

Abstract

Indium-promoted coupling reactions between propargyl aldehydes (1) and α-chloropropargylphenyl sulfide are reported. Although water has been shown to accelerate indium metal promoted reactions, the reverse pattern was observed in this series. Use of N-methylformamide (NMF), which has not previously been a solvent known for use in indium-promoted reactions, afforded an acceleration of these Barbier-style reactions compared to water. Indium-promoted reactions in this study also showed excellent regiocontrol and good stereocontrol, allowing for easy entry into the formation of epoxydiyne and enediyne skeletal structures. This paper also describes use of the Barbier Coupled product (2) as a new, and easy, entry into the formation of enediyne and epoxydiyne skeletal structures.
Indroduction
Many studies over the past 15 years have focused on discovering solvents that help promote efficient C−C bond formation under benign reaction conditions.1,2 New solvents in Diels−Alder(2) and Barbier3,4 reactions have been sought to help streamline the one-pot transformations further, reducing synthetic steps and organic waste. Polar media often accelerate reactions of this type which has led to the use of water as an acceptable solvent for such organic conversions.1−4 Recent contributions from our laboratory to this field have centered on coupling reactions between propargyl aldehydes and allyl or propargyl halides under Barbier conditions to form homoallylic and homopropargylic propargyl alcohols.5a,5b These compounds find wide use as synthetic templates and show a propensity for oxy-Cope rearrangements (Scheme 1).5,6
Scheme 1. Barbier Coupling Reactions of Propargyl Aldehydes and Allyl or Progrargyl Halides.
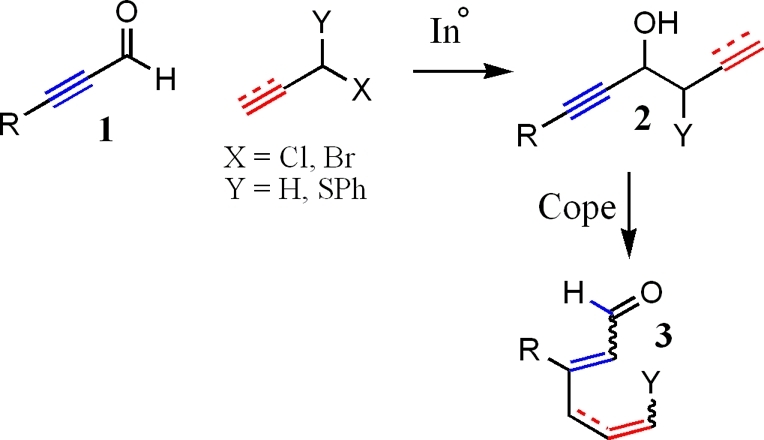
The use of α-chloropropargylphenyl sulfide (4), coupled with a propargyl aldehyde should offer an easy route into formation of enediyne and epoxydiyne skeletal structures (Scheme 2). Previous studies revealed that formation of 2 or 3 under indium-promoted Barbier conditions is solvent dependent.5a,6 Use of an organic solvent for the coupling reaction results in a mixture of 2 and 3 while the use of water as a solvent yields 2 as the sole product.
Scheme 2. Use of 4 To Form Enediyne and Epoxydiyne Skeletal Structures.
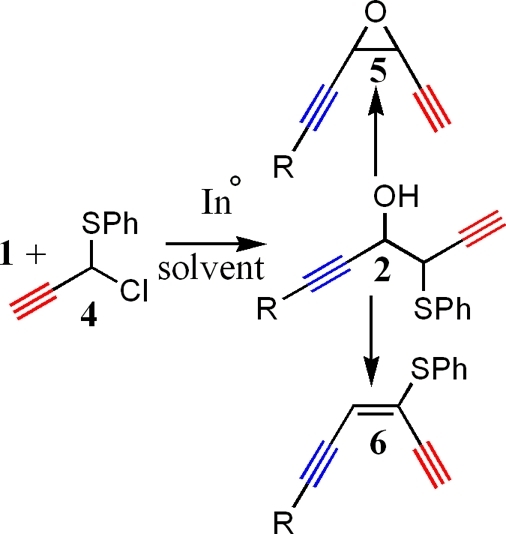
There may be two reasons for this selectivity. It is possible that water protonates the coupled product to form 2 quickly, quenching the alkoxide intermediate, thereby suppressing the possible oxy-Cope pathway shown in Scheme 1. It may also be possible that the increased polarity of the solvent itself stabilizes the anion formed upon coupling, thus raising the relative rate of Cope rearrangement in this system. With a less polar, aprotic solvent, the anion will not be quenched, nor will it be stabilized to the same extent as witnessed in water. A combination of these phenomena may be why an oxy-Cope rearrangement occurs under less polar conditions, forming product 3 (Scheme 1). A coupling reaction between 1 and 4 (Scheme 2) was conducted under aqueous conditions to form 2 (Y = SPh), with good regioselectivity; however, the rate of this reaction is surprisingly slow. Although reactions of the type shown in Scheme 1 are reported in the literature to be accelerated by polar solvents such as water,3−5 coupling reactions between 1 and 4, as shown in Scheme 2, are quite slow. Indeed, when water is used as the solvent, reactions often require 10−36 h for completion. We believe there are two reasons behind the slow rate of reaction. First, when the reagents are mixed, a viscous, orange oil forms. This oil has a density greater than that of water so it is presumed to be an organometallic species formed by interaction of the indium metal with the halide species. We also believe the aldehyde species may be part of the complex as well. Isolation of the oil followed by workup in an acidic THF/H2O mixture leads to isolation of propargylphenyl sulfide and, to a smaller extent, the aldehyde. We surmise that low solubility of this organometallic complex leads to poor reagent mixing, slowing the Barbier coupling under aqueous conditions. Second, indium-promoted coupling reactions conducted in water become more acidic as the reaction proceeds.1,5 We believe it is possible that the aldehyde is in equilibrium with its corresponding hydrate species, reducing the electrophilicity of the carbon center. A combination of these two reasons would lead to a decrease in the rate of reaction between 1 and 4 when conducted in an aqueous solvent. The reaction proceeds more quickly (8−12 h) in DMF, a less polar solvent, due to better solubility of all reagents, however, a mixture of 2 and 3 was isolated. It was originally expected that a mixture of water and DMF would lead to faster reaction times while still maintaining regiocontrol of product formation. The use of a water/DMF solvent system, however, resulted in a mixture of products with water constituting up to 50% v/v of the solvent system. When water exceeds 50% v/v of the solvent system, only product 2 is isolated, but the rate of reaction slows perceptively once again. To help expedite this reaction sequence, we began a search for solvent with polarity similar to, or greater than that of water. It was expected that an extremely polar organic solvent would promote the same type of acceleration normally witnessed in Barbier couplings carried out under aqueous conditions.1−5 An organic solvent could also help solubilize the intermediate and reduce possible formation of the hydrate species, thus allowing for a faster coupling reaction to occur. If increased polarity stabilizes the anion formed by coupling of 1 and 4, oxy-Cope rearrangement should also be suppressed.
With a dielectric constant of 186.9 at 25 °C,7a,7b significantly greater than that of water (78.37(7)), and DMF (38.3(7b)), N-methylformamide (NMF) has found occasional use in previous reactions requiring polar organic solvents; however, it has historically found more widespread use as an organic reagent.7,8 With a pKa of approximately 24,(7d) NMF is reactive under a range of organometallic reagent conditions, limiting its use as a solvent.1−4 Indium-promoted coupling reactions, however, have been shown to be accelerated under aqueous conditions,(1) and the relative acidity of NMF, being higher than that of water, should not hinder the proposed Barbier couplings proposed within this paper. We were especially intrigued by the fact that the dielectric constant of NMF increases to 220 as the temperature is lowered from 25 to 0 °C.6,7 We were interested in examining what effect the increase in dielectric constant at lower temperature could have on a Barbier-style reaction of the type shown in Scheme 2 among others. This paper reports on the use of NMF in Barbier-style reactions with a focus on rates of reaction, stereochemistry, and its use to form enediyne and epoxydiyne skeletal structures.(9)
Results and Discussion
Reaction of Propargyl Aldehydes with α-Chloropropargylphenyl Sulfide
Although NMF has characteristics similar to those of DMF, it also has characteristics similar to water, such as a polar X−H bond.(7) Thus, having found a very polar solvent with properties similar to both water and organic systems, we hypothesized that the 1,2-coupling product could be formed in high yield under short reaction times. A series of reactions was set up to test this hypothesis.
