

Abstract
The host proteome response and molecular mechanisms that drive disease in vivo during infection by a human isolate of the highly pathogenic avian influenza virus (HPAI) and 1918 pandemic influenza virus remain poorly understood. This study presents a comprehensive characterization of the proteome response in cynomolgus macaque (Macaca fascicularis) lung tissue over 7 days of infection with HPAI (the most virulent), a reassortant virus containing 1918 hemagglutinin and neuraminidase surface proteins (intermediate virulence), or a human seasonal strain (least virulent). A high-sensitivity two-dimensional liquid chromatography-tandem mass spectroscopy strategy and functional network analysis were implemented to gain insight into response pathways activated in macaques during influenza virus infection. A macaque protein database was assembled and used in the identification of 35,239 unique peptide sequences corresponding to approximately 4,259 proteins. Quantitative analysis identified an increase in expression of 400 proteins during viral infection. The abundance levels of a subset of these 400 proteins produced strong correlations with disease progression observed in the macaques, distinguishing a “core” response to viral infection from a “high” response specific to severe disease. Proteome expression profiles revealed distinct temporal response kinetics between viral strains, with HPAI inducing the most rapid response. While proteins involved in the immune response, metabolism, and transport were increased rapidly in the lung by HPAI, the other viruses produced a delayed response, characterized by an increase in proteins involved in oxidative phosphorylation, RNA processing, and translation. Proteomic results were integrated with previous genomic and pathological analysis to characterize the dynamic nature of the influenza virus infection process.
The 1918 H1N1 Spanish flu was the most devastating influenza pandemic in recent history, responsible for the deaths of an estimated 50 to 100 million individuals (38). Three other pandemics since then, the 1957 H2N2 Asian flu, 1968 H3N2 Hong Kong flu, and the 2009 H1N1 swine flu, in addition to the highly pathogenic avian H5N1 strains currently circulating in Asia with a reported case fatality rate of >60% (54), highlight the fact that influenza virus remains a significant human pathogen and a potential cause of the next devastating pandemic.
Clearly, the host, the virus, and environmental factors determine the outcome of viral infection (44). Influenza virus virulence factors, such as HA and NA, typically function through interactions with host proteins, producing a response within the host. These viral-host interactions are commonly species specific and do provide a formidable barrier to interspecies viral spread (36, 40). While rodent model systems have been developed to study influenza virus infection (8), genetic and physiological differences compared to humans argue for a more physiologically relevant model (9). In light of this, several macaque species, including Macaca mulatta, Macaca nemestrina, and Macaca fascicularis, currently serve as physiologically relevant model systems for studying pathogenesis induced by human viruses, encompassing agents such as HIV, Ebola, variola major, and influenza A viruses (1, 12). Additionally, the recent completion of the rhesus macaque sequencing project (14) and continuing sequencing efforts for other macaque species now make this primate a particularly amenable system for in-depth global genetic and proteome-based studies.
To date, a limited number of proteomic analyses have been brought to bear against influenza virus-host models. Previous work studying the host proteome response to influenza virus infection has primarily targeted in vitro systems, specifically, human cell lines (30, 35, 52). Vester et al. and Liu et al. both utilized global two-dimensional PAGE (2D-PAGE) separation approaches and reported 16 and 22 differentially abundant proteins, respectively. A more targeted analysis was performed by Mayer et al. (35), who used coimmunoprecipitation studies to identify key cellular factors that copurified with either native viral nucleoprotein or polymerase complexes. Additionally, previous analysis in our laboratory evaluated the global proteome response in macaque (M. nemestrina) lung tissue to a low-pathogenicity influenza virus, A/Texas/36/91 (Tx91) (1). Using a liquid chromatography-tandem mass spectroscopy (LC-MS/MS)-based approach and searching the spectral data against a human protein collection list, 3,548 proteins were identified, the first such proteome investigation into the host-virus response within the macaque model.
Although initially informative, the previous proteome study (1) was limited in many aspects, predominately due to the lack of a robust macaque protein sequence database and with analysis confined to only a single time point for a single viral evaluation. Now with a complete macaque protein sequence for LC-MS/MS data set searching, we have applied similar sensitive proteome analyses toward a more comprehensive survey of the macaque lung tissue in the context of exposure to and infection by human influenza virus. In the full exposure study (3), 32 macaques were infected with the highly pathogenic avian influenza virus H5N1 (HPAI), two Spanish flu 1918 H1N1 reconstruction variants of intermediate virulence, or a relatively mild disease-inducing seasonal human H1N1 virus (Tx91). Macaques were sacrificed 1, 2, 4, and 7 days postinfection (p.i.), and lung tissue was collected to evaluate viral titers, histopathology, transcriptomics, and proteomics. Viral titers and disease pathology were previously described (3), but briefly, unlike the other three viral strains, infectious HPAI viruses were recovered from macaque lung tissue through all time points of the study. While HPAI was clearly the most virulent, causing extensive pulmonary damage and death in one animal between days 6 and 7 p.i., the 1918 reassortant had only slightly greater virulence than the Tx91 virus. The aim of the present study was to integrate and extend observations reported by Baas et al. (1) and Baskin et al. (3) by providing a more comprehensive proteome characterization of the pulmonary response to influenza virus infection in the macaque animal model, by focusing on a subset of viruses and time points that would be most informative. A portion of the response proteins produced distinct expression profiles that distinguished a healthy or basal level for a general core response to influenza virus infection from that of a highly pathogenic infection. Our analysis identified 96 proteins that both increased during infection and showed a strong correlation (r ≥ 0.80) between protein abundance and disease progression.
MATERIALS AND METHODS
Macaque viral infections.
Macaque lung tissue samples were utilized from a previously described study (3). Briefly, 34 macaques matched for age, weight, and sex were inoculated by intratracheal, intranasal, tonsillar, and conjunctival routes with a total of 107 PFU of either A/Vietnam/1203/2004 (HPAI) virus, Tx91 virus or reassortants of Tx91 containing either two genes (HA and NA; here designated 1918/Tx) or three genes [HA, NA, and NS; here designated 1918/Tx (3:5)] from influenza A/South Carolina/1/1918 virus in the Tx91 backbone. Two animals per group were scheduled for sacrifice on days 1, 2, 4, and 7 p.i. Two additional animals were used as uninfected control animals and euthanized on day 7. At necropsy, the lungs were examined and sampled by snap-freezing for either viral titer or proteome analysis. As previously published, a score of pathology was assigned to the lung lobe based on a scale from 0, reflecting no apparent change, to 6, the most severe alteration (3). One macaque infected with HPAI succumbed to infection on day 6 p.i. due to extensive pulmonary damage, and the tissues from this animal were not used in the analysis. Day 7 data in this instance were from two lung specimens from the surviving animal.
Sample preparation.
In the current study, samples from only HPAI, 1918/Tx, or Tx91 virus infections on days 2 and 7 p.i. were processed for proteome analysis. For processing, lung tissue was thawed on ice, and tissues were cut into small pieces and homogenized in 5 ml of 25 mM ammonium bicarbonate buffer with several short (15-s) bursts for approximately 1 min using a rotor stator tissue homogenizer (Omni International, Kennesaw, GA) with soft tissue homogenizer tips. The homogenate was then centrifuged at 5,000 × g for 10 min at 4°C. The supernatant was transferred to a clean tube, and 125 μl of 200 mM tributyl phosphine was added. Solid urea was added to give a final concentration of 8 M (3.74 g/5.125 ml of liquid). The samples were rocked at room temperature for 1 h and then snap-frozen in a dry ice-ethanol bath and stored at −80°C.
