

Abstract
Real-time quantitative PCR (qPCR) for rapid and specific enumeration of microbial agents is finding increased use in aerosol science. The goal of this study was to determine qPCR accuracy, precision, and method detection limits (MDLs) within the context of indoor and ambient aerosol samples. Escherichia coli and Bacillus atrophaeus vegetative bacterial cells and Aspergillus fumigatus fungal spores loaded onto aerosol filters were considered. Efficiencies associated with recovery of DNA from aerosol filters were low, and excluding these efficiencies in quantitative analysis led to underestimating the true aerosol concentration by 10 to 24 times. Precision near detection limits ranged from a 28% to 79% coefficient of variation (COV) for the three test organisms, and the majority of this variation was due to instrument repeatability. Depending on the organism and sampling filter material, precision results suggest that qPCR is useful for determining dissimilarity between two samples only if the true differences are greater than 1.3 to 3.2 times (95% confidence level at n = 7 replicates). For MDLs, qPCR was able to produce a positive response with 99% confidence from the DNA of five B. atrophaeus cells and less than one A. fumigatus spore. Overall MDL values that included sample processing efficiencies ranged from 2,000 to 3,000 B. atrophaeus cells per filter and 10 to 25 A. fumigatus spores per filter. Applying the concepts of accuracy, precision, and MDL to qPCR aerosol measurements demonstrates that sample processing efficiencies must be accounted for in order to accurately estimate bioaerosol exposure, provides guidance on the necessary statistical rigor required to understand significant differences among separate aerosol samples, and prevents undetected (i.e., nonquantifiable) values for true aerosol concentrations that may be significant.
Real-time quantitative PCR (qPCR) is an analytical method for the rapid and potentially sensitive enumeration of broad and specific microbial populations in environmental samples (17). For bioaerosol analysis, this method allows for detection and enumeration independent of culturing, thereby circumventing the significant concerns surrounding the unculturability of environmental microorganisms and loss of culturability due to aerosol sampling (1, 2, 18, 34, 46, 55). Over the last decade, the application of qPCR has advanced research in the human health, environmental, and the national security arenas by enabling the specific measurement of airborne allergenic mold, pathogenic bacteria, and human viruses (6, 7, 9, 13, 37, 45).
The quantitative nature of this technique as well as the documented advantages over culturing provides the potential for integrating microbial measurements with physical and chemical aerosol processes to understand exposure and to describe the fate and sources of biological aerosols in indoor environments and the atmosphere. However, the logarithmic amplification that is the basis of qPCR results in significant standard deviations among repeated qPCRs (25, 50). This variability rarely constrains the use of qPCR in aquatic and terrestrial systems, where biological growth typically dictates concentrations above detection limits and multiple order-of-magnitude differences in microorganism concentrations between treatments. However, the volume concentrations of biological agents in air (103 to 106 per m3 of air) are dramatically more dilute than those measured in environmental waters (1012 to 1014 per m3 of water) (4, 5, 10, 12, 20, 52), and processes that result in indoor and atmospheric bioaerosol concentrations are growth independent. These processes include aerosol infiltration and exfiltration, resuspension, and deposition and typically result in less than an order of magnitude of variability in aerosol or biological particulate matter (PM) concentrations (22, 29, 41). As qPCR becomes more commonly used in indoor and outdoor air quality research, it is necessary to know the analytical variability and method detection limits (MDLs) to determine whether the method is suitable for estimating exposure and delineating the experimental differences observed in aerosol processes.
The goal of this study was to estimate the accuracy, precision, and MDLs associated with qPCR of aerosol samples. These concepts were applied to air sampling filters loaded with three test organisms, including spores of Aspergillus fumigatus and vegetative bacterial cells from the Gram-negative Escherichia coli and Gram-positive Bacillus atrophaeus. The efficiencies associated with DNA extraction and with extraction of whole cells from aerosol filters were measured to describe the statistical accuracy of common qPCR bioaerosol protocols. Overall precision (reproducibility) as well as instrument repeatability were determined, and a binary method for describing MDLs was developed and applied to fungal spores and bacterial cells. Such experimental and statistical treatment of qPCR-based aerosol measurements is expected to guide improved estimates of human exposure, incorporate limits of qPCR precision into experimental design, and provide a context for undetected (i.e., nonquantifiable) values.
MATERIALS AND METHODS
Test organism preparation.
Bacterial test organisms included vegetative cells from E. coli (ATCC 15597) and B. atrophaeus (ATCC 49337). These two organisms have commonly been used in bioaerosol sampling research (3, 21, 32, 33, 36, 49, 54), represent Gram-positive (B. atrophaeus) and Gram-negative (E. coli) cell wall types, and cover a broad range of cell lysis resistances. Spores from A. fumigatus (ATCC 34506) were used as a fungal test organism due to their ubiquity in the environment and their allergenic and infectious potential (30, 31).
To produce bacterial and fungal stocks for qPCR testing, 6 ml of tryptic soy broth (Difco Laboratories, Detroit, MI) was inoculated with pure cultures of E. coli and B. atrophaeus cells and grown overnight at 37°C and 28°C, respectively. Pure cultures were isolated by centrifugation and resuspension in phosphate-buffered saline ([PBS] 10 mM phosphate, 137 mM sodium chloride, pH 7.4) to a concentration of 108 cells ml−1. A. fumigatus was grown on malt extract agar (Difco Laboratories) in the dark at 23°C for 21 days. Spores were removed from hyphae using sterile cotton applicators and then suspended in 50 ml of 70% ethanol and divided into subsamples containing 1.25 × 108 spores per tube. The tubes were centrifuged at 10,000 × g for 3 min to pelletize the spores, the supernatant ethanol was removed, and the remaining spore pellets were stored at −20°C prior to analysis.
To quantify whole-cell concentrations, fungal spores and bacterial cells were counted under a Zeiss AX10 microscope using a 2 × 10−5-ml Coulter counting chamber (Hausser Scientific, Horsham, PA) at magnifications of ×400 and ×1,000 for fungi and bacteria, respectively. For each enumeration, three to five independent replicate volumes of fungal spores or bacterial cells, each including at least 25 fields and 1,000 total counts, were performed. Replicate results were pooled and averaged, and standard deviation microorganism concentrations (ml−1) were determined.
