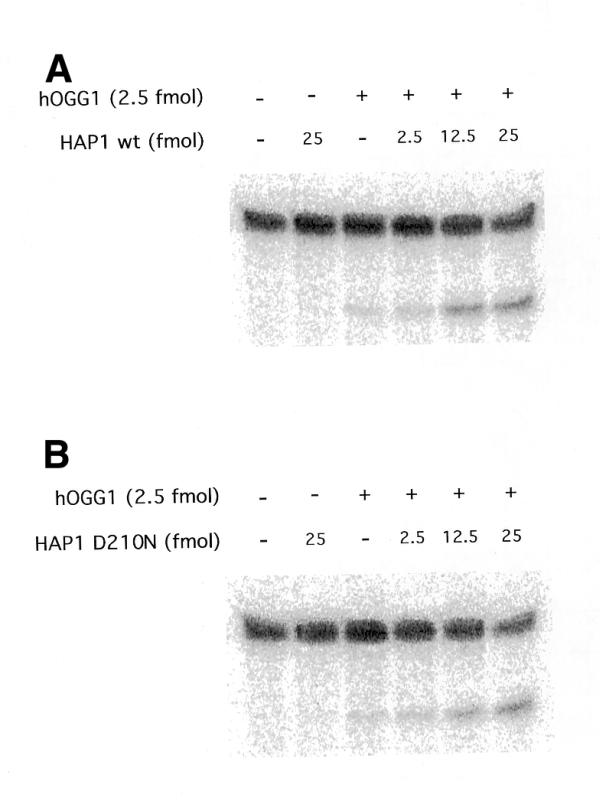
Effect of the inactive HAP1 mutant D210N on the hOGG1 8oxoG DNA glycosylase activity. The reactions were carried out as described in Figure 1 except that increasing amounts of either the wild-type HAP1 (A) or HAP1-D210N (B) were used.
To confirm the results obtained above, the consequences of adding HAP1 to a Fapy DNA glycosylase assay for hOGG1 activity were analysed. In this assay the amounts of base removed are measured, reflecting exclusively the glycosylase activity of hOGG1. Figure 3 shows that HAP1 increased the amount of the formamidopyrimidine liberated by hOGG1. Likewise, the catalytically inactive HAP1-D210N was able to stimulate the DNA glycosylase activity of hOGG1. Similar levels of stimulation were also obtained by the addition of the bacterial AP endonuclease endo IV, the product of the nfo gene (Fig. 3). The ability of the bacterial enzyme, which is structurally unrelated to HAP1, to stimulate hOGG1 suggests that stimulation of hOGG1 activity by AP endonucleases is unlikely to require an intimate physical interaction between these different enzymes.
Figure 3.

Effect of HAP1 upon the Fapy-DNA glycosylase activity of hOGG1. The substrate (2 pmol) was incubated with 250 fmol of hOGG1 for 90 min in the presence of 2.5 pmol of either HAP1, HAP1-D210N or endo IV. The [3H]Me-FapyG excision products were quantified in a scintillation counter. Mean values and standard deviations were calculated from four separate experiments.
HAP1 does not stimulate the AP lyase activity of hOGG1
To test whether the AP lyase activity of hOGG1 is modulated by the human AP endonuclease, a cleavage assay was carried out on a 3′-end-radiolabelled oligonucleotide carrying a single 8-oxoG residue (Fig. 4). No cleavage was found when HAP1 alone was added (lane 2), confirming the absence of preformed AP sites in the 8-oxoG substrate. HAP1, in common with endo IV, cleaves DNA specifically 5′ to an AP site, producing fragments migrating more slowly (lane 8) than those resulting from hOGG1-mediated AP lyase cleavage on the 3′ side of the AP site (lane 3). The results show that no additional DNA migrating at the position of the AP lyase product is generated by HAP1 (lanes 4–6). However, in the presence of an active AP endonuclease there is a new band (*19 nt) corresponding to the cleavage by HAP1 on the 5′ side of the AP site. The new product corresponds to the reaction of HAP1 on the AP sites generated by the glycosylase activity of hOGG1 that have not been cleaved by hOGG1 AP lyase activity. No such product was observed when the mutant HAP1-D210N was used (lane 7). In this case, we conclude that the AP sites generated by the hOGG1 glycosylase activity accumulate without being cleaved by the AP lyase function. These results confirm that there can be uncoupling of the two enzymatic activities of hOGG1.