A bicinchoninic acid assay (Pierce Biotechnology, Inc., Rockford, IL) was performed to determine protein concentrations. Samples were further reduced with 10 mM dithiothreitol (DTT) for 1 h at 37°C and then diluted 10-fold with 50 mM NH4HCO3, pH 7.8. Trypsin digestion was performed with sequencing-grade modified porcine trypsin prepared according to the manufacturer's instructions (Promega, Madison, WI). Trypsin was added to all protein samples at a 1:50 (wt/wt) trypsin-to-protein ratio for 3 h at 37°C and desalted by solid-phase extraction (SPE; Discovery DSC-18; Supelco, Bellefonte, PA). Peptides were eluted from the SPE column using 80% acetonitrile with 0.1% trifluoroacetic acid and dried in a SpeedVac SC 250 Express (Thermo Savant, Holbrook, NY). Peptides were equally pooled between two animals for each condition to 300 μg and separated by strong cation-exchange chromatography (SCX) at a flow rate of 0.2 ml/min using a 200-mm by 2.1-mm, 5 μm, 300-Å polysulfoethyl A column with a 10-mm by 2.1-mm guard column (PolyLC, Inc., Columbia, MD). The details of this SCX fractionation step have previously been described in detail (21, 22). The peptides were resuspended in 900 μl of mobile phase A and separated on an Agilent 1100 system (Palo Alto, CA) equipped with a quaternary pump, degasser, diode array detector, Peltier cooled autosampler, and fraction collector (set at 4°C for all samples). A total of 20 fractions were collected from the 30-to-80 min gradient for each sample, resulting in seven sets of SCX fractions.
RPLC separation and MS/MS acquisition.
The method used in this study, with the coupling of a constant pressure (5,000-lb/in2) reversed-phase capillary liquid chromatography (RPLC) system (75-μm inside diameter, 360-μm outside diameter, 65-cm capillary; Polymicro Technologies Inc., Phoenix, AZ) and a Finnigan LTQ ion trap mass spectrometer (Thermo Scientific, Waltham, MA) using an electrospray ionization source manufactured in-house, has been previously reported (26, 32, 43). Each SCX fraction (from both influenza virus-infected and control lung tissue samples) was analyzed via capillary RPLC-MS/MS, for a total of 7 samples × 20 SCX fractions, or 140 analyses.
LC-MS/MS proteome analysis.
A macaque protein list was generated by compiling Macaca spp. protein sequences from NCBI, SwissProt, and Trembl into a single, nonredundant FASTA protein data list containing 46,344 sequences (http://omics.pnl.gov). An influenza virus protein list was assembled by compiling protein sequences from A/Vietnam/1203/2004, A/Texas/36/91, A/South Carolina/1/18, and A/Brevig_Mission/1/18 into a single, nonredundant FASTA file containing 23 sequences. The criteria selected for filtering followed methods based upon a reverse-database false-positive model (47) that when applied to the macaque data set produced ≥98% confidence for the entire protein data set. Briefly, protein identifications were retained if their identified peptides met the following criteria: (i) SEQUEST DelCN value of ≥0.10 and (ii) a SEQUEST correlation score (Xcorr) of ≥1.6 for charge state +1 and full tryptic peptides, an Xcorr of ≥2.4 for charge state 2+ and full tryptic peptide, an Xcorr of ≥4.3 for charge state 2+ and partial tryptic peptide, an Xcorr of ≥3.2 for charge state 3+ and full tryptic peptide, and an Xcorr of ≥4.7 for charge state 3+ and partial tryptic peptide. Spectral counting was used to measure the relative abundance of individual peptides within a peptide mixture eluting from an LC gradient at any particular moment. The number of times a peptide is selected for fragmentation directly correlates with the relative abundance of that protein (29, 39). Proteins had to pass minimum criteria, which were based on analyses performed for previous reports by our laboratory (21, 46) to be used in a quantitative nature. Briefly, a protein needed to have a minimum of five total peptide identifications in one of the samples, so as to eliminate the inclusion of proteins which are not detected at a sufficient frequency to warrant quantitation, and at least a 6.0-fold increase/decrease in the relative abundance measurements between any influenza virus-infected sample and control in order to be considered differentially expressed or by exclusive detection in either control or at least one of the viral infections. Spectral counts were normalized between experiments by Z-score. The Z-score is a common method of standardizing values within a vector by mean centering relative to the standard deviation (mean [μ] of 0 and standard deviation [σ] of 1). Proteins were clustered using OmniViz (BioWisdom Ltd., Cambridge, United Kingdom) using a K-means algorithm with squared Pearson correlation to measure distances between abundance profiles. Individual clusters were further clustered using a hierarchical algorithm with complete linkage method and squared Pearson correlation as the metric of similarity. Correlations between relative protein abundance and pathology score were performed using the Pearson product-moment correlation coefficient (r).
Pathway analysis.
Human homologs were identified for 393 of the 400 differentially expressed macaque proteins by using INPARANOID (4, 41) and ScalaBlast (42), comparing identified macaque proteins against the human IPI 2009 protein collection list. For pathway analysis, human IPI protein accession numbers were converted to NCBI Entrez gene identifiers by using BRM (48). Human NCBI Entrez gene identifiers corresponding to 374 of the differentially expressed macaque proteins were imported into the Ingenuity Pathways Analysis (Redwood City, CA) and DAVID (5, 20) programs. Differentially expressed proteins within significantly altered pathways were consolidated into an interconnected network.
RESULTS
The macaque protein database improves sensitivity of spectral analysis.
To compare and contrast early and late events during disease, samples from day 2 p.i. were selected as an early time point and day 7 as a late time point (Fig. 1). The HPAI and Tx91 strains were designated for the study since they were the most virulent and least virulent strains, respectively. Because the 1918/Tx (3:5) and 1918/Tx strains produced comparable, intermediate pathology, only the 1918/Tx virus was chosen for the proteomic analysis. Peptides from biological replicates were pooled and characterized using sensitive two-dimensional LC-MS/MS (Fig. 1A). Subsequent data analysis and searching against a combined macaque/virus protein database resulted in the identification of 35,239 unique peptide sequences corresponding to approximately 4,259 proteins. From this total, 37 unique viral peptides from six specific proteins were detected exclusively during HPAI or 1918/Tx infection (Fig. 1B), but not in the parental Tx91 infection or in the control macaques, correlating with the low levels of Tx91 virus replication in these animals (3). The influenza virus protein database used in the SEQUEST analysis represents a compilation of all three viruses used in the study. Conserved genomic regions between viruses resulted in 10 of the 37 observed viral peptides mapping to the same protein sequence in more than one virus. Subsets of the remaining 27 peptides provided complementary information regarding the viral origin of the protein.
FIG. 1.

Overview of cellular and viral peptide observations. Fourteen animals were inoculated with HPAI, 1918/Tx, or Tx91, or mock infected, in groups of two animals per sacrifice day, and the pulmonary proteome response was evaluated on days 2 and 7 p.i. (A) Total peptides and proteins observed and how those peptides and proteins were attributed to either the host or different viruses. A single common peptide was detected in HPAI and 1918/Tx infections. (B) Viral proteins were detected exclusively during HPAI and 1918/Tx infections. Gray indicates the absence of detection. Ctrl, control. (C) Venn diagram comparing macaque peptides identified in the current study (Brown et al.; red) and those from the previous Baas et al. study (yellow), reanalyzed with the macaque protein collection list. While the current study identified 35,202 unique cellular peptides, Baas et al. (1) identified 15,390.