Accuracy, precision, and MDL calculations.
Accuracy is the degree of closeness of a measured value to the true value (35). In aerosol samples collected by filtration, accuracy is limited by efficiencies in DNA extraction from cells and whole-cell extraction from sampling filters. To determine the percent DNA extraction efficiency (ηDNA), the DNA from test microorganisms in aqueous solution was extracted, and the DNA mass was estimated as described below with a PicoGreen assay. This value was compared to the theoretical value of total DNA mass of a selected microorganism (equation 1). The theoretical mass concentration of DNA per cell for each test organism was estimated by the following: DNA mass (in pg) in one cell equals the genome size (in bp) divided by 0.978 × 109 bp/pg (11). No biases have been reported with this method due to relative differences in CG content (48). Five replicate DNA extraction efficiencies were performed for each organism.
 |
(1) |
To determine extraction efficiency of whole cells or spores from sampling filters (ηfilter), a known quantity of cells was spiked onto clean or PM-loaded quartz fiber or polycarbonate track-etched (PCTE) filters and entered into the DNA extraction protocol. The recovered DNA mass (in pg) was divided by the recovered mass of DNA (in pg) from the same number of cells originally spiked into PBS (equation 2). Five independent ηfilter experiments were performed for each of the 12 cases (three test organisms, PM-loaded filters and clean filters, and PCTE filters and quartz glass fiber filters).
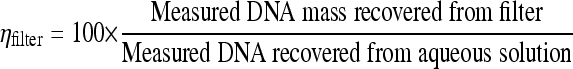 |
(2) |
Precision is the degree to which repeated measurements under unchanged conditions show the same results (35). Here, precision is separated into the total precision (reproducibility) and precision associated with the analytical instrument (instrument repeatability). Reproducibility experiments for A. fumigatus, E. coli, and B. atrophaeus included spiking seven independent PM-loaded quartz fiber and seven PM-loaded PCTE filters and determining the coefficient of variation (COV) as a measure of reproducibility. Reproducibility experiments were conducted with two different operators and at cell concentrations near the limits of detection and included a set of experiments spiked with 103 cells and another set of experiments spiked at 104 cells. This cursory limit of detection of 103 to 104 cells was based on 90% to 95% losses in sample preparation (ηDNA plus ηfilter), assuming that 10% of the DNA extract is used in qPCRs as the template and that 10 to 50 cells are required for positive qPCRs. Instrument repeatability experiments for A. fumigatus, E. coli, and B. atrophaeus included spiking one PM-loaded quartz fiber and one PM-loaded PCTE filter with 103 and 104 microorganisms, extracting nucleic acids, and repeating the qPCR analysis at least seven times for each spiked concentration. Instrument repeatability was also quantified by calculating the COV of replicates.
For MDL, detection by qPCR is based on a logarithmic signal amplification and is binary, i.e., either positive or negative. Thus, a probability distribution was used to estimate the qPCR MDL. For a binominal distribution, the probability of a successful detection (P) of n positive results out of N trials is described by the probability mass function (equation 3):
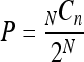 |
(3) |
where NCn represents the number of n factorial combinations from a given set of N elements:
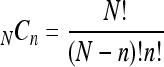 |
(4) |
In order to detect a microorganism concentration at the 99% confidence level (26), at least n = 7 successful detections out of N = 7 trials (P = 1/128) are needed to claim that the detection appears with a probability greater than 0.99%. These qPCR instrument MDLs were performed by producing a serial dilution of standard B. atrophaeus and A. fumigatus DNA samples corresponding to a known cell number and identifying the minimum cell concentration where seven of seven analyses were positive. Estimations of the overall MDLs are the instrument qPCR MDLs (in number of cells) adjusted for the DNA extraction (ηDNA) and whole-cell filter extraction (ηfilter) efficiencies as well as the ratio (F) of total extracted DNA to DNA used as the template in the qPCRs (equation 5):
 |
(5) |
Values of F typically vary from 5 to 25, depending on both the elution volume used to wash off DNA from the spin filter in the last step of DNA extraction and on the volume of DNA used as the qPCR template. Where relevant in equations 1 through 5, background levels of DNA and A. fumigatus, E. coli, and B. atrophaeus gene copies in PM-loaded filters were subtracted from spiked values. For ηDNA and ηfilter calculations, standard deviations of each term were propagated through the efficiency equations by the use of accepted methods for random error propagation in multiplicative expressions (39).
Spiking microorganisms onto filters.
Aerosol samples collected onto filters were considered here as this sampling method is the most commonly used in bioaerosol molecular biology studies (44), provides aerosol collection efficiencies near 100%, and takes advantage of preexisting, well-described particulate matter samplers that allow for a variety of flow rates and control of particle sizes. Aqueous solutions containing known amounts of microorganisms were spiked onto either clean filters or filters preloaded with particulate matter (PM-loaded filters) for whole-cell elution efficiency tests. Filter materials included 81-mm diameter quartz glass fiber filters (New Star Environmental, Inc., Roswell, GA) and 37-mm-diameter, 0.8-μm pore-size PCTE membrane filters (SKC, Inc., Eighty Four, PA). PM-loaded quartz fiber filters were obtained by sampling air for 72 h in an urban location using a high-volume, PM10 (for particulate matter smaller than 10 μm in aerodynamic diameter) sampler (HiVol3000 Ecotech, Enviro Technology Services, PLC, Gloucestershire, United Kingdom) operated at a flow rate of 1.13 m3 min−1 with Whatman EPM2000 quartz fiber filters (Whatman International, Ltd., Maidstone, United Kingdom). Except for the shape of the filters, physical properties of both Whatman and New Star Environment quartz fiber filters were the same (100% pure borosilicate glass, 99% minimum collection efficiency for particles of <0.3 μm). PM-loaded PCTE filters were obtained by loading clean filters onto SKC PM10 personal exposure monitors (PEMs) at a flow rate of 10 ± 0.5 liters min−1 (single-stage impactor) (SKC, Eighty Four, PA) and resuspending floor dust sieved through an 37-μm-pore-size mesh in a cubic aerosol chamber (0.28 m3) until 500 μg of PM10 were collected. To spike clean and loaded PCTE filters with test organisms, a backing filter was loaded onto a sterile filtration gallery, and volumes containing the desired quantity of microorganisms were directly pipetted onto the filters under vacuum. Spiking cells and spores onto glass fiber filters was performed by pipetting directly onto filters placed in sterile petri dishes. A. fumigatus-spiked filters were dried at 23°C for 1 h in sterile petri dishes to evaporate residual ethanol.