Figure 4.
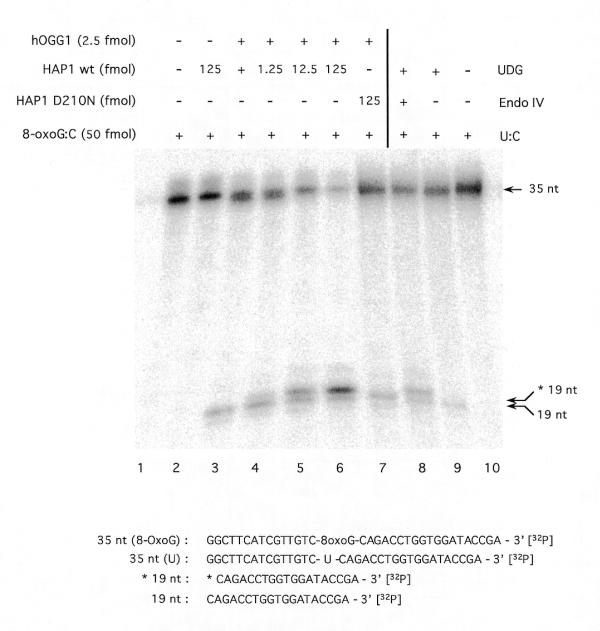
Effect of HAP1 upon the AP lyase activity of human OGG1. A limiting amount (5 fmol) of hOGG1 was incubated under standard assay conditions with 50 fmol of the [8-oxoG:C] duplex 32P-labelled at the 3′-end of the lesion-containing strand and increasing amounts of wild-type HAP1 (lanes 1–6) or HAP1-D210N (lane 7). A 35mer-[U:C] duplex (50 fmol) 32P-labelled at the 3′-end of the lesion-containing strand was incubated under the same conditions with excess amounts of UDG and endo IV, where indicated (lanes 8–10). Reaction products were separated by denaturing 20% PAGE. Arrows indicate the positions of the 3′-radiolabelled substrate and reaction products as follows: 35 nt, non-cleaved 35mer oligonucleotide; 19 nt, 19mer produced by OGG1 strand nicking activity; *19 nt, 19mer with a 5′-deoxyribose-5-phosphate produced by UDG and endo IV treatment of the 35mer [U:C] duplex. Sequences of the radiolabelled DNA species are shown below the figure.
The effect of HAP1 on the complex formed by hOGG1 with AP sites
To gain insight into the nature of the stimulation of hOGG1 by HAP1, EMSAs were performed to analyse the complexes formed between the enzymes and the DNA substrates. hOGG1 and HAP1 are proteins of similar molecular mass (39 and 35 kDa, respectively), hence the fusion protein GST–hOGG1 (68 kDa) (25) was used to distinguish the complexes formed, since the mobility of this protein in a complex with the DNA substrate differs from that of the HAP1–DNA complex (data not shown). HAP1 stimulates GST–hOGG1 enzymatic activity in a similar manner to that of the wild-type hOGG1 protein (data not shown).