From the limited number of previous proteomic investigations of influenza virus-host protein expression (1, 30, 52), the most relevant comparison of results for the current study involves the previous in-depth LC-MS/MS analysis of Baas et al. (1). To update the Baas et al. (1) data set analysis for this comparison, we reanalyzed the original spectra from the study against the macaque protein database, as the original data comparison was performed using a human protein list, which resulted in an increase in the number of peptide identifications from 14,100 to 15,322. While peptide identifications increased, a decrease in the number of proteins from 3,548 to 3,098 was observed. Approximately 66% (10,156) of the peptides identified in the Baas et al. study using the macaque protein database were also identified in the current study (Fig. 1C), corresponding to 2,287 proteins observed in both studies. The average peptide-to-protein ratio was 4.95 in the current data set, compared to 3.98 in the prior study which employed the human database, indicating the macaque protein database is a more specific protein list for analyzing macaque proteomes. These results demonstrate the advantage of obtaining more exact protein identifications by searching against a macaque, rather than a human, database.
Viruses produce distinct protein expression profiles.
Measures of relative protein abundance were evaluated by spectral counts, a commonly used approach in LC-MS/MS analyses for determining relative protein abundance differences between conditions (1, 29, 37, 39). To identify and characterize clear differences in protein abundance at early and late phases of influenza virus infection, substantially altered proteins were conservatively defined by a 6-fold or greater difference in expression in either direction relative to control or by exclusive detection in either control or at least one of the viral infections. These criteria identified 400 increased and 258 decreased proteins (see Tables S1 and S2, respectively, in the supplemental material), from the total of 4,259. Further analysis focused on the 400 proteins that increased in expression relative to controls to investigate the macaque response to influenza virus. Spectral counts were normalized across all conditions by calculating Z-score values for each protein (see Table S3 in the supplemental material), i.e., the mean was standardized to 0 while measures that were 1 standard deviation above or below the mean were standardized to +1 or −1, respectively (Fig. 2 A). The median and interquartile ranges designate the central distribution of 50% of the Z-score values for the 400 proteins in each group at day 2 or day 7. All viral strains caused greater relative abundance of these 400 proteins compared to control, but to various degrees. Tx91 infection caused only a slight increase in abundance values (median Z-scores of −0.59 and −0.17 for days 2 and 7, respectively). In contrast, the greatest response was observed in HPAI infection (median Z-scores of +0.76 and +0.41 for days 2 and 7). The greatest temporal difference was detected for 1918/Tx (median Z-scores of −0.38 and +0.33 for days 2 and 7).
FIG. 2.

Distinct patterns of increased protein abundance in different viral infections. (A) Box plots indicate the interquartile ranges of Z-scores of spectral counts and whiskers designate the minimum and maximum values for the 400 proteins that increased during viral infection. (B to H) Relative levels of abundance for the 400 proteins were binned, based on Z-score of the spectral count, and are depicted in frequency histograms. Protein abundance was quantified for day 2 p.i. (B to E) and day 7 p.i. (F to H) in the control, Tx91, 1918/Tx, and HPAI groups, respectively.
Although a shift toward greater abundance was observed for all viruses compared to control, the structure of these distributions varied across viral strains and times. Tx91 and 1918/Tx produced single peaks on day 2, with slight shifts to the right relative to control (Fig. 2B to D). In contrast, HPAI produced the greatest shift to the right (Fig. 2E). The lower portion of the distribution overlapped with those for Tx91 and 1918/Tx; however, another portion of the distribution was present at higher Z-scores, indicating the greatest level of protein abundances. The frequency histogram elucidated three possible response profiles to influenza virus infection: normal, core, and high responses. The “normal” profile of the control sample represents a basal level of expression for the 400 proteins with Z-scores less than 0. For the “core” response, Z-scores between 0 and 1.5 represent a general, but mild, response to viral infection. In contrast, the “high” response, with Z-scores greater than 1.5, is particularly interesting at the early time point because it is exclusive to HPAI virus. A temporal shift was most prevalent with 1918/Tx as a subset of the core response proteins transitioned to high responders by the late time point (Fig. 2F to H; see also Table S4 in the supplemental material). While the expression levels of the 400 proteins were not increased equally by all viruses, the relative magnitude of the increase appears to correspond to the pathogenicity of the virus.
Clustering response proteins by viral strain and temporal expression.
To characterize individual protein expression patterns, the 400 differentially expressed proteins were grouped into eight distinct bins by using K-means clustering, with experiments ordered by viral strain and time postinfection (Fig. 3; see also Table S5 in the supplemental material). As dictated by the selection method, the control displayed low Z-scores in all clusters. Clusters were color coded to facilitate multidimensional representations of temporal protein expression profiles across viral strains for subsequent analyses (Table 1). The first five bins, maroon through green, represent proteins with predominantly greater abundance during HPAI virus infection, while cyan through purple represent proteins with higher expression during 1918/Tx or Tx91 virus infection. Transiently “high” responders in HPAI produced temporal shifts from “high” on day 2 to “core” responder on day 7 in clusters that are maroon or yellow (see Fig. S1 in the supplemental material); proteins in the green cluster exhibited the opposite behavior, increasing at day 7 in the HPAI infection. In contrast, proteins in the red and orange clusters were persistently “high” responders at both time points. Cyan and blue clusters constitute proteins that were “high” responders in the late time points of 1918/Tx and Tx91, respectively. The purple cluster showed the greatest response at the early time point during Tx91 virus infection.
FIG. 3.

Heat map of Z-scores of spectral counts for the 400 increased proteins, organized into eight clusters by using the K-means algorithm. The eight clusters are color coded relative to the condition(s) in which proteins were most highly expressed. The number of proteins within a cluster is located to the left of the cluster bar.
TABLE 1.