Efficiency studies associated with environmental sample preparation have typically provided comparative measures of efficiency rather than absolute measures of accuracy. Spiking cells onto filters allows for a well-documented starting concentration and enables the absolute estimation of sample preparation losses and method detection levels. A disadvantage associated with spiking filters is that this loading technique may not capture the true cell-filter interactions produced by impaction and interception in an aerosol sampling scenario.
DNA extraction, purification, and qPCR.
A predetermined amount of whole cells or spores was eluted from filters by incorporating 1-cm2 sections of the spiked filters directly into the DNA extraction protocol. DNA was extracted using a modified Mobio kit (Mobio Laboratories, Carlsbad, CA) protocol for high yield. The modifications included supplementing the provided extraction tubes with 300 mg of 0.1-mm-diameter silica beads and 100 mg of 0.5-mm-diameter silica beads and bead-beating the microorganisms on aerosol filters in 2-ml screw-cap tubes at 3,450 oscillations min−1 for 3 min (bacteria) or 5 min (fungi). An additional modification included DNA elution off Mobio spin filters using only 50 μl of prewarmed elution buffer. Total genomic DNA was quantified after extraction using a fluorescent assay (Quant-iT PicoGreen, Invitrogen, Carlsbad, CA). In brief, 50 μl of genomic DNA was mixed with 50 μl of 2× PicoGreen dye in TE buffer (200 mM Tris-HCl, 20 mM EDTA, pH 7.5) and incubated for 5 min at 23°C (48). Fluorescence emission was read at 520 nm with excitation at 497 nm in 96-well plates following the manufacturer's protocol. Corrections for well bias were estimated and accounted for by measuring the fluorescence in different wells containing the same amounts of a standard DNA. Triplicate DNA extraction and measurement experiments were performed for each test organism, and concentrations are reported in pg/cell.
Quantitative PCR was performed using an ABI 7500 Prism sequence detector (Applied Biosystems, Forest City, CA). Specific primers and TaqMan probes selected for each microorganism included the following: for E. coli, uidA primer and probe set (14) (forward [F], 5′-GGGCAGGCCAGCGTATC; reverse [R], 5′-CCCACACTTTGCCGTAATGA; probe, 6-FAM-5′-TGCTGCGTTTCGATGCGGTCA-3′-TAMRA, where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine); for B. atrophaeus, recA gene primer and probe set (8) (F, 5′-ACCAGACAATGCTCGACGTT; R, 5′-CCCTCTTGAAATTCCCGAAT; probe, 6-FAM-5′-ACTGAACAGCTGATCGAGACAGCTGC-3′-TAMRA); and for A. fumigatus, Afumi primer and probe set (53) (F, 5′-GCCCGCCGTTTCGAC; R, 5′-CCGTTGTTGAAAGTTTTAACTGATTAC; probe, 6-FAM-5′-CCCGCCGAAGACCCCAACATG-3′-TAMRA). The amplicon sizes were 293 bp, 131 bp, and 136 bp, respectively. Commercial TaqMan probes were labeled with 5′ fluorophore FAM and 3′ Black Hole Quencher dye BHQ-1 (Biosearch Technologies, Novato, CA). For E. coli and B. atrophaeus, 20-μl qPCR mixtures were prepared including 10 μl of 2× TaqMan Universal PCR master mix with 6-carboxy-X-rhodamine (ROX) passive reference dye (Roche Diagnostics, Indianapolis, IN), 2 μl of 0.4 mg ml−1 bovine serum albumin, 0.4 μl of each 10 μM primer, 0.8 μl of 5 μM probe, and 5 μl of DNA template. For A. fumigatus, 50-μl qPCR mixtures included 25 μl of 2× TaqMan Universal PCR master mix, 1.67 μl of each 30 μM primer, 0.4 μl of 10 μM probe, and 2 to 5 μl of DNA template. Quantitative PCR was performed under thermal cycling conditions consisting of an initial 2-min denaturation at 50°C and 10 min of further denaturation at 95°C, followed by 45 cycles of 15 s of denaturation at 95°C and 60 s of annealing/extension. Standard curves were developed for each qPCR bacterial and fungal species using dilutions from a known concentration of genomic DNA. To test for inhibition, extracts from PM-loaded quartz fiber and PCTE filters were also added to subsets of diluted A. fumigatus DNA, and standard curves were produced. No significant inhibition was observed. Molecular water blanks were run on each qPCR 96-well plate for negative control, and all qPCR measurements were replicated a minimum of three times.
RESULTS
Accuracy.
When filters are used for efficient aerosol collection, the difference between the measured value and true value is related to efficiencies of DNA extraction from cells and extracting cells from aerosol collection filters. DNA extraction efficiencies were measured as the ratio of extracted DNA per cell or spore to the theoretical DNA mass per cell or spore. The percent extraction efficiency, ηDNA, and standard deviations for five independent experiments, from highest to lowest, included the following: for E. coli, ηDNA of 16.3% ± 2.1%; for A. fumigatus, ηDNA of 8.8% ± 0.7%; and for B. atrophaeus, ηDNA of 8.3% ± 1.0%. These values were based on the E. coli, B. atrophaeus, and A. fumigatus DNA masses per cell or spore of 4.74 × 10−3 pg, 4.31 × 10−3 pg, and 3 × 10−2 pg, using genome lengths of 4,639,221 bp, 4,214,810 bp, and 29,384,958 bp, respectively (27, 28, 42).
The efficiencies associated with extracting whole cells from filters (ηfilter) were determined for clean and PM-loaded quartz and PCTE filters for each test microorganism. Whole-cell extraction efficiencies for these test organisms are presented in Fig. 1 and ranged from 40% to 150%, with PM-loaded filters exhibiting a slightly higher average efficiency than clean filters. Two-tailed t tests at 95% confidence comparing clean and PM-loaded filters indicated no significant differences for the three test organisms. While no significant differences in ηfilter values were observed between quartz and PCTE filters for the bacteria, ηfilter values for A. fumigatus were significantly higher than filter efficiencies for bacterial test organisms on quartz fiber filters. Overall, the efficiency associated with DNA extraction and filter extraction can be characterized by multiplying ηDNA by ηfilter. These overall efficiency values for each organism and filter type are reported in Table 1, and their magnitude suggests that without inclusion in sample concentration calculations, measured aerosol concentrations would underestimate the true concentrations by 10 to 16 times for E. coli, 20 to 24 times for B. atrophaeus, and 7 to 20 times for A. fumigatus.