In order to study the equilibrium state in the hOGG1–DNA reaction mixture, a complex was pre-formed by incubating the GST–hOGG1 protein with an oligonucleotide duplex containing a tetrahydrofuranyl residue (F:C) for 30 min at 37°C. This AP site analogue was chosen because it forms a stable complex with OGG1 (17) and cannot be cleaved by its AP lyase activity, thus mimicking the intermediate of the hOGG1 reaction upon which HAP1 was shown to act (Fig. 4). Upon incubation of hOGG1 with the oligonucleotide, >85% of the DNA was in the form of a complex with GST–hOGG1 (Fig. 5, lane 1). At 30 min, the mixtures were diluted and further incubated in the presence or absence of HAP1 protein. In the absence of the AP endonuclease, the hOGG1–F:C complex was very stable, reaching an equilibrium in which 40% of the DNA was present in the complex (Fig. 5, lanes 2–4). However, following addition of wild-type HAP1, the appearance of an HAP1–F:C complex was observed. Under these conditions, after 30 min incubation in the presence of both hOGG1 and HAP1, <6% of the DNA was in a complex with hOGG1. With the inactive HAP1-D210N, there is a similar marked shift of the equilibrium towards a HAP1–F:C complex.
Figure 5.
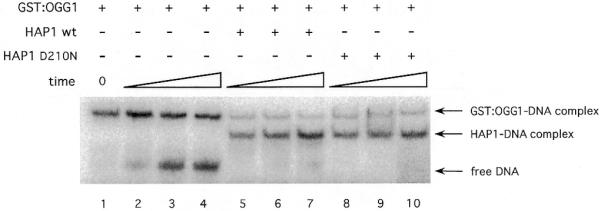
HAP1 effect on the hOGG1–AP site complex. 5′-End-labelled 34mer duplex containing a abasic analogue site (F) (10 fmol) was pre-incubated with an excess of GST–OGG1 protein (215 fmol) to form the GST–OGG1–AP site complex (time 0). After 5, 15 and 30 min incubation at 37°C in the presence or absence of HAP1 or its mutant form (250 fmol), samples were loaded onto a pre-running non-denaturing polyacrylamide gel to separate the protein–DNA complexes from the free DNA. The positions of free DNA, HAP1–DNA and GST–OGG1–DNA complexes are shown by arrows.
Taken together, the results presented thus far suggest two hypotheses regarding the mechanism by which HAP1 can occupy the AP site, permitting the recycling of the glycosylase and therefore inducing stimulation of hOGG1 glycosylase activity. One possibility would be that HAP1 actively displaces hOGG1 from the AP site intermediate. In this case, HAP1 should increase the dissociation rate of hOGG1 from an AP site. Alternatively, HAP1 could be occupying the AP site left following the spontaneous dissociation of hOGG1 from this reaction intermediate. To measure the dissociation constant (koff) for hOGG1, complexes of hOGG1 and the F:C oligonucleotide were pre-formed as described for the experiment shown in Figure 5. At time 0 after formation of the complex, aliquots of the reaction were mixed with a 100-fold excess of unlabelled substrate to trap the glycosylase as it dissociated from the labelled DNA. After incubation for different times at 37°C in the presence or absence of HAP1, samples were loaded onto a non-denaturing gel. The fraction of the radioactivity present in the complex was quantified as a function of time and the data were fitted to an exponential curve to obtain the dissociation rate constants (Table 1). The dissociation parameters show that the AP site–hOGG1 complex has a half-life of a little over 2 min under the conditions tested. Surprisingly, the presence of HAP1, while stimulating the hOGG1 reaction and displacing it from the complex at equilibrium, did not alter the koff of the glycosylase from the AP site. This result favours the second hypothesis, by which HAP1 would occupy the AP sites liberated after hOGG1 spontaneously dissociates from these sites, thus avoiding the glycosylase reassociation to its product.