Representative biological processes and protein members, identified using the Ingenuity Pathways program, within the eight clusters shown in Fig. 3
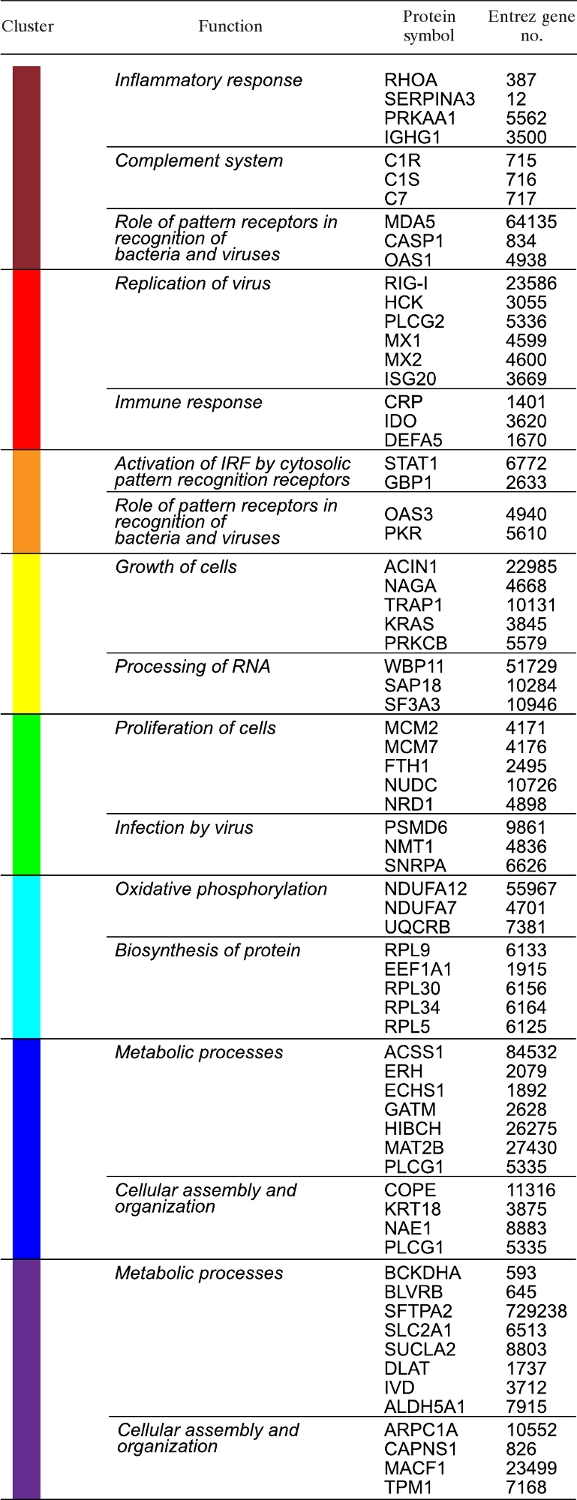 |
Clusters were functionally characterized using a ingenuity pathways analysis (Table 1). With the exception of the yellow cluster, the primary biological processes of HPAI “high” responder clusters (maroon through green) were immune response and inflammation. The yellow cluster displayed high expression levels across all viruses but with temporal differences. Proteins in the yellow cluster are involved in cell growth and fundamental processes for proliferation. While these proteins were increased by all viruses, the increase was delayed in 1918/Tx and Tx91 compared to HPAI virus.
The cyan, blue, and purple clusters represent proteins that were most highly expressed during infections where the virus was cleared within 4 days (1918/Tx and Tx91). These clusters consist of proteins that perform protein biosynthesis, numerous metabolic processes (notably oxidative phosphorylation), and those impacting cellular organization. Although HPAI did not express the proteins in the cyan, blue, and purple clusters to the relative magnitude of the other two viruses, these proteins were increased by HPAI relative to the control.
Response proteins correlate with disease progression.
To identify a subset(s) within the response proteins that correlate with disease progression and may potentially contribute to the “high” response, a linear regression was performed on the median Z-scores within individual clusters against the pathology score assigned to the macaques (Fig. 4). At the time of the necropsy, macaque lung lobes were examined and judged on several features, including inflammation and pneumocyte reaction. A pathology score between 0 and 6 was assigned, with 0 representing healthy lung tissue and 6 the most significantly altered (see Fig. S2 in the supplemental material). Comparing days 2 and 7, the pathology score was highest on day 7 for all infections. While Tx91 produced the least pulmonary damage, with both day 2 and 7 scores ranging between 1 and 2, HPAI induced the most severe pathology, resulting in pathology scores of >5 on days 2 and 7.
FIG. 4.

Linear regression of median Z-scores of spectral counts for proteins within individual clusters relative to pathology score following viral infection. Regression lines are color coded to indicate the representative cluster from Fig. 3 and as shown at the top left corner of each graph. Box plots represent interquartile ranges, and whiskers designate minimum and maximum values. The Pearson product-moment correlation coefficent values (r) are indicated on the upper right corner of each plot. The day 7 groups received the greatest pathology score during all infections. Tx91 scores were between 1 and 2, the 1918/Tx group received scores between 3 and 4, and HPAI caused the greatest damage, with scores between 5 and 6.
The highest correlation between protein abundance and disease was observed for proteins in the red (Fig. 4B) and orange (Fig. 4C) clusters (r = 0.9468 and 0.9401, respectively). A positive, yet modest, correlation was observed for proteins of the clusters in maroon (Fig. 4A), yellow (Fig. 4D), and green (Fig. 4E). In contrast, cyan (Fig. 4F), blue (Fig. 4G), and purple (Fig. 4H) clusters displayed relatively little if any correlation with disease pathology observed in macaque lung tissue, suggesting proteins in these clusters are independent of disease progression. While there are challenges in distinguishing between cause and effect in these types of studies, the direct correlation between increased abundance and disease severity could indicate that the expression of proteins in the red and orange clusters may drive the pathology observed during influenza virus infection. On an individual protein level, 96 proteins produced a correlation with disease greater than 0.80 (see Table S6 in the supplemental material). While the most highly correlated proteins included classic responders to viral infection, such as PKR (EIF2AK2; 0.98), MX1 (0.95), and RIG-I (DDX58; 0.93), proteins involved in other processes, such as cell cycle and metabolism also displayed strong correlations with disease, for example, cell cycle-involved proteins GSPT1 (0.94), PSMC2 (0.94), and RPA1 (0.90) and metabolism-involved proteins OAS2 (0.90), HK3 (0.92), and SYK (0.93). Many of these proteins were observed in our previous proteome analysis (1), including PKR, MX1, RIG-I, PSMC2, OAS2, and HK3. In considering other recent systems approaches to investigating the cellular course of influenza virus infection, we observe functional overlap in the categories of innate and adaptive immunity but quite limited overlap in the specific protein/genes identified in these studies (see Table S7 in the supplemental material) (18, 24, 27, 49, 55). The correlation with proteins involved in cell cycle and metabolism may represent infiltrating, activated lymphocytes and neutrophils in the alveolar spaces during viral infection (3, 51) and the proliferation of local pneumocytes to repair damaged tissues.
Macaque inflammatory response to influenza virus infection.
Several pathway analysis tools, including InGenuity, DAVID, and KEGG, were used to identify functional signaling pathways significantly impacted by the 400 differentially expressed proteins. Considering all clusters, the immune response pathway was recognized as significantly impacted by virus in all analyses, with variations within more specific pathways of the immune response. Increased levels of expression across all viruses were found for proteins involved in RNA-binding, host-virus interaction, and antiviral response pathways. In contrast to the RNA-binding proteins, proteins of the acute-phase response were increased exclusively by HPAI.