FIG. 1.

Filter extraction efficiencies associated with recovery of E. coli, B. atrophaeus, and A. fumigatus spiked onto clean and PM-loaded quartz fiber and PCTE filters. Spikes contained 108 cells or spores. Error bars represent standard deviation (n = 5 experiments).
TABLE 1.
Overall efficiencies associated with preparing aerosol samples for qPCR
| Organism | Filter type | Overall efficiency (%)a |
|---|---|---|
| E. coli | Quartz fiber | 9.55 ± 2.4 |
| PM-loaded quartz fiber | 10.5 ± 2.3 | |
| PCTE | 6.4 ± 2.7 | |
| PM-loaded PCTE | 8.1 ± 0.5 | |
| B. atrophaeus | Quartz fiber | 4.2 ± 1.6 |
| PM-loaded quartz fiber | 4.3 ± 1.5 | |
| PCTE | 3.4 ± 2.2 | |
| PM-loaded PCTE | 4.8 ± 0.7 | |
| A. fumigatus | Quartz fiber | 13.3 ± 1.6 |
| PM-loaded quartz fiber | 10.4 ± 2.7 | |
| PCTE | 6.4 ± 1.0 | |
| PM-loaded PCTE | 5.0 ± 1.2 |
These efficiencies were calculated as the product of ηDNA and ηfilter (mean ± standard deviation).
Precision.
Precision can be stratified into the subjects of reproducibility and instrument repeatability. Reproducibility is the overall variation and incorporates errors from sample loading, sample preparation, and the error inherent in analytical instruments. For instrument repeatability, only the error in analytical equipment is measured. Figure 2 shows plots demonstrating the reproducibility (circles) of qPCR results from samples near the detection limit. The detected numbers were adjusted for ηDNA and ηfilter using the overall values listed in Table 1 and demonstrate that inclusion of these efficiencies results in measured values appropriately adjusts them to the true values (dashed lines). The COVs of the detected gene copy quantities were calculated to assess reproducibility of E. coli, A. fumigatus, and B. atrophaeus qPCR results and are presented in Table 2. Reproducibility COVs ranged from 28% to 79%, averaging 61% for microorganism concentration of ∼103 per filter and from 47% to 70% averaging 58% at concentrations of ∼ 104 microorganisms per filter. Reproducibility experiments were performed on PM-loaded filters and therefore required subtraction of the E. coli, B. atrophaeus, and A. fumigatus concentrations present in the aerosol samples prior to spiking filters. Only A. fumigatus was found in these samples (on the PM loaded onto the quartz fiber filters) at levels above the MDL. In this case, the quantities of the measured spores were above the detection level (total of ηDNA plus ηfilter adjusted average of 1,670 spores/filter) with a COV of 42% (n = 7) and demonstrated reproducibility similarities between naturally occurring and spiked A. fumigatus samples.
FIG. 2.

Reproducibility of qPCR near detection levels (∼103 and ∼104 cells). Each data point represents one of seven independent spiked filter replicates. Dashed lines indicated true values of spiked cultures.
TABLE 2.
Reproducibility and instrument repeatability as COV (n = 7) of qPCR measurements of microorganisms spiked onto filters
| Culture | Filter type | Accuracy (%) in terms of no. of organisms spiked on filter |
True difference for 95% confidence (n-fold concn of sample)a | |||
|---|---|---|---|---|---|---|
| Reproducibility |
Instrument repeatability |
|||||
| ∼103 | ∼104 | ∼103 | ∼104 | |||
| E. coli | Quartz | 78 | 60 | 36 | 44 | 3.1 |
| PCTE | 79 | 70 | 11 | 26 | 3.2 | |
| B. atrophaeus | Quartz | 64 | 47 | 57 | 41 | 2.4 |
| PCTE | 60 | 57 | 58 | 51 | 2.2 | |
| A. fumigatus | Quartz | 61 | 67 | 17 | 61 | 2.5 |
| PCTE | 28 | 49 | 15 | 21 | 1.3 | |
For 95% confidence, n = 7 replicates.
Using the largest measured reproducibilities for each of the three organisms and a 95% confidence interval (n = 7 replicates), the dissimilarity in the microbial quantities of two samples cannot be resolved unless the true difference is greater than 3.2 times for E. coli, 2.5 times for B. atrophaeus, and 2.5 times for A. fumigates. For a given reproducibility COV, this value increases significantly as the number of replicates decreases. For example, for E. coli with a COV of 79%, the two true values must be dissimilar by at least 3.2 times (n = 7), 5.8 times (n = 5), and 35 times (n = 4) to ensure that these differences can be significantly identified by qPCR.
The COV values for instrument repeatability are also presented in Table 2 for all species. Average COV values for instrument repeatability for the ∼103 and ∼104 cases were 32% and 40% for all organisms. Instrument repeatability accounted for more than 50% of the reproducibility.
Method detection limit.
In order to detect a microorganism concentration with 99% confidence (26), at least n = 7 successful detections out of N = 7 trials (P = 1/128) are required. The smallest amount of DNA at which seven successful detections can be observed out of seven trials was measured to be the genomic DNA from five cells of B. atrophaeus (1 qPCR target gene/genome) and the genomic DNA from 0.05 spores for A. fumigatus (average of 54 qPCR target genes per genome) (Table 3). Overall MDL values that account for extraction efficiencies ηDNA and ηfilter as well as the ratio of extracted DNA to the DNA used as template (F) in the qPCRs are also presented in Table 3 for an F of 25. Figure 3 integrates sampling into MDL estimates by demonstrating the volume of air that must be sampled to meet detection levels for a particular organism based on the overall MDL values presented in Table 3.
TABLE 3.
MDLs for B. atrophaeus and A. fumigatus
FIG. 3.