As shown in Table 1, use of either water or NMF resulted in formation of the 1,2-product (2) solely with no trace of 3 detected in the product mixture. Although the yield of product was moderate to high in either solvent, product formation in reactions conducted in NMF proceeded at a much greater rate. We believe that there are two possible reasons behind the rate increase of coupling reactions carried out in NMF.
Table 1. Comparison of NMF versus Water in Indium-Promoted Coupling of 1 and 2a.
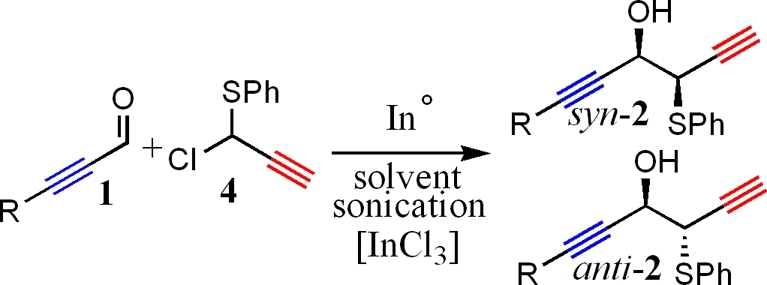
| R | solvent | ratio (syn/anti) | yield (%) | reaction time (h) | |
|---|---|---|---|---|---|
| 1 | n-butyl | NMF | 25:75 | 92 | 3.5 |
| 2 | H2O | 40:60 | 83 | 20.0 | |
| 3 | NMF/InCl3 | 85:15 | 93 | 4.0 | |
| 4 | H2O/InCl3 | 69:31 | 77 | 20.0 | |
| 5 | phenyl | NMF | 20:80 | 91 | 5.0 |
| 6 | H2O | 20:80 | 80 | 32.0 | |
| 7 | NMF/InCl3 | 85:15 | 93 | 5.5 | |
| 8 | H2O/InC13 | 75:25 | 75 | 28.0 | |
| 9 | TMS | NMF | 40:60 | 94 | 4.5 |
| 10 | H2O | 50:50 | 70 | 17.5 | |
| 11 | NMF/InC13 | 70:30 | 91 | 4.5 | |
| 12 | H2O/InC13 | 60:40 | 72 | 20.0 | |
| 13 | TBSO(CH2)3 | NMF | 15:85 | 87 | 5.0 |
| 14 | H2O | 40:60 | 77 | 32.5 | |
| 15 | NMF/InCl3 | 90:10 | 89 | 5.5 | |
| 16 | H2O/InC13 | 70:30 | 71 | 36.0 |
Reactions were run from 3 to 36 h under sonication. The ratio of reagent was as follows: 1.0:1.5:1.1 (aldehyde/halide/indium). The reaction was run at 0.1 M with respect to indium. In the Lewis acid catalyzed reaction, 10 molar % of the Lewis acid was used.
First, there is better mixing of reagents in NMF. All species completely dissolved in NMF, unlike what was observed for reactions in water. As such, we believe mixing is much more efficient, allowing the reaction to proceed at a faster rate. Second, there is increased solvent polarity. As discussed previously, increased solvent polarity has also been noted in many literature examples to lead to rate enhancement in indium-promoted Barbier reactions.3−5 We believe a mixture of better solubility coupled with greater solvent polarity results in an additive effect toward increasing the rate of reactions conducted in NMF when compared to water. H-bonding capabilities of NMF may explain the greater regioselectivity for reactions conducted in NMF when compared to those observed in DMF and THF;1,5,7 however, the H-bonding capacity of NMF is much lower than that of water.(7d) Increased solvent polarity, thus increasing the stability of the intermediate anion and raising the relative energy of the oxy-Cope rearrangement, may seem more likely at this time. In an effort to exclude the solvent as a direct participant in intermediate formation when NMF is used, NMR spectroscopy studies were performed to determine if a possible hemiaminal or imminium species was forming in situ (Figure 1). The imminium functionality carries a positive charge and would increase the electrophilicity of the reactive carbon. Mixing of 1 and NMF under sonication for periods of up to 24 h, however, revealed the presence of only 1 and NMF by NMR spectroscopy.
Figure 1.
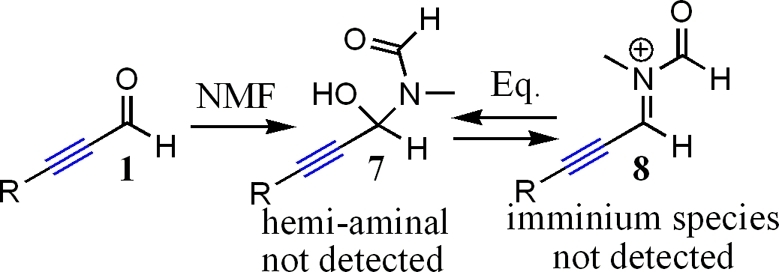
Possible formation of hemiaminal or imminium species.
Stereoselectivity of Reactions
The presence or absence of a Lewis acid has shown to help control stereoselectivity in indium-promoted C−C bond-forming reactions.5b,5c,10 Previous studies in our laboratory have shown that the addition of indium(III) chloride as a Lewis acid favors formation of the syn-product in indium-promoted C−C bond-forming reactions between 4 and aldehyde functional groups.
In the absence of the Lewis acid, the anti-product is favored (Figure 2).(5b) It was our hope that similar effects would be witnessed in the present system, especially while using NMF as the reaction solvent, and we were pleased to see this trend continue. Absence of a Lewis acid in the reagent mixture favored a nonchelated pathway, leading to formation of anti-2.
Figure 2.

Use of indium chloride to control stereochemistry.(5b)
The presence of a Lewis acid in the reaction mixture favored a chelated pathway, leading to formation of syn-2 (Figure 3).
Figure 3.

Nonchelated and chelated pathways.
Diastereoselectivities were determined either by direct measurement of vicinal coupling constants in the hydroxyl sulfide or by corresponding conversion to the epoxide compounds as shown in Figure 4.(11)
Figure 4.
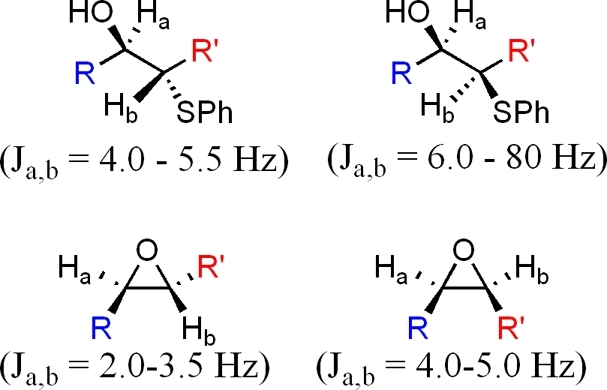
General J values for hydroxyl sulfides and epoxide structures.
Entries 1, 5, and 13 (Table 1) describe reactions run in NMF in the absence of a Lewis acid. Good anti-selectivity is witnessed with ratios as high as 15:85 (syn/anti). In all cases, with the exception of R = phenyl, systems, with and without InCl3, run in NMF gave better selectivity than systems run in water. Both systems showed poor selectivity when R = TMS for reasons of which we are not quite sure. When InCl3 was added as a Lewis acid, the chelated route was favored in ratios as high as 90:10 (syn:anti, entry 15). N-Methylformamide was shown to increase the rate of reaction compared with water and offered better stereocontrol in the systems studied.
Formation of Epoxydiyne and Enediyne Skeletal Structures
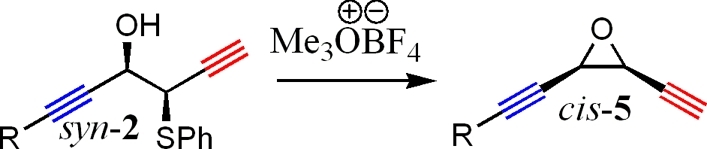
Enediyne and epoxydiyne functionalities contain either a (Z)-3-ene-1,5-diyne or (Z)-3-epoxy-1,5-diyne unit which leads to their noted activity.(9) These unique functional groups cyclize to form a p-benzyne- or p-benzyne-like intermediate that interacts with DNA by abstracting hydrogen.(9) The DNA diradical then either couples with itself, or cleaves, stopping replication. Natural product enediyne and epoxydiynes have found use as anticarcinogenic compounds.(9) This area of study is slowed, however, by complicated access into the skeletal systems required.(9) Literature procedures that find use in epoxy alkyne formation such as halohydrin cylization,(12)m-CPBA,(13) or Oxone(14) have been shown to be successful, but limited examples are known with these systems at present.12−15 Under the Barbier coupling conditions described in this paper, formation of compound 2 offers an efficient and controlled method for the formation of either an enediyne or epoxydiyne skeletal structure under benign conditions by allowing easy conversion to either the cis-epoxydiyne (from syn-2, Scheme 3) or the cis-enediyne (from anti-2, Scheme 4). Facile synthesis of epoxydiyne and enediyne functional groups under the reported conditions would allow for their use as template structures in organic synthesis, opening access to more sophisticated units for further exploration. In the following pages, we describe the transformation of 1 into either an epoxydiyne (5) or enediyne (6) skeletal structure in two steps with high regio- and stereocontrol using NMF as a new solvent to form 2 under Barbier conditions, followed by a one-step transformation to yield either 5 or 6.