Table 1. Kinetic parameters for the dissociation of the hOGG1–[F:C] complex.
| |
–HAP1 |
+HAP1 |
| koff | 5.00 × 10–3 s–1 | 4.83 × 10–3 s–1 |
| Half-life of the complex | 2.3 min | 2.4 min |
The amount of protein–DNA complex as a function of time was determined using EMSA (see Materials and Methods) and the kinetic parameters were derived by fitting the data to an exponential curve.
hOGG1 does not bind to the product of the AP endonuclease
Because it has been shown that, in the presence of Mg2+, HAP1 can rapidly dissociate from DNA after performing its catalytic activity liberating an incised DNA (29), the stimulation model proposed requires that hOGG1 loses affinity for the AP site once it is cleaved on its 5′ side by the endonuclease. To test this proposition, the relative affinity of hOGG1 for intact AP sites and cleaved AP sites was determined. Incised AP site duplex DNA (*F:C) was prepared by treating the F:C oligonucleotide with endo IV as described in Materials and Methods. Two 32P-labelled duplexes (F:C and incised F:C) were incubated with increasing amounts of hOGG1 and the mixtures were analysed by EMSA. The results of the binding reactions showed that hOGG1 bound preferentially to the non-cleaved (F:C) site and poorly to the incised product of HAP1 (incised F:C) (Fig. 6). These findings support the hypothesis that once HAP1 has performed the cleavage on the product of the hOGG1 glycosylase activity, hOGG1 will not re-bind to the incised DNA, thus favouring turnover.
Figure 6.
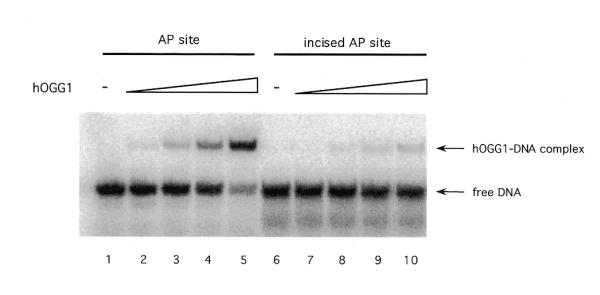
Binding of HAP1 protein to a duplex DNA containing an AP site or a 5′ incised AP site. Binding reaction mixtures containing 0, 25, 125, 250 and 500 fmol of hOGG1 protein and 5′-32P-labelled [F:C] (AP site) or endo IV-treated [F:C] (incised AP site) duplexes were carried out as indicated in Materials and Methods. The positions of free DNA and hOGG1–DNA complexes are shown by arrows.
hOGG1 remains tightly bound to the product of its AP lyase activity
The mechanism suggested by the above experiments implies that most reactions initiated by hOGG1 on 8-oxoG-containing substrates would proceed through the glycosylase step and then hOGG1 would dissociate to allow HAP1 access to the resulting AP site. Following cleavage of the AP site by HAP1, a suitable substrate for the next step, performed by DNA polymerase β, would be provided. To gain insight into the scheme of events in those cases where the hOGG1 enzyme acts both as a DNA glycosylase and an AP lyase, the stability of the complex between the enzyme and the final DNA product (a DNA carrying a 3′ cleaved AP site) was studied. Such a complex was pre-formed by incubating the labelled 34mer carrying a single 8-oxoG residue with an excess of GST–hOGG1. Denaturing gel analysis of the complex showed that >95% of the DNA was cleaved by the AP lyase activity of the enzyme on the 3′ side of the AP site (data not shown). As before, the dissociation constants for hOGG1 were calculated in the presence or absence of HAP1 by adding a 100-fold excess of unlabelled substrate DNA to sequester the dissociated enzyme. Samples were analysed by EMSA at various times (Fig. 7). A koff value of 5.3 × 10–5 was calculated from the data. The dissociation rate for hOGG1 from the incised DNA was much lower than that observed for the dissociation from a non-cleaved AP site. The values obtained indicate that hOGG1 dissociates very slowly from its product when the AP lyase activity has been employed (half-life of the complex = 131 min). Interestingly, the presence of HAP1 did not alter the dissociation rate of hOGG1 from the nicked AP site substrate (koff = 4.8 × 10–5, half-life of the complex = 141 min).
Figure 7.