Volume of air that must be sampled to meet MDLs for B. atrophaeus and A. fumigatus. Upper and lower dashed lines represent ranges due to sample processing. Upper dashed lines represent overall MDL using an F of 25 and lowest value for ηDNA and ηfilter; the lower dashed lines represent an F of 10 and highest value of ηDNA and ηfilter.
DISCUSSION
The rapidly expanding field of environmental molecular biology brings the promise of describing how physical, chemical, and biological processes influence human exposure to allergenic, toxic, or pathogenic microorganisms in air. The comprehensive treatment of accuracy, precision, and MDLs presented here provides guidelines enabling researchers to prevent underestimation of exposure due to sample processing inefficiencies, to define a level of statistical rigor for aerosol qPCR experimental design such that real differences are not obscured by experimental error, and to choose sampling strategies to ensure that MDLs are below relevant aerosol concentrations. Such information is required if qPCR measurements are to advance exposure science and enable mechanistic investigations of the fate and sources of indoor and ambient biological aerosols. An inherent limitation of this study was that accuracy, precision, and MDL estimates are influenced by organism and sampling processing protocols. Thus, these values cannot be directly interpreted for every aerosol study but, rather, are meant to demonstrate the need for a more statistically rigorous qPCR approach and to provide a template for these approaches. While laboratories that perform qPCR on aerosol samples must determine these parameters with their particular equipment, personnel, sample types, and target organisms, many general concepts can be taken from the results presented here. The estimates here included Gram-positive and Gram-negative bacteria and a common airborne fungus as well as readily available and common DNA extraction methods.
Results from DNA and whole-cell filter extraction efficiency demonstrated that these values are significant and that excluding them may cause a dramatic underrepresentation of airborne concentrations or exposure. DNA extraction was the most pronounced source of these inefficiencies. DNA extraction methods used here were included because they are commonly used in environmental analysis and because many aerosol labs have limited expertise and resources in molecular biology and rely on kits for DNA extraction. More rigorous custom methods for extracting and recovering DNA from aerosols have been published (15, 43) although direct comparisons with the ηDNA presented here are complicated by the fact that these literature values are based on comparative efficiencies rather than on absolute efficiencies. Beyond DNA, extraction efficiencies of whole cells from common aerosol filter material have not been systematically reported in the literature (16, 24, 40, 44).
Collection efficiencies for aerosol sampling devices may also contribute to inaccurate aerosol qPCR measurements. At common aerodynamic diameters for bacteria and fungi, bioaerosol sampling efficiencies associated with filtration are near 100%; thus, sampling was not included in this study. However, many bioaerosol samplers used for viability sampling have variable collection efficiencies (23), and a firm knowledge of these efficiencies is requisite for use in bioaerosol studies that seek to quantify absolute exposure. An alternate method for obtaining accurate aerosol qPCR values is to calibrate qPCR output with samples collected at a known aerosol concentration (3). This calibration technique thus incorporates all of the unknown efficiencies but requires specialized equipment for aerosolizing a known stream of pure culture organisms, and this may not be practical for the diverse group of researchers and practitioners engaged in bioaerosol quantification. Results here also suggest that a standard curve produced by spiking a known content of target organisms onto the relevant sampling substrate will also produce an accurate strategy for aerosol qPCR-based measurements. Finally, while PCR inhibition was not observed here, inhibition from particulate matter has been previously demonstrated to reduce accuracy and precision associated with qPCR and must be reduced or removed when relevant (15, 38).
The results presented here also contribute to bioaerosol quantification by defining the precision that can be expected in qPCR analysis of PM-loaded filters. The experimental variations inherent in qPCR place practical limits on resolving the differences among different aerosol samples or treatments. Using the highest COV values (n = 7) for reproducibility, samples that were different in concentrations by less than 3.2 times could not be statistically distinguished. This value increased when fewer replicates were used (Table 3). While including a large amount of replication should be standard practice in qPCR aerosol investigations, there are additional opportunities to improve precision. The largest uncertainties in reproducibility were embedded in the instrument repeatability, which accounted for greater than 50% of the total variation. Instrument repeatability, however, can be improved if the quantity of sample template is significantly greater than the MDLs (50). The MDL experiments performed here on seven replications for different concentrations demonstrated that the instrument repeatability (as COV) decreases by 1.6 to 2.5 times at approximately 2 orders of magnitude above the qPCR limits of detection (Fig. 4). More generally, others have suggested that threshold cycle (CT) values below 30 results in improved instrumental repeatability (25).
FIG. 4.

Instrument repeatability as a function of cell quantity COV (n = 7). The qPCR MDLs are 5 cells for B. atrophaeus and 0.05 cells for A. fumigatus.
The MDL concept has added importance in bioaerosol research. The constant exposure of humans through inhalation results in health effects (allergenic response, infection, etc.) at low aerosol concentration levels. Thus, impacts may occur at levels that go undetected in aerosol samples when collection time is limited. Where health-relevant or other regulatory guideline exposure levels can be defined, knowledge of the MDL can be used to determine the volume of air that must be sampled to reach these relevant levels. In cases where agents are not detected, MDLs and sample volume information provide insight into the importance of values below the limits of detection. The overall MDLs reported for the test bacteria B. atrophaeus and test fungi A. fumigatus ranged from 2,000 to 3,000 bacteria and 10 to 25 spores, respectively. The major differences in these two values was due to the number of target gene copies in each organism, with one copy per cell of the recA gene for B. atrophaeus and an average of 54 gene copies per spore of the 18S rRNA gene in A. fumigatus. Detection limits are also strongly influenced by the ratio (F) of total DNA extracted to the DNA that is used in the final qPCRs. This ratio and the overall MDL can be reduced by concentrating the DNA in the final extract by either evaporation or ethanol precipitation although losses in these processes are possible from inefficient precipitation and collection or from increased PCR inhibition caused by concentrating contaminants in the extract.
Finally, E. coli was not included in MDL determinations due to the presence of contaminant DNA from recombinant E. coli DNA polymerase production. When E. coli or universal bacterial primers are applied to no-template controls, qPCR typically produces a positive signal ranging from 101 to 102 gene copies. In these cases, custom qPCR master mixes may be produced in order to independently dilute the DNA polymerase concentration (51), which, in turn, reduces the signal in no-template controls. This reduction, however, changes the shape of qPCR curves plotting fluorescence versus cycle number and requires that full standard curves be produced at these reduced DNA polymerase amounts. Such concerns with positive no-template controls are also associated with qPCR using universal primers and have been reported for other specific primer sets (50). In these cases, qPCR MDLs are more appropriately defined by identifying a template quantity that is significantly greater than the response generated by a no-template control. These MDLs will be greater than those reported for assays where no-template controls consistently produce a negative response.