Scheme 3. Pathway of Conversion of cis-Epoxydiyne (5) from syn-2.
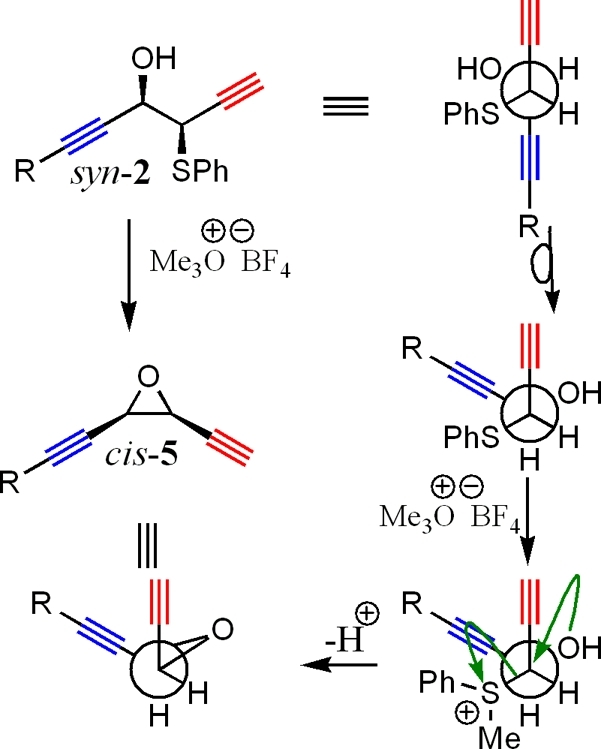
Scheme 4. Pathway of Conversion of anti-2 to (E)-Enediyne 6.
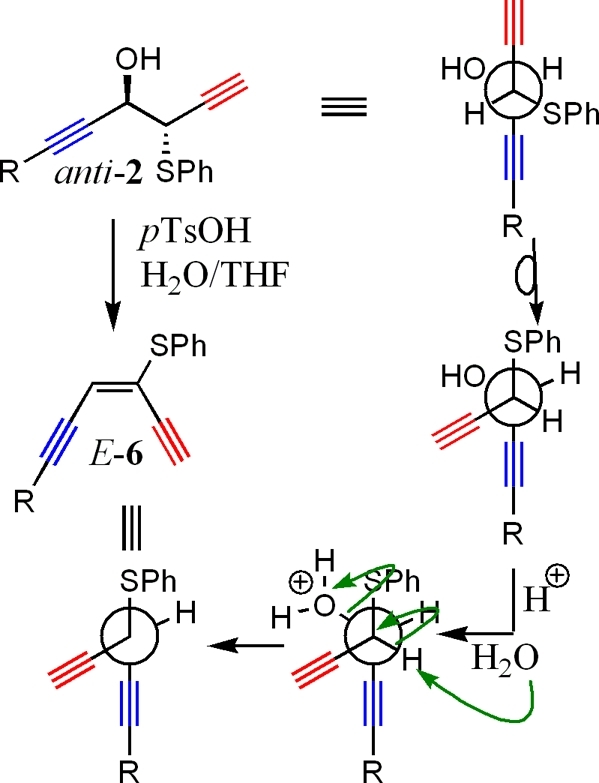
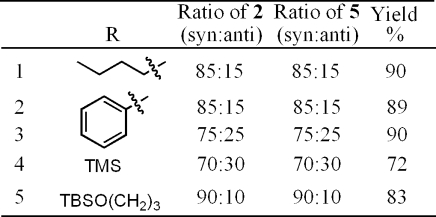
As shown in Scheme 3, syn-2 can adopt the correct conformation to form a cis-oxirane upon reaction with either Me3OBF4 or TlOEt. Conversion of the hydroxy sulfide starting material (2) to the epoxyalkyne product (5) was quite successful as shown in Table 2. In all cases, the propargyl group derived from the halide is preserved. This fact is worth mentioning as Barbier reactions with propargyl halides can lead to rearrangement of the organometallic intermediate into an allenyl product.4,5,16 Reagent 4 has been previously shown to offer one of the few examples of a Barbier reaction with a propargyl halide where rearrangement does not occur.(5b) Use of TlOEt(17) gave yields similar to that of Me3OBF4(18) in all cases of oxirane formation, with stereoselectivity preserved under both sets of conditions.
Table 2. Conversion of syn-2 to syn-Epoxydiynes 5.
 |
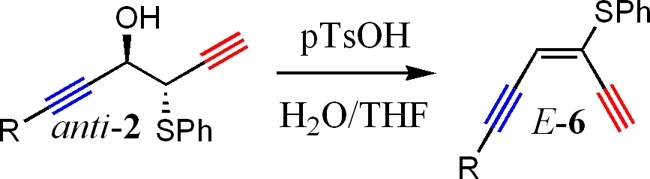
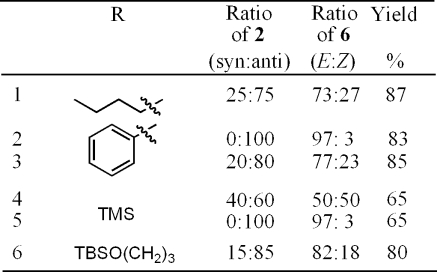
Use of the anti-2 allows formation of the (E)-alkene (the alkyne functional groups are cis to each other) functional group by the pathway shown in Scheme 4.
Formation of the enediyne product was possible under slightly acidic conditions. A catalytic amount of p-TsOH is added to anti-2 in a mixture of water and THF (10% water) with stirring for 3−7 h. The reaction was also conducted under basic conditions after transformation of the OH group to a tosylate. Yields were quite low under basic conditions, presumably due to the competing acidity of the alkynyl proton, leading to decomposition and side product formation. Due to low yields, a transformation under basic conditions was not further explored during this study. Results of the acid-catalyzed conversion of anti-2 to (E)-6 are shown in Table 3. Yields for most systems were in the low 80% range. A slight amount of isomerization was witnessed under acidic conditions (about 3% of the product mixture), but overall, good stereocontrol was realized in these conversions, allowing for easy entry into the cis-enediyne ((E)-alkene) skeletal structure in a two-step sequence.
Table 3. Conversion of anti-2 into (E)-6 (cis-Enediyne).
 |
The assignment of E- and Z-isomers was made by comparison of the β-vinyl proton shift. Alignment of the β-vinyl proton and the heteroatom cis to each other gives rise to a lower field proton resonance than trans alignment of these two groups (Figure 5).(19) The assignments for the enediyne products are compared correctly with the stereochemistry of compounds 2 and 3 within this study.
Figure 5.

Representative 1H NMR shift when aligned cis and/or trans to the heteroatom.
Coupled with the original indium-promoted Barbier reaction, the transformation of starting materials 1 and 4 into enediyne or epoxydiyne compounds proceeded in two steps in an overall 60−80% overall yield, offering an efficient, benign, and good-yielding formation of these important skeletal structures.
Conclusion
Barbier reactions have become increasingly popular as a method form C−C bonds more efficiently. The use of N-methylformamide is shown to be a nice alternative to aqueous conditions in indium promoted coupling reactions between propargyl aldehydes (1) and α-chloropropargylphenyl sulfide (4). This paper introduces the use of NMF as s viable solvent in a Barbier coupling reaction, revealing increased rates and better selectivity when compared to reactions conducted in water and other organic solvents. The increased efficiency of the Barbier coupling reaction as described in this paper allows for facile formation of epoxy-and enediyne skeletal structures in an overall two-step procedure. The introduction of NMF as an alternative solvent in Barbier reactions will open new pathways to organic synthetic chemists. Further studies to test other halides and aldehyde functional groups under Barbier coupling reaction conditions in NMF are currently underway and will be reported in due time.
Experimental Section
General Experimental Procedures
Tetrahydrofuran was purified by distillation under an argon atmosphere over sodium and benzophenone prior to use. Dimethylformamide and CCl4 were purified by distillation over CaH2 prior to use. All other solvents and reagents were used as purchased. Radial chromatography was performed using a Chromatatron.