Dissociation rates of the complex between hOGG1 and DNA containing an incision 3′ of an AP site. The complex between GST–hOGG1 and its AP lyase activity product was obtained by incubation of an 8-oxoG-containing oligonucleotide (10 fmol) with 215 fmol of GST–hOGG1 fusion protein. After confirming the complete cleavage of the substrate, a 100-fold excess of unlabelled (8-oxoG:C) oligonucleotide was added, in the absence (circles) or presence (squares) of 2.5 pmol of HAP1 (time = 0). The fraction of labelled DNA present in the complex was determined by EMSA.
DISCUSSION
The generation of ROS in cells either by incomplete reduction of oxygen to H2O during normal metabolism or by exposure to exogenous genotoxic agents can generate potentially toxic or mutagenic lesions in the DNA. In the case of oxidised bases, a major product is 8-oxoG, which has a strong mutagenic effect. This oxidised guanine is recognised by specific DNA glycosylases; Fpg in bacteria and OGG1 in eukaryotes. BER of the lesion is initiated with the excision of the base by the DNA glycosylases, generating an AP site. These enzymes can perform a second reaction, the incision of the AP site through a so-called AP lyase activity. In the case of OGG1, a β elimination reaction takes place, the final product being a 3′-α,β-unsaturated aldehyde residue that needs to be removed in order to allow the action of a DNA polymerase. In human cells, it is postulated that the removal of this fragmented deoxyribose moiety is carried out by HAP1. The product of this reaction is a 1 nt gap with a 3′-OH terminus that can be used as a primer by the DNA polymerase.
We show here that the complex formed between hOGG1 and its final product, a 3′-cleaved AP site, is extremely stable (half-life of >2 h). This can lead to a lack of enzyme turnover that would explain the reported inefficiency of the overall reaction of hOGG1 on 8-oxoG substrates (24,28,30). It is likely that hOGG1 activity in vivo is boosted by the action of other factors in order to counteract the mutagenic effects of this oxidised purine. Here, the efficiency of the first reaction carried out by hOGG1, the excision of the modified base, was found to be stimulated by HAP1. Similar stimulatory effects of HAP1 have been reported on the activity of two other human DNA glycosylase enzymes, UDG (18) and TDG (20). These two enzymes are mono-functional DNA glycosylases and can initiate both short and long patch BER, while the repair of 8-oxoG is almost exclusively done through the 1 nt gap filling reaction (13,31). The activation of hOGG1 presented in this work is the first example of the stimulation of a bi-functional DNA glycosylase by HAP1. However, HAP1 is unable to stimulate the DNA glycosylase activity of Fpg (data not shown). This difference could be due to the lack of structural homology between the OGG1 and Fpg (32,33) or to the different coordination levels of their DNA glycosylase and AP lyase activities. It would be interesting to analyse whether the S3 protein from Drosophila, which is structurally unrelated to OGG1 but catalyses the same reactions (34), is also stimulated by HAP1.
Our experiments suggest a model by which the DNA glycosylase activity stimulation can be achieved. In this model, hOGG1 would, in most cases, spontaneously dissociate from the AP site intermediate generated by its DNA glycosylase activity. As shown by the dissociation rate constants, this step is not accelerated by HAP1. The occupancy of the AP site by wild-type HAP1 or a catalytically inactive mutant would avoid the reassociation of the glycosylase to its product, thus increasing the turnover of hOGG1. The fact that the bacterial endo IV is also able to stimulate hOGG1 activity suggests that protein–protein interactions are not absolutely required for the hOGG1 activity stimulation. However, we cannot rule out that in the presence of the other components of the BER machinery, an active coordination of the reaction could be taking place. The loss of affinity of hOGG1 for the AP site once this substrate has been incised by the AP endonuclease would further favour a more efficient turnover of hOGG1. This model could also be applied to the data presented in a recent report showing stimulation of the E.coli double strand UDG by endo IV (35). Moreover, a similar mechanism of relief from product inhibition has been proposed for the stimulation of HAP1 by DNA polymerase β (27).