Conclusion.
Application of quantitative molecular methods to indoor and outdoor aerosols shows promise for elucidating the physical, chemical, and biological processes that influence human exposure to infectious and noninfectious agents. Variables that influence accuracy, precision, and method detection limits of qPCR for bioaerosols include sample collection efficiency, efficiencies associated with extracting target cells from sampling medium and extracting DNA from cells, the number of target gene copies per genome, PCR inhibition, and the inherent variable in the qPCR instrument (especially at low template concentrations). The results presented here are meant to provide guidance on the experimental and statistical rigor that must accompany qPCR aerosol measurements, which are unlike water and terrestrial matrices in that differences between aerosol samples are smaller and concentrations typically hover near MDLs. This guidance includes recommendations on the importance of accounting for sample preparation efficiencies, requirements for sample replication, opportunities for improving precision, and approaches for estimating and meeting method detection limits. With these controls in place, the continued use of qPCR in aerosol science and engineering is expected to lead to more robust estimations of exposure and contribute to elucidating the processes that influence biological concentrations and fate in indoor air and the atmosphere.
Acknowledgments
Primary funding for this project was provided by the Alfred P. Sloan foundation. N. Yamamoto currently receives a postdoctoral fellowship grant from the Japan Society for the Promotion of Science.
Footnotes
Published ahead of print on 3 September 2010.
REFERENCES
- 1.Amann, R. I., W. Ludwig, and K. H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amato, P., M. Menager, M. Sancelme, P. Laj, G. Mailhot, and A. M. Delort. 2005. Microbial population in cloud water at the Puy de Dôme: implications for the chemistry of clouds. Atmos. Environ. 39:4143-4153. [Google Scholar]
- 3.An, H. R., G. Mainelis, and L. White. 2006. Development and calibration of real-time PCR for quantification of airborne microorganisms in air samples. Atmos. Environ. 40:7924-7939. [Google Scholar]
- 4.Bauer, H., M. Claeys, R. Vermeylen, E. Schueller, G. Weinke, A. Berger, and H. Puxbaum. 2008. Arabitol and mannitol as tracers for the quantification of airborne fungal spores. Atmos. Environ. 42:588-593. [Google Scholar]
- 5.Bergh, O., K. Y. Borsheim, G. Bratbak, and M. Heldal. 1989. High abundance of viruses found in aquatic environments. Nature 340:467-468. [DOI] [PubMed] [Google Scholar]
- 6.Blachere, F. M., W. G. Lindsley, T. A. Pearce, S. E. Anderson, M. Fisher, R. Khakoo, B. J. Meade, O. Lander, S. Davis, R. E. Thewlis, I. Celik, B. T. Chen, and D. H. Beezhold. 2009. Measurement of airborne influenza virus in a hospital emergency department. Clin. Infect. Dis. 48:438-440. [DOI] [PubMed] [Google Scholar]
- 7.Booth, T. F., B. Kournikakis, N. Bastien, J. Ho, D. Kobasa, L. Stadnyk, Y. Li, M. Spence, S. Paton, B. Henry, B. Mederski, D. White, D. E. Low, A. McGeer, A. Simor, M. Vearncombe, J. Downey, F. B. Jamieson, P. Tang, and F. Plummer. 2005. Detection of airborne severe acute respiratory syndrome (SARS) coronavirus and environmental contamination in SARS outbreak units. J. Infect. Dis. 191:1472-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buttner, M. P., P. Cruz, L. D. Stetzenbach, A. K. Klima-Comba, V. L. Stevens, and T. D. Cronin. 2004. Determination of the efficacy of two building decontamination strategies by surface sampling with culture and quantitative PCR analysis. Appl. Environ. Microbiol. 70:4740-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, P. S., and C. S. Li. 2005. Quantification of airborne Mycobacterium tuberculosis in health care setting using real-time qPCR coupled to an air-sampling filter method. Aerosol Sci. Technol. 39:371-376. [Google Scholar]
- 10.DeLong, E. F., L. T. Taylor, T. L. Marsh, and C. M. Preston. 1999. Visualization and enumeration of marine planktonic archaea and bacteria by using polyribonucleotide probes and fluorescent in situ hybridization. Appl. Environ. Microbiol. 65:5554-5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doležel, J., J. Bartos, H. Voglmayr, J. Greilhuber, and R. A. Thomas. 2003. Nuclear DNA content and genome size of trout and human. Cytometry A 51:127-129. [DOI] [PubMed] [Google Scholar]
- 12.Elbert, W., P. E. Taylor, M. O. Andreae, and U. Pöschl. 2007. Contribution of fungi to primary biogenic aerosols in the atmosphere: wet and dry discharged spores, carbohydrates, and inorganic ions. Atmos. Chem. Phys. 7:4569-4588. [Google Scholar]
- 13.Fabian, P., J. J. McDevitt, W. H. DeHaan, R. O. P. Fung, B. J. Cowling, K. H. Chan, G. M. Leung, and D. K. Milton. 2008. Influenza virus in human exhaled breath: an observational study. PLoS One 3:e2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haugland, R. A., and S. J. Vesper. May 2002. Method for identifying and quantifying specific fungi and bacteria. U.S. patent 6,387,652.