General Formation of Propargyl Aldehyde Compounds(20a)
To a solution of 30 mmol of alkyne in 80 mL of THF that had been cooled to − 60 °C under nitrogen was added 15 mL of n-BuLi (2.0 M in THF) with stirring. To this solution was added N,N-dimethylformamide (4.66 mL, 60 mmol) dropwise over 10 min. The cooling bath was removed and the solution allowed to warm to 23 °C. After 20 min of further stirring, the solution was added to a mixture of 75 mL of tert-butyl methyl ether and 160 mL of 10% KH2PO4 solution which had been previously cooled to 0 °C. The entire solution was stirred at 0 °C for 30 min and then allowed to warm to room temperature. The aqueous and organic layers were separated. The organic layer was dried over MgSO4, and the solvent removed under vacuum to give a clear oil.
All propargyl aldehydes were used without further purification and could be stored at 0 °C for 2−3 weeks.
Yields for this reaction ranged from 85 to 95%.
I. Coupling Procedures for Propargyl Aldehyde (1) with α-Chloropropargylphenyl Sulfide (4)
I.a. General Procedure Using Water as Solvent
A magnetically stirred solution of aldehyde (1) (2.0 mmol) in deionized water (22 mL) was treated with α-chloropropargylphenyl sulfide (702 mg, 3.0 mmol) and indium powder (228 mg, 2.0 mmol). The solution was allowed to proceed at room temperature until no 1 was seen (TLC analysis). Dichloromethane was added, and stirring was maintained for 30 min. The layers were separated, and the aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic layers were dried over MgSO4 and evaporated to leave a dark yellow to brown oil. Purification was accomplished by flash chromatography or radial chromatography on silica gel (hexanes−ethyl acetate) to give a mixture of hydroxy sulfides (2)(11) as a light yellow oil.
I.a.1. Use of 2-Heptynal (R = n-C4H9) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.a. Purification was accomplished using radial chromatography on silica gel (40:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield = 471 mg (1.76 mmol) = 83%; 1H NMR (300 MHz, CDCl3) δ 0.9−1.5 (m, 7H), 1.82 (d, J = 1.7 Hz, 0.2H), 1.87 (d, J = 1.6 Hz, 0.8H), 2.13 (t, J = 6.5 Hz, 2H), 4.09 (m, 1H), 4.86 (m, 1H), 7.1−7.8 (m, 5H), the OH proton was not detected; 13C NMR (75 MHz, CDCl3) δ 15.5, 18.2, 21.3, 34.2, (41.5, 42.9), (65.0, 66.1), 71.3, 78.5, 80.2, 84.7, 125.4, 126.1, 126.3, 127.6, 128.0, 136.4; MS m/z (M+) calcd 258.1078, obsd 258.0998. Anal. Calcd for C16H18OS: C, 74.38; H, 7.02. Found: C, 74.61; H, 6.99.
I.a.2. Use of 3-Phenylpropynal (R = Phenyl) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in part I.a. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 20:80 syn/anti ratio: total yield = 489 mg (1.78 mmol) = 80%. Anti isomer: yield = 391 mg (1.41 mmol); 1H NMR (300 MHz, CDCl3) δ 1.94 (d, J = 1.8 Hz, 1H), 4.10 (dd, J = 1.8, 4.7 Hz, 1H), 5.11 (d, J = 4.7 Hz, 1H), 7.2−7.7 (m, 10H), the OH proton was not detected; 13C NMR (75 MHz, CDCl3) δ 43.8, 66.0, 68.0, 81.3, 87.2, 93.6, 121.7, 125.8 (2), 127.3 (3), 128.1, 128.3(2), 129.3, 129.5, 136.1. Anal. Calcd for C18H14OS: C, 77.66; H, 5.07. Found: C, 77.28; H, 5.18. Syn isomer: yield = 98 mg (0.37 mmol); 1H NMR (300 MHz, CDCl3) δ 1.83 (d, J = 1.8 Hz, 1H), 3.95 (dd, J = 1.8, 8.1 Hz, 1H), 4.95 (d, J = 8.1 Hz, 1H), 7.2−7.7 (m, 10H), the OH proton was not detected; 13C NMR (75 MHz, CDCl3) δ 44.1, 65.8, 68.0, 81.3, 87.2, 93.6, 121.7, 125.8 (2), 127.3 (3), 128.1, 128.3(2), 129.3, 129.5, 136.1.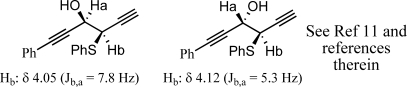
I.a.3. Use of 3-Trimethylsilypropynal (R = TMS) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.a. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 50:50 syn/anti ratio: total yield = 418 mg (1.54 mmol) = 70%; Anti isomer: yield = 209 mg (7.7 mmol); 1H NMR (300 MHz, CDCl3) δ 0.24 (s, 9H), 1.84 (d, J = 1.6 Hz, 1H), 4.23 (dd, J = 1.6, 5.3 Hz, 1H), 4.80 (d, J = 5.3 Hz, 1H), 7.1−7.4 (m, 5H), the OH proton was not detected; 13C NMR (75 MHz, CDCl3) δ 0.28(3), 44.4, 66.5, 67.5, 74.1, 84.3, 86.4, 125.0, 125.3, 126.5 (2), 127.3, 136.1; MS m/z (M+) calcd 274.0848, obsd 274.0819. Anal. Calcd for C15H18OSSi: C, 65.64; H, 6.61. Found: C, 65.17; H, 6.55. Syn isomer: yield = 209 mg (7.7 mmol) = 70%; 1H NMR (300 MHz, CDCl3) δ 0.24 (s, 9H), 1.79 (d, J = 1.7 Hz, 1H), 4.15 (dd, J = 1.7, 7.8 Hz, 1H), 4.69 (d, J = 7.8 Hz, 1H), 7.1−7.4 (m, 5H), the OH proton was not detected; 13C NMR (75 MHz, CDCl3) δ 0.28(3), 44.1, 67.9, 68.1, 75.0, 83.2, 86.9, 125.0, 125.3, 126.5 (2), 127.3, 136.1.
I.a.4. Use of 6-(tert-Butyldimethylsiloxy)-2-hexynal (R = (CH2)3OTBS) with α-Chloropropargylphenyl Sulfide (1)
Processing and analysis of this reaction proceeded as described in section I.a. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides: yield = 633 mg (1.69 mmol) = 77%; 1H NMR (300 MHz, CDCl3) δ 0.25 (s, 6H), 0.97 (s, 9H), 1.25 (m, 2H), 1.80−2.00 (m, 3H), 3.83 (t, J = 6.5, 2H), 4.11 (m, 1H), 4.99 (m, 1H), 7.1−7.4 (m, 5H), the OH proton was not detected; 13C NMR (75 MHz, CDCl3) δ 0.28 (2), 14.5, 15.1, 21.8 (2), 21.9 33.4, 47.0, 64.6, 65.1, 67.9, 75.2, 81.9, 87.3, 125.4, 126.6 (2), 127.1, 128.4, 135.9; MS m/z (M+) calcd 374.1736, obsd 365.1633 (loss of H2O). It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product.
I.b. General Procedure Using Water as Solvent with InCl3 Added
The general procedure was followed as described in section I.a with the exception that 1 mmol of InCl3 was added to the reaction mixture for every 1 mmol of aldehyde used.
I.b.1. Use of 2-Heptynal (R = n-C4H9) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in part I.b. Separation was accomplished using radial chromatography on silica gel (40:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield = 437 mg (1.63 mmol) = 77%. Analysis as shown in section I.a.1.
I.b.2. Use of 3-Phenylpropynal (R = Phenyl) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in part I.b. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 75:25 syn/anti ratio: yield = total = 458 mg (1.65 mmol) = 75%. Anti isomer: yield = 115 mg (0.41 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 1.94 (d, J = 1.8 Hz, 1H), 4.12 (dd, J = 1,8, 4.5 Hz, 1H), 5.14 (d, J = 4.5 Hz, 1H). Syn isomer: yield = 343 mg (1.24 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 1.83 (d, J = 1.8 Hz, 1H), 3.92 (dd, J = 1.8, 8.0 Hz, 1H), 4.94 (d, J = 8.1 Hz, 1H)
I.b.3. Use of 3-Trimethylsilypropynal (R = TMS) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.b. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 60:40 syn/anti ratio: yield = total = 434 mg (1.58 mmol) = 72%. Anti isomer: yield = 174 mg (0.63 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.25 (dd, J = 1.7, 5.5 Hz, 1H), 4.77 (d, J = 5.5 Hz, 1H). Syn isomer: yield = 260 mg (0.95 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.15 (dd, J = 1.5, 7.8 Hz, 1H), 4.71 (d, J = 7.8 Hz, 1H).