It is interesting to note that in the case of the repair of oxidised pyrimidines by hNTH1, which is also a bi-functional DNA glycosylase, stimulation was obtained by the XPG protein (16). In this case, the effect of XPG was shown to be through stimulation of the binding of the hNth1 to its substrate. However, XPG failed to stimulate the activity of hOGG1 on 8-oxoG-containing substrates (16). These results define a difference between the mechanism of repair of oxidised purines and pyrimidines, which are both initiated by DNA glycosylases/AP lyases. Oxidised pyrimidines, such as thymine glycol, cause a distortion of the helix, resembling a small bubble (36). It was proposed that XPG would then bind the DNA, bending it, exposing the lesion and thereby facilitating the binding of hNth1 (16). In the case of an 8-oxoG, the distortion caused by the lesion is likely to be very limited and insufficient to direct the binding of XPG.
Besides the stimulation of hOGG1 DNA glycosylase activity, HAP1 can have an additional, and perhaps more important, role in improving the repair efficiency of 8-oxoG. As discussed above, the results presented in this work suggest a model by which, after excision of the oxidised base, hOGG1 would in most cases dissociate from the AP site intermediate without engaging in the AP lyase reaction. In the absence of HAP1, hOGG1 will rebind to the AP site and cleave the DNA. As shown here, hOGG1 will then remain tightly bound to its product, the half-life of the complex being >2 h. This would result in a lack of enzyme turnover. However, in the presence of equimolar or higher amounts of HAP1, a situation likely to occur in the cell given the known abundance of HAP1, the AP site intermediate is occupied by the endonuclease and cleaved 5′ of the deoxyribose moiety to generate a nick with a 5′-deoxyribosephosphate moiety. Such a DNA intermediate can now serve as a substrate for the dRPase function of the DNA polymerase β. This model suggests that, under physiological conditions, hOGG1 would essentially act as a mono-functional DNA glycosylase. These results open the question of the biological relevance of the conservation of the hOGG1 AP lyase capability. One possible explanation would be that the AP lyase activity conservation is a secondary consequence of the formation of the Schiff base that has to be hydrolysed to yield either an intact abasic site (probably the preferred reaction as shown in our results) or the cleaved AP site. Stimulation of glycosylase activity by AP endonuclease, without an increase in the rate of AP lyase function, will accentuate the dissociation of the hOGG1 activities, directing most of the products of the glycosylase towards cleavage by HAP1, which has a greater efficiency to cleave an abasic site rather than at a 3′ terminus (37). This mechanism will tend to preclude the AP lyase reaction, which otherwise would lead to the formation of a very stable hOGG1–DNA complex that could considerably slow down the overall rate of repair of 8-oxoG in DNA. Such a mechanism would also have the virtue of avoiding the exposure of the potentially toxic intermediate generated by the AP lyase activity (15). Thus, the requirement of HAP1 to improve the efficiency of the reactions initiated by hOGG1 would extend what is already known about the degree to which the various steps in BER are coordinated (38). Our data also suggest that HAP1 plays a hitherto unrecognised role in the suppression of mutations induced by agents that generate oxidised purines in DNA.
NOTE ADDED AT REVISION
While this work was going through the review process similar results regarding the stimulation of hOGG1 by the AP endonuclease were published by Hill et al. (39).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Marc Audebert for the preparation of purified Ogg1 enzymes, Dominic Rothwell for the HAP1 and HAP1-D210N proteins, and Dr Jean Cadet for the 8-oxoG-containing oligonucleotides. We are also grateful to T.Begley for his suggestions, and Claudine Dhérin and Patricia Auffret van der Kemp for their technical assistance. A.E.V. is a recipient of a postdoctoral fellowship from the Ministerio de Educacion y Cultura (Spain). This work was supported by the Commissariat à l’Energie Atomique (CEA), Centre National pour la Recherche Scientifique (CNRS) and Electricité de France.