- 15.Haugland, R. A., N. Brinkman, and S. J. Vesper. 2002. Evaluation of rapid DNA extraction methods for the quantitative detection of fungi using real-time PCR analysis. J. Microbiol. Methods 50:319-323. [DOI] [PubMed] [Google Scholar]
- 16.Haugland, R. A., J. L. Heckman, and L. J. Wymer. 1999. Evaluation of different methods for the extraction of DNA from fungal conidia by quantitative competitive PCR analysis. J. Microbiol. Methods 37:165-176. [DOI] [PubMed] [Google Scholar]
- 17.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 18.Heidelberg, J. F., M. Shahamat, M. Levin, I. Rahman, G. Stelma, C. Grim, and R. R. Colwell. 1997. Effect of aerosolization on culturability and viability of Gram-negative bacteria. Appl. Environ. Microbiol. 63:3585-3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrera, M. L., A. C. Vallor, J. A. Gelfond, T. F. Patterson, and B. L. Wickes. 2009. Strain-dependent variation in 18S ribosomal DNA copy numbers in Aspergillus fumigatus. J. Clin. Microbiol. 47:1325-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurst, C. J., R. L. Crawford, G. R. Knudsen, M. J. McInerney, and L. D. Stetzenbach. 2002. Introduction to aerobiology, p. 801-813. In C. J. Hurst and R. L. Crawford (ed.), Manual of environmental microbiology, 2nd ed. ASM Press, Washington, DC.
- 21.Jensen, P. A., W. F. Todd, G. N. Davis, and P. V. Scarpino. 1992. Evaluation of eight bioaerosol samplers challenged with aerosols of free bacteria. AIHA J. 53:660-667. [DOI] [PubMed] [Google Scholar]
- 22.Jones, N. C., C. A. Thornton, D. Mark, and R. M. Harrison. 2000. Indoor/outdoor relationships of particulate matter in domestic homes with roadside, urban and rural locations. Atmos. Environ. 34:2603-2612. [Google Scholar]
- 23.Juozaitis, A., K. Willeke, S. A. Grinshpun, and J. Donnelly. 1994. Impaction onto a glass slide or agar versus impingement into a liquid for the collection and recovery of airborne microorganisms. Appl. Environ. Microbiol. 60:861-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabir, S., N. Rajendran, T. Amemiya, and K. Itoh. 2003. Quantitative measurement of fungal DNA extracted by three different methods using real-time polymerase chain reaction. J. Biosci. Bioeng. 96:337-343. [DOI] [PubMed] [Google Scholar]
- 25.Karlen, Y., A. McNair, S. Perseguers, C. Mazza, and N. Mermod. 2007. Statistical significance of quantitative PCR. BMC Bioinformatics 8:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keith, L. H., W. Crummett, J. Deegan, Jr., R. A. Libby, J. K. Taylor, and G. Wentler. 1983. Principles of environmental analysis. Anal. Chem. 55:2210-2218. [Google Scholar]
- 27.Kohara, Y., K. Akiyama, and K. Isono. 1987. The physical map of the whole E. coli chromosome: application of a new strategy for rapid analysis and sorting of a large genomic library. Cell 50:495-508. [DOI] [PubMed] [Google Scholar]
- 28.Kunst, F., N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessiéres, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S. K. Choi, J. J. Codani, I. F. Connerton, N. J. Cummings, R. A. Daniel, F. Denizot, K. M. Devine, A. Düsterhöft, S. D. Ehrlich, P. T. Emmerson, K. D. Entian, J. Errington, C. Fabret, E. Ferrari, D. Foulger, C. Fritz, M. Fujita, Y. Fujita, S. Fuma, A. Galizzi, N. Galleron, S. Y. Ghim, P. Glaser, A. Goffeau, E. J. Golightly, G. Grandi, G. Guiseppi, B. J. Guy, K. Haga, J. Haiech, C. R. Harwood, A. Hénaut, H. Hilbert, S. Holsappel, S. Hosono, M. F. Hullo, M. Itaya, L. Jones, B. Joris, D. Karamata, Y. Kasahara, M. Klaerr-Blanchard, C. Klein, Y. Kobayashi, P. Koetter, G. Koningstein, S. Krogh, M. Kumano, K. Kurita, A. Lapidus, S. Lardinois, J. Lauber, V. Lazarevic, S. M. Lee, A. Levine, H. Liu, S. Masuda, C. Mauël, C. Médigue, N. Medina, R. P. Mellado, M. Mizuno, D. Moestl, S. Nakai, M. Noback, D. Noone, M. O'Reilly, K. Ogawa, A. Ogiwara, B. Oudega, S. H. Park, V. Parro, T. M. Pohl, D. Portetelle, S. Porwollik, A. M. Prescott, E. Presecan, and P. Pujic. 1997. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249-256. [DOI] [PubMed] [Google Scholar]
- 29.Kuo, H. W., and H. Y. Shen. 2009. Indoor and outdoor PM2.5 and PM10 concentrations in the air during a dust storm. Build. Environ. 45:610-614. [Google Scholar]
- 30.Lacey, J., and B. Crook. 1988. Fungal and actinomycete spores as pollutants of the workplace and occupational allergens. Ann. Occup. Hyg. 32:515-533. [DOI] [PubMed] [Google Scholar]
- 31.Latge, J. P. 2001. The pathobiology of Aspergillus fumigatus. Trends Microbiol. 9:382-389. [DOI] [PubMed] [Google Scholar]
- 32.Lee, B. U., and S. S. Kim. 2003. Sampling E. coli and B. subtilis bacteria bioaerosols by a new type of impactor with a cooled impaction plate. J. Aerosol Sci. 34:1097-1100. [Google Scholar]
- 33.Li, C. S., M. L. Hao, W. H. Lin, C. W. Chang, and C. S. Wang. 1999. Evaluation of microbial samplers for bacterial microorganisms. Aerosol Sci. Technol. 30:100-108. [Google Scholar]
- 34.Lin, X., T. Reponen, K. Willeke, Z. Wang, S. A. Grinshpun, and M. Trunov. 2000. Survival of airborne microorganisms during swirling aerosol collection. Aerosol Sci. Technol. 32:184-196. [Google Scholar]
- 35.MacBerthouex, P., and L. C. Brown. 2002. Statistics for environmental engineers, 2nd ed. CRC Press, Boca Raton, FL.
- 36.Mainelis, G., R. L. Gorny, T. Reponen, M. Trunov, S. A. Grinshpun, P. Baron, J. Yadav, and K. Willeke. 2002. Effect of electrical charges and fields on injury and viability of airborne bacteria. Biotechnol. Bioeng. 79:229-241. [DOI] [PubMed] [Google Scholar]
- 37.Makino, S. I., H. I. Cheun, M. Watarai, I. Uchida, and K. Takeshi. 2001. Detection of anthrax spores from the air by real-time PCR. Lett. Appl. Microbiol. 33:237-240. [DOI] [PubMed] [Google Scholar]
- 38.McDevitt, J. J., P. S. J. Lees, W. G. Merz, and J. Schwab. 2007. Inhibition of quantitative PCR analysis of fungal conidia associated with indoor air particulate matter. Aerobiologia 23:35-45. [Google Scholar]
- 39.Miller, J. C., and J. N. Miller. 1993. Statistics for analytical chemistry. Ellis Horwood, Ltd., West Sussex, England.