Differentially expressed proteins were assembled onto a previously annotated functional pathway, generating a global view of the host-pathogen immune response to influenza virus infection (Fig. 5). Proteins in the pathway were color coded according to the clusters shown in Fig. 4 to represent their expression profiles across viral strains and time points. While several members of the immune response were increased during influenza virus infection, these proteins displayed distinct kinetic profiles, such as CRP, which was specific to HPAI virus. The domination of the inflammatory pathway map by maroon, red, and green cluster proteins was specific for HPAI upregulation in the early and/or late time point in response to influenza virus infection. The expression levels of a large arsenal of RNA-binding proteins that function in recognizing foreign RNA were increased in macaque lung tissue. RIG-I and MDA5 (IFIH1) transmit signals through a hub protein on the outer mitochondrial membrane, mitochondrial antiviral signaling protein (also known as MAVS, IPS1, or VISA), resulting in the expression of inflammatory cytokines and type I interferons (IFN). Release of type I IFN produces secondary signaling, both autocrine and paracrine, resulting in the phosphorylation and dimerization of signal transducer and activator of transcription (STAT) molecules that induce the transcription of IFN-stimulated genes (ISG), including ISG15, indole 2,3-dioxygenase (IDO), IFN-induced factor 44 (IFF44), and IFN-induced protein with tetratricopeptide repeats 1, 2, and 3 (IFIT1-3). Influenza virus infection increased the expression of an important cellular inhibitor of host defense, P58ipk (DNAJC3). P58ipk is activated during influenza viral infection and regulates the host defense by inhibiting PKR (28). IFN induces the expression of antiviral RNA receptors, including interferon stimulated exonuclease gene 20 kDa (ISG20), the 2′,5′-oligoadenylate synthetase (OAS) family, and the myxovirus resistance (MX) family of proteins. Particularly interesting is the prevalent antiviral response induced during infection, which strongly correlated with disease progression (Fig. 5), despite continued viral replication by HPAI. Integration of multidimensional data sets onto the host-virus pathogen signaling network provided extensive insight into the spatial-temporal relationships between the host and different viral pathogens during infection.
FIG. 5.

Influenza virus infection induces proteins involved in several areas of the immune response. Proteins are color coded relative to the cluster to which they belong (Fig. 3). White proteins are proteins that were either not observed or not differentially expressed during infection. A large number of RNA binding proteins that function in recognizing viral nucleic acids and initiating a response were induced primarily during HPAI infection. Gray boxes indicate a functionally related class of proteins (to distinguish them from a subcellular localization compartment).
DISCUSSION
This study currently represents the most expansive and in-depth proteome analysis of any macaque disease model. Results from previous research (1) provided a foundation to systematically extend the overall scope and design of the study, allowing for direct comparisons to be made between different strains of influenza virus, with various degrees of virulence in macaques over time. Development and implementation of a macaque protein database in MS/MS search algorithms substantially enhanced proteome coverage and protein specificity. Thorough testing in our laboratory has optimized sample preparation procedures, effectively reducing sample loss and resulting in increased peptide identifications. All these developments have been integrated to facilitate the expanded coverage and identification of unique peptides and proteins, thereby furnishing a distinct, high-throughput data set for exploring the host response in this macaque model of human respiratory pathogens. While a subset of the proteome observations overlapped with the parallel transcriptomics results (3), specifically, with IFN-mediated and antivirus responsive factors, an initial survey of the proteomics and genomics results showed limited correlation with each other (see Fig. S3 in the supplemental material), demonstrating the often divergent natures of the two analysis streams and the importance of proteomic approaches for an orthogonal systems characterization. Specifically, the proteomics analysis provided a unique insight into the molecular pulmonary response to influenza virus infection, including the direct correlation between protein abundance and disease progression and the distinct molecular effects that different influenza viruses produce in the alveolar compartment.
The in vivo molecular response was characterized in the lower lung of macaques during mild, intermediate, and highly pathogenic influenza virus infections. The accessory lobe was selected for proteome analysis because the greatest pathology was observed in the lower lung for all viruses. Our proteomic approach identified several viral proteins at time points where matching plaque assays showed recoverable levels of virus from lung tissue. Although macaques successfully cleared 1918/Tx and Tx91 viral infections, overall pathology continued to progress, suggesting that the host response contributes to disease. Protein abundance differences, when organized by virus and time point, led us to distinguish “core” molecular host response profiles involved in clearing the viral pathogen from profiles, such as the “high” response, that potentially contribute to severe disease in the host and possibly death. Cluster analysis identified eight distinct groups of protein abundance profiles that showed temporal patterns specific to the infecting virus. Those clusters representing proteins with persistently high abundance levels in HPAI infection displayed a strong correlation with disease progression (Fig 4 and 5, red and orange clusters). These proteins contrast with those of greatest abundance on day 2 in HPAI samples, which displayed a positive correlation largely because of decreased protein abundance during late HPAI infection (maroon cluster). This set consists mainly of IFN-responsive, antiviral proteins, including MDA5, ISG15, OAS1, and IFIT1. It is unclear whether the decreased abundances observed during the late stages of HPAI infection represent a suppressive effect initiated by the virus, possibly through NS1-mediated IFN-signaling interference (2, 7, 11, 13), or negative feedback from an initial burst of antiviral and inflammatory processes. A number of other viruses, besides influenza virus, also target IFN signaling to evade or attenuate the host response (10, 17, 33). Further studies will compare influenza virus infection relative to other respiratory viruses to identify common and distinguishing viral signatures in the host response that may represent mechanisms that regulate pathogenicity.
For HPAI infection, it is interesting to contrast the behavior of the proteins in the green cluster to those of the maroon set. Green cluster proteins trended in an opposite manner and increased from day 2 to day 7 in the HPAI infection. Within the green cluster are several proteins that are markers of neutrophil action, specifically, defensins and reactive oxygen generators, including DEFA1, DEFA2, MPO, NCF1, and BP1. The observation of these proteins at this later stage suggests a change in the inflammatory processes: whereas the maroon cluster shows innate signaling as the earliest inflammatory driver, it appears supplanted by the neutrophil-associated processes on day 7. It is also noteworthy that the preponderance of the immunoglobulin proteins were detected in the maroon and green clusters, with significantly higher levels of these proteins for the HPAI day 2 or day 7 specimens compared to the other samples (maroon and green clusters, respectively) (see Table S8 in the supplemental material). Further, considering all the immunoglobulin proteins contained in Fig. 3, there is a very significant association for these to occur in high abundance in the HPAI samples. Comparison of the maroon and green clusters implies an evolution of this antibody response in the HPAI lung samples; for example, the presence of the mu constant region at day 2 shows the significant extent of the primary IgM antibody response in this tissue sample, or it may represent the receptor form on activated B cells (23). By day 7, this response has steeply declined and may reflect the migration of the initially activated B cells to secondary lymphoid organs, accelerated lymphocyte death, or a combination of trafficking and death. Therefore, while the proteomics data have provided novel insights into the dynamics of the antibody response in the lungs of infected animals, we are left with the paradox that the greatest response occurs in the context of highest viral replication and greatest pathology, suggesting that there is still an insufficiency to the quality or quantity of the immune response at early times in the HPAI infection.
Several RNA-binding proteins, including PKR (25), RIG-I (31), and ISG20 (6), have been previously reported to provide protective roles against viral infection. While many of these RNA-binding proteins are constitutively expressed at low levels in several cell types, these proteins can be substantially induced by IFN stimulation (15, 19), so enhanced expression appears to be an indirect or secondary effect of viral infection. P58ipk is a negative regulator of PKR kinase activity, a cytosolic serine/threonine kinase that is activated by recognition of double-stranded RNA. Influenza virus induces expression of P58ipk, and we note the significant levels of P58ipk at day 7 in the HPAI infections (green cluster). P58ipk attenuates many antiviral effects of PKR, including the translational arrest of host and viral proteins and antiviral signaling through IRF3 and NF-κB. However, P58ipk also appears to help attenuate the host inflammatory response inasmuch as knockout mouse models deficient in P58ipk show significantly greater pulmonary damage during influenza virus infection (16). Highly pathogenic viruses, such as HPAI, may be capable of inducing a devastating host response that overwhelms checkpoints and regulators, including P58ipk.