References
- 1.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 2.Michaels M.L. and Miller,J.H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol ., 174, 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boiteux S., O’Connor,T.R. and Laval,J. (1987) Formamidopyrimidine-DNA glycosylase of Escherichia coli: cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J., 6, 3177–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabrera M., Nghiem,Y. and Miller,J.H. (1988) mutM, a second mutator locus in Escherichia coli that generates G.C–T.A transversions. J. Bacteriol., 170, 5405–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nghiem Y., Cabrera,M., Cupples,C.G. and Miller,J.H. (1988) The mutY gene: a mutator locus in Escherichia coli that generates G.C–T.A transversions. Proc. Natl Acad. Sci. USA, 85, 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radicella J.P., Dherin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nash H.M., Bruner,S.D., Scharer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 8.van der Kemp P.A., Thomas,D., Barbey,R., de Oliveira,R. and Boiteux,S. (1996) Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N- methylformamidopyrimidine. Proc. Natl Acad. Sci. USA, 93, 5197–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas D., Scot,A.D., Barbey,R., Padula,M. and Boiteux,S. (1997) Inactivation of OGG1 increases the incidence of G. C→T.A transversions in Saccharomyces cerevisiae: evidence for endogenous oxidative damage to DNA in eukaryotic cells. Mol. Gen. Genet., 254, 171–178. [DOI] [PubMed] [Google Scholar]
- 10.Klungland A., Rosewell,I., Hollenbach,S., Larsen,E., Daly,G., Epe,B., Seeberg,E., Lindahl,T. and Barnes,D.E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl Acad. Sci. USA, 96, 13300–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minowa O., Arai,T., Hirano,M., Monden,Y., Nakai,S., Fukuda,M., Itoh,M., Takano,H., Hippou,Y., Aburatani,H., Masumura,K., Nohmi,T., Nishimura,S. and Noda,T. (2000) Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl Acad. Sci. USA, 97, 4156–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dianov G., Bischoff,C., Piotrowski,J. and Bohr,V.A. (1998) Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem., 273, 33811–33816. [DOI] [PubMed] [Google Scholar]
- 13.Fortini P., Parlanti,E., Sidorkina,O.M., Laval,J. and Dogliotti,E. (1999) The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J. Biol. Chem., 274, 15230–15236. [DOI] [PubMed] [Google Scholar]
- 14.Kubota Y., Nash,R.A., Klungland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 15.Sobol R.W., Prasad,R., Evenski,A., Baker,A., Yang,X.P., Horton,J.K. and Wilson,S.H. (2000) The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature, 405, 807–810. [DOI] [PubMed] [Google Scholar]
- 16.Klungland A., Hoss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (1999) Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell, 3, 33–42. [DOI] [PubMed] [Google Scholar]
- 17.Guibourt N., Castaing,B., van der Kemp,P.A. and Boiteux,S. (2000) Catalytic and DNA binding properties of the ogg1 protein of Saccharomyces cerevisiae: comparison between the wild-type and the K241R and K241Q active-site mutant proteins. Biochemistry, 39, 1716–1724. [DOI] [PubMed] [Google Scholar]
- 18.Parikh S.S., Mol,C.D., Slupphaug,G., Bharati,S., Krokan,H.E. and Tainer,J.A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J., 17, 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scharer O.D., Nash,H.M., Jiricny,J., Laval,J. and Verdine,G.L. (1998) Specific binding of a designed pyrrolidine abasic site analog to multiple DNA glycosylases. J. Biol. Chem., 273, 8592–8597. [DOI] [PubMed] [Google Scholar]
- 20.Waters T.R., Gallinari,P., Jiricny,J. and Swann,P.F. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem., 274, 67–74. [DOI] [PubMed] [Google Scholar]