I.b.4. Use of 6-(tert-Butyldimethylsiloxy)-2-hexynal (R = (CH2)3OTBS) with α-Chloropropargylphenyl Sulfide (1)
Processing and analysis of this reaction proceeded as described in section I.b. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield 583 mg (1.55 mmol) = 71%. Analysis as shown in section I.a.4.
I.c. General Procedure Using N-Methylformamide as Solvent
A magnetically stirred solution of aldehyde 1 (2.0 mmol) in NMF (22 mL) was treated with α-chloropropargylphenyl sulfide (702 mg, 3.0 mmol) and indium powder (228 mg, 2.0 mmol). The solution was allowed to proceed at room temperature until no 1 was seen (TLC analysis). Dichloromethane (20 mL) and water (30 mL) were added. Stirring was maintained for 30 min. The layers were separated, and the aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with 1 N HCl (3 × 20 mL), dried over MgSO4, and evaporated to leave a dark yellow to brown oil. Purification was accomplished by flash chromatography or radial chromatography on silica gel (hexanes−ethyl acetate) to give a mixture of hydroxy sulfides (2)(11) as a light yellow oil.
I.c.1. Use of 2-Heptynal (R = n-C4H9) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.c. Separation was accomplished using radial chromatography on silica gel (40:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield = 522 mg (2.02 mmol) = 92%. Analysis as shown in section I.a.1.
I.c.2. Use of 3-Phenylpropynal (R = Phenyl) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.c. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 20:80 syn/anti ratio: yield = total = 556 mg (2.00 mmol) = 91%. Anti isomer: yield = 445 mg (1.60 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.10 (dd, J = 1.8, 4.7 Hz, 1H), 5.11 (d, J = 4.7 Hz, 1H). Syn isomer: yield = 111 mg (0.40 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 3.95 (dd, J = 1.8, 8.1 Hz, 1H), 4.95 (d, J = 8.1 Hz, 1H).
I.c.3. Use of 3-Trimethylsilypropynal (R = TMS) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.c. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 40:60 syn/anti ratio: yield = total = 566 mg (2.06 mmol) = 94%. Anti isomer: yield = 340 mg (1.24 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.25 (dd, J = 1.7, 5.5 Hz, 1H), 4.77 (d, J = 5.5 Hz, 1H). Syn isomer: yield = 226 mg (0.82 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.15 (dd, J = 1.5, 7.8 Hz, 1H), 4.71 (d, J = 7.8 Hz, 1H).
I.c.4. Use of 6-(tert-Butyldimethylsiloxy)-2-hexynal (R = (CH2)3OTBS) with α-Chloropropargylphenyl Sulfide (1)
Processing and analysis of this reaction proceeded as described in section I.c. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield = 717 mg (1.91 mmol) = 87%. Analysis as shown in section I.a.4.
I.d. General Procedure Using NMF as Solvent with InCl3 Added
The general procedure was followed as described in section I.c with the exception that 1 mmol of InCl3 was added to the reaction mixture for every 1 mmol of aldehyde used.
I.d.1. Use of 2-Heptynal (R = n-C4H9) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.d. Separation was accomplished using radial chromatography on silica gel (40:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield = 527 mg (2.05 mmol) = 93%. Analysis as shown in section I.a.1.
I.d.2. Use of 3-Phenylpropynal (R = Phenyl) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.d. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 85:15 syn/anti ratio: yield = total = 568 mg (2.05 mmol) = 93%. Anti isomer: yield = 85 mg (0.31 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.13 (dd, J = 1.8, 4.7 Hz, 1H), 5.08 (d, J = 4.7 Hz, 1H). Syn isomer: yield = 483 mg (1.74 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 3.92 (dd, J = 1.8, 8.1 Hz, 1H), 4.97 (d, J = 8.1 Hz, 1H).
I.d.3. Use of 3-Trimethylsilypropynal (R = TMS) with α-Chloropropargylphenyl Sulfide (4)
Processing and analysis of this reaction proceeded as described in section I.d. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) allowing isolation of two hydroxyl sulfide diastereomers in a 70:30 syn/anti ratio: yield = total = 548 mg (2.00 mmol) = 91%. Anti isomer:: yield = 164 mg (0.60 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.23 (dd, J = 1.7, 5.5 Hz, 1H), 4.80 (d, J = 5.5 Hz, 1H). Syn isomer: yield = 384 mg (1.40 mmol); relevant 1H NMR shifts (300 MHz, CDCl3) δ 4.15 (dd, J = 1.5, 7.8 Hz, 1H), 4.68 (d, J = 7.8 Hz, 1H).
I.d.4. Use of 6-(tert-Butyldimethylsiloxy)-2-hexynal (R = (CH2)3OTBS) with α-Chloropropargylphenyl Sulfide (1)
Processing and analysis of this reaction proceeded as described in section I.d. Separation was accomplished using radial chromatography on silica gel (35:1 hexanes−ethyl acetate) to give a mixture of diastereomeric hydroxy sulfides. It was not possible to determine stereoselectivity directly. Stereoselectivity was determined upon transformation to the epoxide product: yield = 732 mg (1.96 mmol) = 89%. Analysis as shown in section I.a.4.
II. Formation of Epoxy Diyne Compounds from Hydroxy Sulfides
II.a. General Procedure Using Trimethyloxonium Tetrafluoroborate Reagent
II.a.1. Formation of 2-Ethynyl-3-hex-1-ynyloxirane
A solution of the hydroxy sulfide mixture (85:15 syn/anti) (100 mg, 0.39 mmol) in dichloromethane (10 mL) was treated with trimethyloxonium tetrafluoroborate (90 mg (0.60 mmol). The solution was stirred at room temperature for 8 h and diluted with 7% sodium hydroxide solution (aqueous, 8 mL). After 20 min of stirring, the separated organic layer was dried and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 80:1 hexanes−ethyl acetate) to give a mixture of diastereomers as a colorless oil in an 85:15 syn/anti ratio: yield = 52 mg (0.35 mmol) = 90%; 1H NMR (300 MHz, CDCl3) δ 0.9 − 1.5 (m, 7H), 1.82 (d, J = 1.8 Hz, 0.15H), 1.86 (d, J = 1.9 Hz, 0.85H), 2.17 (m, 2H), 3.25 (d, J = 4.6 Hz, 0.85H), 3.40 (d, J = 2.3 Hz, 0.15H), 3.50 (dd, J = 1.8, 2.3 Hz, 0.15H), 3.61 (dd, J = 1.9, 4.6 Hz, 0.85H); 13C NMR (75 MHz, CDCl3) δ (14.0, 14.1), (17.2, 17.4), 21.6, (31.7, 31.8), 44.5, (48.2, 48.3), 66.7, (77.5, 77.9), (79.5, 79.7), (85.2, 85.5). Anal. Calcd for C10H12O: C, 81.04; H, 8.16. Found: C, 80.99; H, 8.19.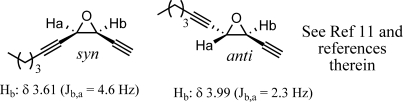
II.a.2. Formation of 2-Ethynyl-3-phenylethylyloxirane
A solution of the hydroxy sulfide mixture (85:15 syn/anti) (200 mg 0.72 mmol) in dichloromethane (20 mL) was treated with trimethyloxonium tetrafluoroborate (180 mg (1.20 mmol). The solution was stirred at room temperature for 12 h and diluted with 7% sodium hydroxide solution (aqueous, 15 mL). After 20 min of stirring, the separated organic layer was dried and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 100:1 hexanes−ethyl acetate) allowing isolation of two diastereomers as colorless oils in an 85:15 syn/anti ratio: yield = total = 110 mg (0.64 mmol) = 89%. Anti isomer: yield = 12 mg (0.07 mmol); 1H NMR (300 MHz, CDCl3) δ 2.44 (d, J = 1.8 Hz, 1H), 3.51 (dd, J = 1.8, 2.4 Hz, 1H), 3.97 (d, J = 2.4 Hz, 1H), 7.15−7.25 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 48.2, 53.1, 66.2, 80.3, 87.3, 91.7, 123.5, 128.4 (2), 129.0, 133.1 (2). Anal. Calcd for C12H8O = C, 85.69 H, 4.79. Found: C, 86.00 H, 4.71. Syn isomer: yield = 98 mg (0.57 mmol); 1H NMR (300 MHz, CDCl3) δ 2.43 (d, J = 1.8 Hz, 1H), 3.46 (dd, J = 1.8, 4.7 Hz, 1H), 4.15 (d, J = 4.7 Hz, 1H), 7.15−7.23 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 48.7, 53.1, 66.5, 80.1, 87.3, 92.0, 123.0, 128.3 (2), 128.7, 132.3(2).