- 40.Mumy, K. L., and R. H. Findlay. 2004. Convenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. J. Microbiol. Methods 57:259-268. [DOI] [PubMed] [Google Scholar]
- 41.Nazaroff, W. W. 2004. Indoor particle dynamics. Indoor Air 14(Suppl. 7):175-183. [DOI] [PubMed] [Google Scholar]
- 42.Nierman, W. C., A. Pain, M. J. Anderson, J. R. Wortman, H. S. Kim, J. Arroyo, M. Berriman, K. Abe, D. B. Archer, C. Bermejo, J. Bennett, P. Bowyer, D. Chen, M. Collins, R. Coulsen, R. Davies, P. S. Dyer, M. Farman, N. Fedorova, T. V. Feldblyum, R. Fischer, N. Fosker, A. Fraser, J. L. García, M. J. García, A. Goble, G. H. Goldman, K. Gomi, S. Griffith-Jones, R. Gwilliam, B. Haas, H. Haas, D. Harris, H. Horiuchi, J. Huang, S. Humphray, J. Jiménez, N. Keller, H. Khouri, K. Kitamoto, T. Kobayashi, S. Konzack, R. Kulkarni, T. Kumagai, A. Lafton, J. P. Latgé, W. Li, A. Lord, C. Lu, W. H. Majoros, G. S. May, B. L. Miller, Y. Mohamoud, M. Molina, M. Monod, I. Mouyna, S. Mulligan, L. Murphy, S. O'Neil, I. Paulsen, M. A. Peñalva, M. Pertea, C. Price, B. L. Pritchard, M. A. Quail, E. Rabbinowitsch, N. Rawlins, M. A. Rajandream, U. Reichard, H. Renauld, G. D. Robson, S. Rodriguez De Cordoba, J. M. Rodríguez-Peña, C. M. Ronning, S. Rutter, S. L. Salzberg, M. Sanchez, J. C. Sánchez-Ferrero, D. Saunders, K. Seeger, R. Squares, S. Squares, M. Takeuchi, F. Tekaia, G. Turner, C. R. Vazquez De Aldana, J. Weidman, O. White, J. Woodward, J. H. Yu, C. Fraser, J. E. Galagan, K. Asai, M. Machida, N. Hall, B. Barrell, and D. W. Denning. 2005. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151-1156. [DOI] [PubMed] [Google Scholar]
- 43.Paez-Rubio, T., E. Viau, S. Romero-Hernandez, and J. Peccia. 2005. Source bioaerosol concentration and rRNA gene-based identification of microorganisms aerosolized at a flood irrigation wastewater reuse site. Appl. Environ. Microbiol. 71:804-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peccia, J., and M. Hernandez. 2006. Incorporating polymerase chain reaction-based identification, population characterization, and quantification of microorganisms into aerosol science: a review. Atmos. Environ. 40:3941-3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pyankov, O. V., I. E. Agranovski, O. Pyankova, E. Mokhonova, V. Mokhonov, A. S. Safatov, and A. A. Khromykh. 2007. Using a bioaerosol personal sampler in combination with real-time PCR analysis for rapid detection of airborne viruses. Environ. Microbiol. 9:992-1000. [DOI] [PubMed] [Google Scholar]
- 46.Radosevich, J. L., W. J. Wilson, J. H. Shinn, T. Z. DeSantis, and G. L. Andersen. 2002. Development of a high-volume aerosol collection system for the identification of air-borne micro-organisms. Lett. Appl. Microbiol. 34:162-167. [DOI] [PubMed] [Google Scholar]
- 47.Saikaly, P. E., M. A. Barlaz, and F. L. De Los Reyes III. 2007. Development of quantitative real-time PCR assays for detection and quantification of surrogate biological warfare agents in building debris and leachate. Appl. Environ. Microbiol. 73:6557-6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singer, V. L., L. J. Jones, S. T. Yue, and R. P. Haugland. 1997. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal. Biochem. 249:228-238. [DOI] [PubMed] [Google Scholar]
- 49.Sislian, P. R., D. Pham, X. Zhang, M. Li, L. Mädler, and P. D. Christofides. 2009. Bacterial aerosol neutralization by aerodynamic shocks using an impactor system: experimental results for E. coli and analysis. Chem. Eng. Sci. 65:1490-1502. [Google Scholar]
- 50.Smith, C. J., D. B. Nedwell, L. F. Dong, and A. M. Osborn. 2006. Evaluation of quantitative polymerase chain reaction-based approaches for determining gene copy and gene transcript numbers in environmental samples. Environ. Microbiol. 8:804-815. [DOI] [PubMed] [Google Scholar]
- 51.Spangler, R., N. L. Goddard, and D. S. Thaler. 2009. Optimizing Taq polymerase concentration for improved signal-to-noise in the broad range detection of low abundance bacteria. PLoS One 4:e7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor, J. P., B. Wilson, M. S. Mills, and R. G. Burns. 2002. Comparison of microbial numbers and enzymatic activities in surface soils and subsoils using various techniques. Soil Biol. Biochem. 34:387-401. [Google Scholar]
- 53.U.S. Environmental Protection Agency. June 2010, posting date. EPA technology for mold identification and enumeration. National Exposure Research Laboratory, U.S. Environmental Protection Agency, Cincinnati, OH. http://www.epa.gov/microbes/moldtech.htm#license.
- 54.Wang, Z., T. Reponen, S. A. Grinshpun, R. L. Górny, and K. Willeke. 2001. Effect of sampling time and air humidity on the bioefficiency of filter samplers for bioaerosol collection. J. Aerosol Sci. 32:661-674. [Google Scholar]
- 55.Yamamoto, N., M. Kimura, H. Matsuki, and Y. Yanagisawa. 2010. Optimization of a real-time PCR assay to quantitate airborne fungi collected on a gelatin filter. J. Biosci. Bioeng. 109:83-88. [DOI] [PubMed] [Google Scholar]