Several other antiviral, IFN-stimulated proteins, including ISG15, IFIT1/2/3, IDO, and IFI44, were also increased during HPAI infection, likely as secondary responses to IFN. It is confounding that despite the increased abundance of such a large number of antiviral, RNA-binding, and interferon-stimulated proteins, particularly during the HPAI infection, virus continued to replicate in lung tissue through day 7 p.i. Infiltrating CD8+ effector/memory cells play a prominent role in producing a state commonly referred to as “heterologous antiviral immunity” via IFN-γ production during influenza virus infection (34), which further emphasizes the aggressive host defense that HPAI virus must overcome to successfully continue to replicate. Because influenza virus has evolved a number of mechanisms to evade the innate host response (17), the reason why influenza virus can continue to replicate may be due to evasive mechanisms that attenuate host response exclusively within infected cells, while the continued, prominent antiviral and inflammatory response originates from neighboring cells and leukocyte infiltrates. Assessment of antiviral protein abundances on a cell-by-cell basis may identify key inflammatory differences between infected and noninfected cells. Alternatively, HPAI may have evolved much stronger resistance, relative to other viruses used in this study, against many host antiviral proteins that could provide a replication advantage.
HPAI infection specifically increased the expression of several secreted proteins, including CRP and complement proteins C1S, C1R, and C7. Although these proteins can be synthesized in the lungs (50), they are primarily produced in the liver (45, 53). Transcriptome analysis from the lungs of these macaques failed to detect significant differences in the RNA levels of CRP or these complement factors, suggesting HPAI induced systemic inflammation during viral infection, resulting in the transport of these exogenous immune mediators from the liver. Extrapulmonary viral replication and the inflammation-associated effects induced specifically by HPAI virus on multiple organs throughout these macaques could contribute to this systemic response (51).
Proteins in the cyan, blue, and purple clusters, while increased in the HPAI-exposed group relative to control, were expressed at higher levels in macaques that effectively cleared virus. These proteins are involved in oxidation phosphorylation, RNA processing, and metabolism. We suggest that these proteins are related to early onset of homeostasis and repair and that the expression of these proteins facilitates control of virus-induced cell death, resulting in less cell-mediated killing. Gene expression analysis demonstrated that both 1918/Tx and Tx91 viral infections induced transcripts, reflecting a strong IFN response, but lack of recoverable viral titers indicated the response was sufficient to blunt viral replication without significant immunopathology (3). Within the cyan, blue, and purple clusters, there may be proteins responsible for maintaining the balance between inhibiting viral spread and regulating host response, including tissue repair. A better understanding of how these proteins function during viral infection and why they are regulated during disease could lead to an alternative approach to ameliorating the detrimental host response to infection.
Proteins of the yellow cluster appear to follow opposite kinetics, depending on the pathogenicity of the virus. The yellow cluster includes a large number of proteins involved in growth of cells and the processing and modification of RNA as well as proteins involved in oxidative phosphorylation. Proteins related to growth of cells include transcription regulators and kinases, but also a large number of transporters associated with metabolic pathways. For the HPAI infection, proteins in the yellow cluster were upregulated on day 2 and declined by day 7. In this context it could represent a proliferative burst of immune cells that have entered the tissues, or a very early proliferation of pneumocytes following the loss of such cells by either viral cytopathic effects or cell-mediated killing affected by NK cells or macrophages. However, this proliferative response cannot be sustained to day 7, perhaps reflecting the extensive necrotic damage observed in the lung tissues at this stage in the HPAI infection. The block in the expression of these specific proteins shows an opposite temporal behavior for less-pathological viral infections, where it increases in abundance from day 2 to day 7. This increase in cell proliferation processes may be part of the same homeostatic processes that are being supported by the upregulated metabolic pathways representing the aforementioned cyan, purple, and blue clusters.
There are many interesting commonalities between the previously published gene expression analysis, pathology findings (3), and the current proteomic study. The highest level of antiviral protein expression was observed during HPAI viral infection, which is consistent with viral replication measurements. Transcript levels for several IFN-induced proteins, including IFITs, IFIs, and ISG15, were also increased early in the HPAI group. The coordinated induction of these defense pathways corresponds to pathogenic evidence of inflammation during HPAI virus infection. Inflammation was caused, in part, by preferential infection of type II pneumocytes, the most abundant cell type in respiratory epithelium. Type II pneumocytes are responsible for respiratory epithelium repair and surfactant production. Coincidentally, the proteome analysis revealed that production of several surfactant proteins, including A1 and A2, were increased by HPAI virus infection early, followed by a sharp decrease at day 7. This decrease is consistent with the reduction, by 3- to 10-fold, in gene expression observed at day 2 p.i. in the same group. The proteomic analysis failed to detect increased levels of cytokines and chemokines observed in the genomics analysis, which was likely due to the low abundance levels of these proteins relative to other proteins. Finally, in the current proteomic study we observed an interesting depression at day 2 and recovery at day 7 of production of several proteosomes and major histocompatibility complex proteins in the HPAI group which are important in processing of viral antigens to mount an adaptive immune response. The later recovery is consistent with the increased gene expression of proteins in the same family at day 2 p.i. This finding mirrors the inadequacy of the early cell-mediated immune response in the form of depletion of dendritic cells and increased margination of lymphocytes during HPAI virus infection, as detected by immunohistochemistry and flow cytometry, respectively. Clearly, not all findings were consistent between genomic, proteomic, and pathological analyses. Discrepancies which cannot always be easily explained are likely to yield critical answers regarding influenza virus pathogenesis, and future efforts focused on integrating these approaches into a molecular systems biology examination will refine our understanding of the dynamic nature of the influenza virus infection process.
Supplementary Material
Acknowledgments
We thank Kevin Coty for excellent technical assistance.
Portions of this research were performed at the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. Pacific Northwest National Laboratory is operated by Battelle Memorial Institute for the U.S. Department of Energy under contract nos. DEAC06-76RL0. Portions of this work were supported by Battelle Internal Research and Development funds and by National Institutes of Health grants R01AI46954, P01AI58113, U54AI057158, and U19AI083025 (to A.G.-S.); R24 RR16354-04, P51 RR00166-45, and R01 AI022646-20A1 (to M.G.K.); K08 AI059106-02 (to Cambridge Research Biochemicals); R03 AI075019-01 (to H.B.-O.); and RR018522 (to R.D.S.).
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the funding agencies.