- 21.Petronzelli F., Riccio,A., Markham,G.D., Seeholzer,S.H., Stoerker,J., Genuardi,M., Yeung,A.T., Matsumoto,Y. and Bellacosa,A. (2000) Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J. Biol. Chem., 275, 32422–32429. [DOI] [PubMed] [Google Scholar]
- 22.Girard P.M., Guibourt,N. and Boiteux,S. (1997) The Ogg1 protein of Saccharomyces cerevisiae: a 7,8-dihydro-8-oxoguanine DNA glycosylase/AP lyase whose lysine 241 is a critical residue for catalytic activity. Nucleic Acids Res., 25, 3204–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boiteux S., Belleney,J., Roques,B.P. and Laval,J. (1984) Two rotameric forms of open ring 7-methylguanine are present in alkylated polynucleotides. Nucleic Acids Res., 12, 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Audebert M., Radicella,J.P. and Dizdaroglu,M. (2000) Effect of single mutations in the OGG1 gene found in human tumors on the substrate specificity of the ogg1 protein. Nucleic Acids Res., 28, 2672–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dherin C., Radicella,J.P., Dizdaroglu,M. and Boiteux,S. (1999) Excision of oxidatively damaged DNA bases by the human α-hOgg1 protein and the polymorphic α-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res., 27, 4001–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rothwell D.G., Hang,B., Gorman,M.A., Freemont,P.S., Singer,B. and Hickson,I.D. (2000) Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilises substrate binding. Nucleic Acids Res., 28, 2207–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda Y., Bennett,R.A. and Demple,B. (1998) Dynamics of the interaction of human apurinic endonuclease (Ape1) with its substrate and product. J. Biol. Chem., 273, 30352–30359. [DOI] [PubMed] [Google Scholar]
- 28.Asagoshi K., Yamada,T., Terato,H., Ohyama,Y., Monden,Y., Arai,T., Nishimura,S., Aburatani,H., Lindahl,T. and Ide,H. (2000) Distinct repair activities of human 7,8-dihydro-8-oxoguanine DNA glycosylase and formamidopyrimidine DNA glycosylase for formamidopyrimidine and 7,8-dihydro-8-oxoguanine. J. Biol. Chem., 275, 4956–4964. [DOI] [PubMed] [Google Scholar]
- 29.Masuda Y., Bennett,R.A. and Demple,B. (1998) Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. J. Biol. Chem., 273, 30360–30365. [DOI] [PubMed] [Google Scholar]
- 30.Zharkov D.O., Rosenquist,T.A., Gerchman,S.E. and Grollman,A.P. (2000) Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem., 275, 28607–28617. [DOI] [PubMed] [Google Scholar]
- 31.Klungland A. and Lindahl,T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J., 16, 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruner S.D., Norman,D.P.G. and Verdine,G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859–866. [DOI] [PubMed] [Google Scholar]
- 33.Sugahara M., Mikawa,T., Kumasaka,T., Yamamoto,M., Kato,R., Fukuyama,K., Inoue,Y. and Kuramitsu,S. (2000) Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8. EMBO J., 19, 3857–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yacoub A., Augeri,L., Kelley,M.R., Doetsch,P.W. and Deutsch,W.A. (1996) A Drosophila ribosomal protein contains 8-oxoguanine and abasic site DNA repair activities. EMBO J., 15, 2306–2312. [PMC free article] [PubMed] [Google Scholar]
- 35.Sung J.S. and Mosbaugh,D.W. (2000) Escherichia coli double-strand uracil-DNA glycosylase: involvement in uracil-mediated DNA base excision repair and stimulation of activity by endonuclease IV. Biochemistry, 39, 10224–10235. [DOI] [PubMed] [Google Scholar]
- 36.Kung H.C. and Bolton,P.H. (1997) Structure of a duplex DNA containing a thymine glycol residue in solution. J. Biol. Chem., 272, 9227–9236. [DOI] [PubMed] [Google Scholar]
- 37.Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- 38.Wilson S.H. and Kunkel,T.A. (2000) Passing the baton in base excision repair. Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]
- 39.Hill J.W., Hazra,T.K., Izumi,T. and Mitra,S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res., 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]