II.a.3. Formation of 2-Ethynyl-3-(trimethylsilyl)ethylyloxirane
A solution of the hydroxy sulfide mixture (70:30 syn/anti) (200 mg 0.73 mmol) in dichloromethane (20 mL) was treated with trimethyloxonium tetrafluoroborate (180 mg (1.20 mmol). The solution was stirred at room temperature for 12 h and diluted with 7% sodium hydroxide solution (aqueous, 15 mL). After 20 min of stirring, the separated organic layer was dried and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 100:1 hexanes−ethyl acetate) allowing isolation of two diastereomers as colorless oils in an 70:30 syn/anti ratio: yield = total = 108 mg (0.66 mmol) = 90%; Anti isomer: yield = 32 mg (0.30 mmol); 1H NMR (300 MHz, CDCl3) δ 0.13 (s, 9H); 2.21 (d, J = 2.0 Hz, 1H), 3.25 (dd, J = 2.0, 2.3 Hz, 1H), 3.55 (d, J = 2.3 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 0.10 (3), 44.9, 49.9, 69.9, 81.7, 86.0, 102.0; MS m/z (M+) calcd = 164.0657, obsd = 164.0656. Syn isomer: yield = 76 mg (0.46 mmol); 1H NMR (300 MHz, CDCl3) δ 0.13 (s, 9H), 2.19 (d, J = 2.0 Hz, 1H), 3.32 (dd, J = 2.0, 4.9 Hz, 1H), 3.63 (d, J = 4.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 0.10 (3), 45.1, 49.3, 70.1, 81.5, 85.6, 101.2.
II.a.4. Formation of 2-Ethynyl-3-(tert-butyldimethylpropoxysilyl)ethylyloxirane
A solution of the hydroxy sulfide mixture (90:10 syn/anti) (250 mg 0.67 mmol) in dichloromethane (20 mL) was treated with trimethyloxonium tetrafluoroborate (150 mg (1.00 mmol). The solution was stirred at room temperature for 12 h and diluted with 7% sodium hydroxide solution (aqueous, 12 mL). After 20 min of stirring, the separated organic layer was dried and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 100:1 hexanes−ethyl acetate) to give a mixture of two diastereomers as a colorless oil in a 90:10 syn/anti ratio: yield = 148 mg (0.56 mmol) = 83%; 1H NMR (300 MHz, CDCl3) δ 0.22 (s, 6H); 0.97 (s, 9H), 1.27 (m, 2H), 1.8−2.0 (m, 3H), 3.23 (d, J = 4.4 Hz, 0.9H), 3.31 (d, J = 2.5 Hz, 0.1H), 3.53 (dd, J = 1.9, 2.5 Hz, 0.1 Hz), 3.69 (dd, J = 1.9, 4.4 Hz, 0.9H), 3.83 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 0.28 (2), 14.5, 14.7, 21.8 (2), (22.0, 22.3), (33.9, 34.2), (43.8, 44.0), (47.1, 47.9), (64.7, 65.0), 67.3, (78.7, 79.1), 81.8, (85.4, 85.8); MS m/z (M+) calcd = 264.1546, obsd = 264.1545. Anal. Calcd for C15H24OSi: C, 68.13; H, 9.15. Found: C, 67.79; H, 9.4.
II.b. General Procedure Using TlOEt. Formation of 2-Ethynyl-3-phenylethylyloxirane
II.b.1. Formation of 2-Ethynyl-3-phenylethylyloxirane
A solution of the hydroxy sulfide mixture (85:15 syn/anti) (200 mg 0.72 mmol) in chloroform (20 mL) was treated with TlOEt (234 mg, 0.93 mmol). The solution was stirred at room temperature for 15 h and diluted with ether. After 20 min of stirring, the insolubles were removed by filtration through a short Celite pad. The resulting organic solution was washed with a saturated NaHCO3 solution (aq) and a saturated NaCl solution (aq). The organic layer was dried and concentrated. Purification and analysis of products proceeded in the manner previously described (section II.a).
Subsequent hydroxy sulfides were treated in a fashion similar to that in II.b.1 and analyzed in the manner previously described in section II.a.
III. Formation of Enediyne Compounds from Hydroxy Sulfides
III.a. General Procedure: Acid Catalyzed Conditions
III.a.1. Formation of 6-Butylhex-3-ene-1,5-diyne-3-phenyl Sulfide
A solution of the hydroxy sulfide mixture (25:75 syn/anti) (100 mg, 0.39 mmol) in a 9:1 THF/H2O mixture (10 mL) was treated with a catalytic amount of p-toluenesulfonic acid . The solution was stirred at room temperature for 8−12 h and diluted with 10% sodium bicarbonate solution (aqueous, 10 mL). After 5 min of stirring, 20 mL of dichloromethane was added to the solution with stirring for 10 min. The separated aqueous layer was extracted with 3 × 10 mL of dichloromethane. The organic layers were combined, washed with 3 × 10 mL of water, dried over MgSO4, and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 25:1 hexanes−ethyl acetate) to give a mixture of diastereomers as a colorless oil in a 73:27 cis/trans ratio: yield = 82 mg (0.34 mmol) = 87%; 1H NMR (300 MHz, CDCl3) δ 0.90−1.51 (m, 7H), 2.07 (m, 2H), 2.63 (s, 0.27H), 2.71 (s, 0.73H), 5.90 (s, 0.73H), 6.21 (s, 0.27H) 7.10−7.25 (m, 5H); 13C NMR (75 MHz, CDCl3) δ (13.9, 14.1), 17.1, 22.9, 31.6, 77.2, (79.5, 80.3), (83.1, 83.7), (90.5, 91.7), (113.3, 113.9), 125.1, 128.4(2), 129.3(2), 134.0, (145.9, 146.2); MS m/z (M+) calcd 240.0973, obsd 240.0973. Anal. Calcd for C16H16S: C, 79.95; H, 6.71. Found: C, 80.09; H, 6.60.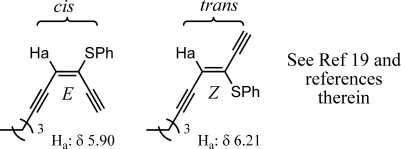
III.a.2. Formation of 6-Phenylhex-3-ene-1,5-diyne-3-phenyl Sulfide
III.a.2.a. Anti-Isomer Only
A solution of the hydroxy sulfide (200 mg 0.72 mmol) in a 9:1 THF/H2O mixture (20 mL) was treated with a catalytic amount of p-toluenesulfonic acid. The solution was stirred at room temperature for 10−15 h and diluted with 10% sodium bicarbonate solution (aqueous, 20 mL). After 5 min of stirring, 30 mL of dichloromethane was added to the solution with stirring for 10 min. The separated aqueous layer was extracted with 3 × 10 mL of dichloromethane. The organic layers were combined, washed with 3 × 10 mL of water, dried over MgSO4, and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 25:1 hexanes−ethyl acetate) to give the product as a colorless oil in a 97:3 E/Z ratio (the minor isomer was detected by GC−MS; the minor isomer was not seen by NMR): ield = 156 mg (0.60 mmol) = 83%; 1H NMR (300 MHz, CDCl3) δ 0.2.95 (s, 1H), 6.01 (s, 1H), 7.07−7.30 (m, 10H): 13C NMR (75 MHz, CDCl3) δ 81.7, 82.9, 89.3, 92.6, 114.0, 122.0, 125.7, 128.1 (2), 128.3, 128.8 (2), 129.5 (2), 133.6 (2), 134.0, 144.6. Anal. Calcd for C18H12S: C, 83.04; H, 4.65. Found: C, 82.91; H, 4.80.
III.a.2.b. Mixture of Isomers
A solution of the hydroxy sulfide mixture (20:80 syn/anti) (200 mg, 0.72 mmol) in a 9:1 THF/H2O mixture (20 mL) was treated with a catalytic amount of p-toluenesulfonic acid. The solution was stirred at room temperature for 10−15 h and diluted with 10% sodium bicarbonate solution (aqueous, 20 mL). After 5 min of stirring, 30 mL of dichloromethane was added to the solution with stirring for 10 min. The separated aqueous layer was extracted with 3 × 10 mL of dichloromethane. The organic layers were combined, washed with 3 × 10 mL of water, dried over MgSO4, and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 25:1 hexanes−ethyl acetate) to give a mixture of diastereomers as a colorless oil in a 77:23 E/Z ratio: yield = 160 mg (0.61 mmol) = 85%; 1H NMR (300 MHz, CDCl3) δ 0.2.95 (s, 0.77H), 3.01 (s, 0.23H), 6.04 (s, 0.77H), 6.35 (s, 0.27H), 7.07−7.30 (m, 10H): 13C NMR (75 MHz, CDCl3) δ (81.5, 81.7), 83.0, 89.3, (92.6, 93.0), (114.0, 119.2), 122.0, 125.7, 128.1 (2), (128.3, 128.5), 128.8 (2), 129.5 (2), 133.6 (2), (134.0, 134.5), (144.6, 145.1).