Footnotes
Published ahead of print on 15 September 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Baas, T., C. R. Baskin, D. L. Diamond, A. Garcia-Sastre, H. Bielefeldt-Ohmann, T. M. Tumpey, M. J. Thomas, V. S. Carter, T. H. Teal, N. Van Hoeven, S. Proll, J. M. Jacobs, Z. R. Caldwell, M. A. Gritsenko, R. R. Hukkanen, D. G. Camp II, R. D. Smith, and M. G. Katze. 2006. Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. J. Virol. 80:10813-10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baskin, C. R., H. Bielefeldt-Ohmann, A. Garcia-Sastre, T. M. Tumpey, N. Van Hoeven, V. S. Carter, M. J. Thomas, S. Proll, A. Solorzano, R. Billharz, J. L. Fornek, S. Thomas, C. H. Chen, E. A. Clark, K. Murali-Krishna, and M. G. Katze. 2007. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J. Virol. 81:11817-11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baskin, C. R., H. Bielefeldt-Ohmann, T. M. Tumpey, P. J. Sabourin, J. P. Long, A. Garcia-Sastre, A. E. Tolnay, R. Albrecht, J. A. Pyles, P. H. Olson, L. D. Aicher, E. R. Rosenzweig, K. Murali-Krishna, E. A. Clark, M. S. Kotur, J. L. Fornek, S. Proll, R. E. Palermo, C. L. Sabourin, and M. G. Katze. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berglund, A. C., E. Sjolund, G. Ostlund, and E. L. Sonnhammer. 2008. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 36:D263-D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis, G., Jr., B. T. Sherman, D. A. Hosack, J. Yang, W. Gao, H. C. Lane, and R. A. Lempicki. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4:P3. [PubMed] [Google Scholar]
- 6.Espert, L., G. Degols, C. Gongora, D. Blondel, B. R. Williams, R. H. Silverman, and N. Mechti. 2003. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 278:16151-16158. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Sesma, A. 2007. The influenza virus NS1 protein: inhibitor of innate and adaptive immunity. Infect. Disord. Drug Targets 7:336-343. [DOI] [PubMed] [Google Scholar]
- 8.Fornek, J. L., L. Gillim-Ross, C. Santos, V. Carter, J. M. Ward, L. I. Cheng, S. Proll, M. G. Katze, and K. Subbarao. 2009. A single-amino-acid substitution in a polymerase protein of an H5N1 influenza virus is associated with systemic infection and impaired T-cell activation in mice. J. Virol. 83:11102-11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fornek, J. L., M. J. Korth, and M. G. Katze. 2007. Use of functional genomics to understand influenza-host interactions. Adv. Virus Res. 70:81-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frieman, M., M. Heise, and R. Baric. 2008. SARS coronavirus and innate immunity. Virus Res. 133:101-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Sastre, A., A. Egorov, D. Matassov, S. Brandt, D. E. Levy, J. E. Durbin, P. Palese, and T. Muster. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324-330. [DOI] [PubMed] [Google Scholar]
- 12.Gardner, M. B., and P. A. Luciw. 2008. Macaque models of human infectious disease. ILAR J. 49:220-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geiss, G. K., M. Salvatore, T. M. Tumpey, V. S. Carter, X. Wang, C. F. Basler, J. K. Taubenberger, R. E. Bumgarner, P. Palese, M. G. Katze, and A. Garcia-Sastre. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl. Acad. Sci. U. S. A. 99:10736-10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibbs, R. A., J. Rogers, M. G. Katze, R. Bumgarner, G. M. Weinstock, E. R. Mardis, K. A. Remington, R. L. Strausberg, J. C. Venter, R. K. Wilson, M. A. Batzer, C. D. Bustamante, E. E. Eichler, M. W. Hahn, R. C. Hardison, K. D. Makova, W. Miller, A. Milosavljevic, R. E. Palermo, A. Siepel, J. M. Sikela, T. Attaway, S. Bell, K. E. Bernard, C. J. Buhay, M. N. Chandrabose, M. Dao, C. Davis, K. D. Delehaunty, Y. Ding, H. H. Dinh, S. Dugan-Rocha, L. A. Fulton, R. A. Gabisi, T. T. Garner, J. Godfrey, A. C. Hawes, J. Hernandez, S. Hines, M. Holder, J. Hume, S. N. Jhangiani, V. Joshi, Z. M. Khan, E. F. Kirkness, A. Cree, R. G. Fowler, S. Lee, L. R. Lewis, Z. Li, Y. S. Liu, S. M. Moore, D. Muzny, L. V. Nazareth, D. N. Ngo, G. O. Okwuonu, G. Pai, D. Parker, H. A. Paul, C. Pfannkoch, C. S. Pohl, Y. H. Rogers, S. J. Ruiz, A. Sabo, J. Santibanez, B. W. Schneider, S. M. Smith, E. Sodergren, A. F. Svatek, T. R. Utterback, S. Vattathil, W. Warren, C. S. White, A. T. Chinwalla, Y. Feng, A. L. Halpern, L. W. Hillier, X. Huang, P. Minx, J. O. Nelson, K. H. Pepin, X. Qin, G. G. Sutton, E. Venter, B. P. Walenz, J. W. Wallis, K. C. Worley, S. P. Yang, S. M. Jones, M. A. Marra, M. Rocchi, J. E. Schein, R. Baertsch, L. Clarke, M. Csuros, J. Glasscock, R. A. Harris, P. Havlak, A. R. Jackson, H. Jiang, et al. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316:222-234. [DOI] [PubMed] [Google Scholar]
- 15.Gongora, C., G. David, L. Pintard, C. Tissot, T. D. Hua, A. Dejean, and N. Mechti. 1997. Molecular cloning of a new interferon-induced PML nuclear body-associated protein. J. Biol. Chem. 272:19457-19463. [DOI] [PubMed] [Google Scholar]
- 16.Goodman, A. G., J. L. Fornek, G. R. Medigeshi, L. A. Perrone, X. Peng, M. D. Dyer, S. C. Proll, S. E. Knoblaugh, V. S. Carter, M. J. Korth, J. A. Nelson, T. M. Tumpey, and M. G. Katze. 2009. P58(IPK): a novel “CIHD” member of the host innate defense response against pathogenic virus infection. PLoS Pathog. 5:e1000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haller, O., and F. Weber. 2009. The interferon response circuit in antiviral host defense. Verh. K Acad. Geneeskd. Belg. 71:73-86. [PubMed] [Google Scholar]
- 18.Hao, L., A. Sakurai, T. Watanabe, E. Sorensen, C. A. Nidom, M. A. Newton, P. Ahlquist, and Y. Kawaoka. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horisberger, M. A. 1992. Virus-specific effects of recombinant porcine interferon-gamma and the induction of Mx proteins in pig cells. J. Interferon Res. 12:439-444. [DOI] [PubMed] [Google Scholar]
- 20.Huang Da, W., B. T. Sherman, and R. A. Lempicki. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44-57. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs, J. M., D. L. Diamond, E. Y. Chan, M. A. Gritsenko, W. Qian, M. Stastna, T. Baas, D. G. Camp II, R. L. Carithers, Jr., R. D. Smith, and M. G. Katze. 2005. Proteome analysis of liver cells expressing a full-length hepatitis C virus (HCV) replicon and biopsy specimens of posttransplantation liver from HCV-infected patients. J. Virol. 79:7558-7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs, J. M., H. M. Mottaz, L. R. Yu, D. J. Anderson, R. J. Moore, W. N. Chen, K. J. Auberry, E. F. Strittmatter, M. E. Monroe, B. D. Thrall, D. G. Camp II, and R. D. Smith. 2004. Multidimensional proteome analysis of human mammary epithelial cells. J. Proteome Res. 3:68-75. [DOI] [PubMed] [Google Scholar]
- 23.Jones, P. D., and G. L. Ada. 1986. Influenza virus-specific antibody-secreting cells in the murine lung during primary influenza virus infection. J. Virol. 60:614-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlas, A., N. Machuy, Y. Shin, K. P. Pleissner, A. Artarini, D. Heuer, D. Becker, H. Khalil, L. A. Ogilvie, S. Hess, A. P. Maurer, E. Muller, T. Wolff, T. Rudel, and T. F. Meyer. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818-822. [DOI] [PubMed] [Google Scholar]
- 25.Katze, M. G. 1995. Regulation of the interferon-induced PKR: can viruses cope? Trends Microbiol. 3:75-78. [DOI] [PubMed] [Google Scholar]
- 26.Kelly, R. T., J. S. Page, R. Zhao, W. J. Qian, H. M. Mottaz, K. Tang, and R. D. Smith. 2008. Capillary-based multi nanoelectrospray emitters: improvements in ion transmission efficiency and implementation with capillary reversed-phase LC-ESI-MS. Anal. Chem. 80:143-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konig, R., S. Stertz, Y. Zhou, A. Inoue, H. H. Hoffmann, S. Bhattacharyya, J. G. Alamares, D. M. Tscherne, M. B. Ortigoza, Y. Liang, Q. Gao, S. E. Andrews, S. Bandyopadhyay, P. De Jesus, B. P. Tu, L. Pache, C. Shih, A. Orth, G. Bonamy, L. Miraglia, T. Ideker, A. Garcia-Sastre, J. A. Young, P. Palese, M. L. Shaw, and S. K. Chanda. 2010. Human host factors required for influenza virus replication. Nature 463:813-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, T. G., J. Tomita, A. G. Hovanessian, and M. G. Katze. 1992. Characterization and regulation of the 58,000-dalton cellular inhibitor of the interferon-induced, dsRNA-activated protein kinase. J. Biol. Chem. 267:14238-14243. [PubMed] [Google Scholar]
- 29.Liu, H., R. G. Sadygov, and J. R. Yates III. 2004. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76:4193-4201. [DOI] [PubMed] [Google Scholar]
- 30.Liu, N., W. Song, P. Wang, K. Lee, W. Chan, H. Chen, and Z. Cai. 2008. Proteomics analysis of differential expression of cellular proteins in response to avian H9N2 virus infection in human cells. Proteomics 8:1851-1858. [DOI] [PubMed] [Google Scholar]
- 31.Loo, Y. M., J. Fornek, N. Crochet, G. Bajwa, O. Perwitasari, L. Martinez-Sobrido, S. Akira, M. A. Gill, A. Garcia-Sastre, M. G. Katze, and M. Gale, Jr. 2008. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82:335-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marginean, I., R. T. Kelly, R. J. Moore, D. C. Prior, B. L. LaMarche, K. Tang, and R. D. Smith. 2009. Selection of the optimum electrospray voltage for gradient elution LC-MS measurements. J. Am. Soc. Mass Spectrom. 20:682-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall, E. E., and A. P. Geballe. 2009. Multifaceted evasion of the interferon response by cytomegalovirus. J. Interferon Cytokine Res. 29:609-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsland, B. J., N. L. Harris, M. Camberis, M. Kopf, S. M. Hook, and G. Le Gros. 2004. Bystander suppression of allergic airway inflammation by lung resident memory CD8+ T cells. Proc. Natl. Acad. Sci. U. S. A. 101:6116-6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayer, D., K. Molawi, L. Martinez-Sobrido, A. Ghanem, S. Thomas, S. Baginsky, J. Grossmann, A. Garcia-Sastre, and M. Schwemmle. 2007. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J. Proteome Res. 6:672-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehle, A., and J. A. Doudna. 2009. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc. Natl. Acad. Sci. U. S. A. 106:21312-21316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metz, T. O., J. M. Jacobs, M. A. Gritsenko, G. Fontes, W. J. Qian, D. G. Camp II, V. Poitout, and R. D. Smith. 2006. Characterization of the human pancreatic islet proteome by two-dimensional LC/MS/MS. J. Proteome Res. 5:3345-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morens, D. M., and A. S. Fauci. 2007. The 1918 influenza pandemic: insights for the 21st century. J. Infect. Dis. 195:1018-1028. [DOI] [PubMed] [Google Scholar]
- 39.Nesvizhskii, A. I., O. Vitek, and R. Aebersold. 2007. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4:787-797. [DOI] [PubMed] [Google Scholar]
- 40.Neumann, G., T. Noda, and Y. Kawaoka. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Brien, K. P., M. Remm, and E. L. Sonnhammer. 2005. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 33:D476-D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oehmen, C., and J. Nieplocha. 2006. ScalaBLAST: a scalable implementation of BLAST for high-performance data-intensive bioinformatics analysis. IEEE Trans. Parallel Distrib. Syst. 17:740-749. [Google Scholar]
- 43.Page, J. S., R. T. Kelly, K. Tang, and R. D. Smith. 2007. Ionization and transmission efficiency in an electrospray ionization-mass spectrometry interface. J. Am. Soc. Mass Spectrom. 18:1582-1590. [DOI] [PubMed] [Google Scholar]
- 44.Palese, P. 2004. Influenza: old and new threats. Nat. Med. 10:S82-S87. [DOI] [PubMed] [Google Scholar]
- 45.Pepys, M. B., and G. M. Hirschfield. 2003. C-reactive protein: a critical update. J. Clin. Invest. 111:1805-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian, W. J., J. M. Jacobs, D. G. Camp II, M. E. Monroe, R. J. Moore, M. A. Gritsenko, S. E. Calvano, S. F. Lowry, W. Xiao, L. L. Moldawer, R. W. Davis, R. G. Tompkins, and R. D. Smith. 2005. Comparative proteome analyses of human plasma following in vivo lipopolysaccharide administration using multidimensional separations coupled with tandem mass spectrometry. Proteomics 5:572-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian, W. J., T. Liu, M. E. Monroe, E. F. Strittmatter, J. M. Jacobs, L. J. Kangas, K. Petritis, D. G. Camp II, and R. D. Smith. 2005. Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J. Proteome Res. 4:53-62. [DOI] [PubMed] [Google Scholar]
- 48.Shah, A. R., M. Singhal, K. R. Klicker, E. G. Stephan, H. S. Wiley, and K. M. Waters. 2007. Enabling high-throughput data management for systems biology: the Bioinformatics Resource Manager. Bioinformatics 23:906-909. [DOI] [PubMed] [Google Scholar]
- 49.Shapira, S. D., I. Gat-Viks, B. O. Shum, A. Dricot, M. M. de Grace, L. Wu, P. B. Gupta, T. Hao, S. J. Silver, D. E. Root, D. E. Hill, A. Regev, and N. Hacohen. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strunk, R. C., D. M. Eidlen, and R. J. Mason. 1988. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J. Clin. Invest. 81:1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tolnay, A. E., C. R. Baskin, T. M. Tumpey, P. J. Sabourin, C. L. Sabourin, J. P. Long, J. A. Pyles, R. A. Albrecht, A. Garcia-Sastre, M. G. Katze, and H. Bielefeldt-Ohmann. 2010. Extrapulmonary tissue responses in cynomolgus macaques (Macaca fascicularis) infected with highly pathogenic avian influenza A (H5N1) virus. Arch. Virol. 155:905-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vester, D., E. Rapp, D. Gade, Y. Genzel, and U. Reichl. 2009. Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics 9:3316-3327. [DOI] [PubMed] [Google Scholar]
- 53.Wheat, W. H., R. Wetsel, A. Falus, B. F. Tack, and R. C. Strunk. 1987. The fifth component of complement (C5) in the mouse. Analysis of the molecular basis for deficiency. J. Exp. Med. 165:1442-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.WHO. 2010. Global alert and response (GAR): avian influenza. World Health Organization, Geneva, Switzerland. http://www.who.int/csr/disease/avian_influenza/.
- 55.Zhang, L., J. M. Katz, M. Gwinn, N. F. Dowling, and M. J. Khoury. 2009. Systems-based candidate genes for human response to influenza infection. Infect. Genet. Evol. 9:1148-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.