III.a.3. Formation of 6-Trimethylsilylhex-3-ene-1,5-diyne-3-phenyl Sulfide
III.a.3.a. Anti-Isomer Only
A solution of the hydroxy sulfide (200 mg 0.73 mmol) in a 9:1 THF/H2O mixture (20 mL) was treated with a catalytic amount of p-toluenesulfonic acid. The solution was stirred at room temperature for 10−15 h and diluted with 10% sodium bicarbonate solution (aqueous, 20 mL). After 5 min of stirring, 30 mL of dichloromethane was added to the solution with stirring for 10 min. The separated aqueous layer was extracted with 3 × 10 mL of dichloromethane. The organic layers were combined, washed with 3 × 10 mL of water, dried over MgSO4, and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 15:1 hexanes−ethyl acetate) to give the product as a colorless oil in a 97:3 E/Z ratio (the minor isomer was detected by GC−MS; the minor isomer was not seen by NMR): yield = 123 mg (0.0.48 mmol) = 65%; 1H NMR (300 MHz, CDCl3) δ 0.0.11 (s, 9H), 2.87 (s, 1H), 6.20 (s, 1H), 7.10−7.29 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 0.09 (3), 78.7, 81.2, 95.3, 100.9, 112.5, 124.8, 127.2(2), 128.7(2), 131.8, 142.1. Anal. Calcd for C15H16SSi: C, 70.25; H, 6.29. Found: C, 69.99; H, 6.43.
III.a.3.b. Mixture of Isomers
A solution of the hydroxy sulfide mixture (40:60 syn/anti) (200 mg 0.73 mmol) in a 9:1 THF/H2O mixture (20 mL) was treated with a catalytic amount of p-toluenesulfonic acid. The solution was stirred at room temperature for 10−15 h and diluted with 10% sodium bicarbonate solution (aqueous, 20 mL). After 5 min of stirring, 30 mL of dichloromethane was added to the solution with stirring for 10 min. The separated aqueous layer was extracted with 3 × 10 mL of dichloromethane. The organic layers were combined, washed with 3 × 10 mL of water, dried over MgSO4, and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 15:1 hexanes−ethyl acetate) to give a mixture of diastereomers as a colorless oil in a 50:50 E/Z ratio: yield = 120 mg (0.48 mmol) = 65%; 1H NMR (300 MHz, CDCl3) δ 0.09 (s, 4.5H) 0.10 (s, 4.5H) 2.81 (s, 0.5H), 2.87 (s, 0.5H), 5.77 (s, 0.5H), 6.20 (s, 0.5H), 7.10−7.29 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 0.09 (3), (78.7, 79.1), (81.2, 81.8), (95.3, 95.9), 100.9, (112.5, 116.1), 124.8, 127.2(2), 128.7(2), (131.8, 132.9), (142.1, 143.5).
III.a.4. Formation of 6-(tert-Butyldimethylpropoxysilyl)hex-3-ene-1,5-diyne-3-phenyl Sulfide
A solution of the hydroxy sulfide mixture (15:85 syn/anti) (250 mg 0.67 mmol) in a 9:1 THF/H2O mixture (10 mL) was treated with a catalytic amount of p-toluenesulfonic acid. The solution was stirred at room temperature for 8−12 h and diluted with 10% sodium bicarbonate solution (aqueous, 10 mL). After 5 min of stirring, 20 mL of dichloromethane was added to the solution with stirring for 10 min. The separated aqueous layer was extracted with 3 × 10 mL of dichloromethane. The organic layers were combined, washed with 3 × 10 mL of water, dried over MgSO4, and concentrated. Purification was accomplished by radial chromatography on silica gel (elution with 10:1 hexanes−ethyl acetate) to give a mixture of diastereomers as a colorless oil in an 82:18 cis/trans ratio: yield = 192 mg (0.54 mmol) = 80%; 1H NMR (300 MHz, CDCl3) δ 0.15 (s, 6H), 0.93 (s, 9H), 1.27 (m, 2H), 2.11(m, 2H), 2.71 (s, 0.82H), 2.80 (s, 0.18H), 3.75 (m, 2H), 5.93 (s, 0.82H), 6.16 (s, 0.18H), 7.07−7.26 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 0.13(2), (13.9, 14.1), (15.0, 15.1), 21.3(3), 33.9, (64.9, 65.3), (77.3, 78.1), (79.7, 80.1), (83.2, 84.1), (93.1, 93.7), (114.1, 115.0), 125.4, 127.7(2), 129.1, 129.3, (133.7, 134.0), (144.9, 145.4); MS m/z (M+) calcd 356.1630, obsd 355.9891. Anal. Calcd for C21H28OSSi: C, 70.73; H, 7.91. Found: C, 70.81; H, 8.01.
Acknowledgments
Financial support received from an NSF-REU grant, Pfizer Undergraduate Research Award Grant, the Howard Hughes Medical Institute, and the Trinity College Internal Grant Foundation is greatly acknowledged.
Supporting Information Available
1H and 13C NMR spectral data are available for each hydroxy sulfide (2), epoxydiyne (5), and ene diyne (6). Spectral data for each isomer are shown where separation of the isomers was possible. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- For reviews and examples, see: ; a Auge J.; Lubin-Germain N.; Uziel J. Synthesis 2007, 1739. [Google Scholar]; b Li C.-J. Chem. Rev. 2005, 105, 3095. [DOI] [PubMed] [Google Scholar]; c Araki S.; Hirashita T. In Main Group Metals in Organic Synthesis; Yamamoto H., Oshima K., Eds.; Wiley-VCH: Weinheim, 2004; Vol. 1, pp 323−386. [Google Scholar]; d Paquette L. A. In Green Chemistry: Frontiers in Benign Chemical Synthesis and Processing; Anastas P., Williamson T., Eds.; Oxford University Press: New York, 1998. [Google Scholar]; e Li C. J.; Chan T. H. Organic Reactions in Aqueous Media: John Wiley & Sons: New York, 1997. [Google Scholar]
- a Girotti R.; Marrocchi A.; Minuti L.; Piermati O.; Pizzo F.; Vaccaro L J. Org. Chem. 2006, 71, 70. [DOI] [PubMed] [Google Scholar]; b Qian J.; Timko M. T.; Allen A. J.; Russell C. J.; Winnik B.; Buckley B.; Steinfeld J. I.; Tester J. W. J. Am. Chem. Soc. 2004, 126, 5465. [DOI] [PubMed] [Google Scholar]; c Meijer A.; Otto S.; Engberts J. B. F. N. J. Org. Chem. 1998, 63, 8989–8994. [Google Scholar]; d Rideout D. C.; Breslow R. J. Am. Chem. Soc. 1980, 102, 7816. [Google Scholar]; e Grieco P. A.; Garner P.; He Z. Tetrahedron Lett. 1983, 24, 1897. [Google Scholar]
- a Shen Z.-L.; Yeo Y.-L.; Loh T.-P.. J. Org. Chem., 2008, 73, 3922 and references cited therein. [DOI] [PubMed] [Google Scholar]; b Araki S.; Ito H.; Butsugan Y.. J. Org. Chem. 1988, 53, 1831. [Google Scholar]; c Li C. J.; Chan T. H. Tetrahedron Lett. 1991, 113, 6674. [Google Scholar]; d Blomberg C.; Hartog F. A. Synthesis 1977, 18. [Google Scholar]
- Recent indium-promoted barbier reaction of conjugated systems: ; a Lin M. J.; Loh T. P. J. Am. Chem. Soc. 2003, 125, 13042. [DOI] [PubMed] [Google Scholar]; b Yi X. H.; Meng Y.; Hua X. G.; Li C. J. J. Org. Chem. 1998, 63, 7472. [DOI] [PubMed] [Google Scholar]; c Isaac M. B.; Chan T. H. Chem. Commun. 1995, 1003. [Google Scholar]; d Chan T. H.; Yang Y. J. Am. Chem. Soc. 1999, 121, 3228. [Google Scholar]
- a Mitzel T. M.; Wzorek J.; Troutman A.; Allegue K. J. Org. Chem. 2007, 72, 3042. [DOI] [PubMed] [Google Scholar]; b Mitzel T. M.; Palomo C.; Jendza K. J. Org. Chem. 2002, 67, 136. [DOI] [PubMed] [Google Scholar]; c Paquette L. A.; Mitzel T. M.. J. Am. Chem. Soc. 1996, 118, 1931 and references therein. [Google Scholar]
- For reviews and examples, see: ; a Evans D. A.; Golob A. M. J. Am. Chem. Soc. 1975, 97, 4765. [Google Scholar]; b Clayden J.; Greeves N.; Warren S.; Wothers P.. Organic Chemistry; Oxford University Press: New York, 2001; pp 947−951. [Google Scholar]; c Gagosz F. Org. Lett. 2005, 7, 4129. [DOI] [PubMed] [Google Scholar]; d Miao W.; Chung W. C.; Wu Y.-D.; Chan T. H. J. Am. Chem. Soc. 2004, 125, 13326. [DOI] [PubMed] [Google Scholar]; e White B. H.; Snapper M. L. J. Am. Chem. Soc. 2003, 135, 14901–14904. [DOI] [PubMed] [Google Scholar]; f Santora V. J.; Moore H. W. J. Org. Chem. 1996, 61, 7976. [DOI] [PubMed] [Google Scholar]; g Viola A.; MacMillan J. H. J. Am. Chem. Soc. 1970, 92, 2404. [Google Scholar]
- a Leader G. R.; Gormley J. F. J. Am. Chem. Soc. 1951, 73, 5731. [Google Scholar]; b Bass S. J.; Nathan W. I.; Meighan R. M.; Cole R. H. J. Phys. Chem. 1964, 68, 509. [Google Scholar]; c Mitzel T. M.Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons, Ltd: New York, 2006; www.mrw.interscience.wiley.com/eros/ (accessed March 2009). [Google Scholar]; d Fawcett W. R. J. Phys. Chem. 1993, 97, 9540. [Google Scholar]
- A few representative examples of NMF use as a solvent: ; a Camerel F.; Gabriel J-C. P.; Batail P.; Davidson P.; Lemaire B.; Schnutz M.; Gulik-Krzywicki T.; Bourgaux C. Nano Lett. 2002, 2, 403. [Google Scholar]; b Chassin C.; Schmidt E. A.; Hoffmann H. M. R. J. Am. Chem. Soc. 1974, 96, 606. [Google Scholar]; c Berger S. K.; Powell C. R.; Henning B. D.; Furman G. S.; Loffredo W. M.; Rydberg E. M.; Neubert R. A.; Shoop C. E.; Blauch D. N. J. Phys. Chem. 1981, 85, 1236. [Google Scholar]
- Epoxy diynes: ; a Baker J. R.; Britton T. H.; Caddick S. Org. Lett. 2007, 9, 45. [DOI] [PubMed] [Google Scholar]; b Myers A. G.; Glatthar R.; Hamond M.; Harrington P. M.; Kuo Y.; Liang J.; Schaus S. E.; Wu Y.; Xiang J.-N. J. Am. Chem, Soc. 2002, 124, 5380. [DOI] [PubMed] [Google Scholar]; c Kobayashi S.; Reddy R. S.; Sugiura Y.; Sasaki D.; Miyagawa N.; Hirama M. J. Am. Chem. Soc. 2001, 123, 2887. [DOI] [PubMed] [Google Scholar]; d Ando T.; Ishii M.; Kajiura T.; Kameyama T.; Miwa K.. 38th Symposium on the Chemistry of Natural Products, Japan, Symposium Papers, 1996, p 487. Enediynes: ; e Johnson H. D.; Thorson J. S. J. Am. Chem. Soc. 2008, 130, 17662. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Van Lanen S. G.; Shen B. Curr. Top. Med. Chem. 2008, 8, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]; g DeVoss J. J.; Townsend C. A.; Ding W.0D.; Morton G. O.; Ellestad G. A.; Zein N.; Tabor A. B.; Schreiber S. L. J. Am. Chem. Soc. 1990, 112, 9669. [Google Scholar]; h Zein N.; Sinha A. M.; McGahren W. J.; Ellestad G. A. Science 1988, 240, 1198. [DOI] [PubMed] [Google Scholar]
- a Das B.; Damodar K.; Shashikaath B.; Srinivas Y.; Kalavalhi I. Synlett 2008, 3133. [Google Scholar]; b Engstrom G.; Morelli M.; Mitzel T. M. Tetrahedron Lett. 1999, 40, 5967. [Google Scholar]
- a Paquette L. A.; Mitzel T. M.; Isaac M. B.; Crasto C. F.; Schomer W. M. J. Org. Chem. 1997, 62, 4293. [DOI] [PubMed] [Google Scholar]; b Sata T.; Otera J.; Nozaki H. J. Org. Chem. 1990, 55, 6116. [Google Scholar]; c Shimagaki M.; Maeda T.; Matsuzaki Y.; Hori I.; Nakata T.; Oishi T. Tetrahedron Lett. 1984, 25, 4775. [Google Scholar]
- Chemla F.; Bernard N.; Normant J. F. Tetrahedron Lett. 1999, 40, 75. [Google Scholar]
- Examples: Normant J. F.Tetrahedron, 1991, 47, 1677 and ref. therein. [Google Scholar]
- Cao G.-A.; Wang Z. X.; Tu Y.; Shi Y.. Tetrahedron Lett. 1998, 39, 4425 and ref. therein. [Google Scholar]
- a Kanger T.; Piret J.; Müürisep A. M.; Pek T.; Lopp M. Tetrahedron: Asymmetry 1998, 9, 2499. [Google Scholar]; b Wu S. H.; Huang B. Z.; Gao X. Synth. Commun. 1990, 20, 1279. [Google Scholar]; c Bohlmann F.; Burkhardt T.; Zdero C.. Naturally Occurring Acetylenes; Academic Press: New York, 1973. [Google Scholar]
- a Kwon J. S.; Pae A. N.; Choi K. I.; Koh H. Y.; Kim Y.; Cho Y. S. Tetrahedron Lett. 2001, 42, 1957. [Google Scholar]; b Kirihara M.; Takuwa T.; Takizawa S.; Takefumi M.; Nemoto H. Tetrahedron 2000, 56, 8275. [Google Scholar]; c Cho Y. S.; Lee J. E.; Pae A. N.; Choi K. I.; Koh H. Y. Tetrahedron Lett. 1999, 40, 1725. [Google Scholar]; d Isaac M. B.; Chan T.-H. Tetrahedron Lett. 1995, 36, 8957. [Google Scholar]
- Sato T.; Otera J.; Nozaki H. J. Org. Chem. 1990, 55, 6116. [Google Scholar]
- a Pirkle W. H.; Rinaldi P. L. J. Org. Chem. 1978, 43, 3803. [Google Scholar]; b Sato T.; Otera J.; Nozaki H. J. Org. Chem. 1990, 55, 6116. [Google Scholar]
- NMR shifts in cis- vs trans-enol ethers: ; a Giessert A. J.; Brazis N. J.; Diver S. T. Org. Lett. 2003, 5, 3819. [DOI] [PubMed] [Google Scholar]; b Choi J.; Imai E.; Mihara M.; Oderaotoshi Y.; Minakata S; Komatsu M. J. Org. Chem. 2003, 68, 6164. [DOI] [PubMed] [Google Scholar]; c Trost B. M.; Lavoie A. J. Am. Chem. Soc. 1983, 105, 5075. [Google Scholar]; d House H. O.; Czuba L. J.; Gall M.; Olmstead H. D.. J. Org. Chem. 1969, 34, 2324 and references therein. [Google Scholar]
- Examples of formation of propargyl aldehydes: ; a Journet M.; Cai D.; DiMichele L. M.; Larsen R. D. Tetrahedron Lett. 1998, 39, 6427. [Google Scholar]; b Jackson M. M.; Leverett C.; Toczko J. F.; Roberts J. C. J. Org. Chem. 2002, 67, 5032. [DOI] [PubMed] [Google Scholar]; c Dixon D. J.; Ley S. V.; Tate E. W. Synlett 1998, 1093. [Google Scholar]; d Trost B. M.; Schmidt T. J. Am. Chem. Soc. 1988, 110, 2301. [Google Scholar]; e Molander G. A.; McWilliams J. C.; Noll B. C. J. Am. Chem. Soc. 1997, 119, 1265. [Google Scholar]; f Nahm S.; Weinreb S. M. Tetrahedron Lett. 1981, 22, 3815. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.