

Abstract
The innate immune receptor Toll-like receptor 3 (TLR3) can be present on the surface of the plasma membranes of cells and in endolysosomes. The Unc93b1 protein has been reported to facilitate localization of TLR7 and 9 and is required for TLR3, -7, and -9 signaling. We demonstrate that siRNA knockdown of Unc93b1 reduced the abundance of TLR3 on the cell surface without altering total TLR3 accumulation. In addition, siRNA to Unc93b1 reduced the secretion of the TLR3 ectodomain (T3ECD) into the cell medium. Furthermore, two human single nucleotide polymorphisms that affected herpesvirus and influenza virus encephalopathy as well as a natural isoform generated by alternative splicing were found to be impaired for T3ECD secretion and decreased the abundance of TLR3 on the cell surface. The locations of the SNP P554S and the deletion in the isoform led to the identification of a loop in the TLR3 ectodomain that is required for secretion and a second whose presence decreased secretion. Finally, a truncated protein containing the N-terminal 10 leucine-rich repeats of T3ECD was sufficient for secretion in an Unc93b1-dependent manner.
Keywords: Innate Immunity, Ribonuclear Protein (RNP), Signal Transduction, Toll-like Receptors (TLR), Trafficking
Introduction
Toll-like receptors (TLRs)2 are a family of receptors that recognize pathogen-associated molecular patterns (PAMPs) and initiate signal transduction pathways to activate innate immune responses and to modulate adaptive immunity responses. There are more than 10 known human TLRs that collectively recognize a remarkable variety of ligands (1). The subcellular location of the different TLRs is related to ligand recognition. TLR3, -7, -8, and -9 recognize nucleic acids in endolysosomal compartments, whereas TLR1, -2, -4, -5, and -6 mainly recognize bacterial products and are located primarily on the cell surface (2–4).
TLR3 binds viral double-stranded (ds) RNAs and dsRNA mimics such as poly(I:C), annealed homopolymers of inosinic acid and cytidylic acid. Like other TLRs, TLR3 is a type I transmembrane glycoprotein with a large N-terminal ectodomain (ECD) for ligand binding, a single transmembrane helix, and a C-terminal cytoplasmic Toll-interleukin-1 receptor (TIR) domain. The TLR3 ectodomain (T3ECD) consists of 23 conserved leucine-rich repeats (LRRs) capped at the termini by cysteine-rich motifs (5, 6). The T3ECD has 15 predicted N-linked glycosylation sites and two short loops that reside in LRR12 (Loop 1) and LRR20 (Loop 2), respectively (7, 8). The crystal structure of the T3ECD complexed with dsRNA revealed two ligand binding sites, one within the region from LRR19 to LRR21 and the other within the N terminus to LRR3, as well as a dimerization domain in the C-terminal cysteine-rich cap (9, 10).
Single nucleotide polymorphisms (SNPs) in TLRs have been correlated with disease conditions such as asthma, endotoxin hyporesponsiveness, sepsis, immunodeficiencies, and atherosclerosis (11–18). For TLR3, SNP P554S within LRR20 is linked with partial penetrance of herpes simplex virus-associated encephalitis in children (19). SNP F303S within LRR11 is linked to pathologies of influenza virus infection (20). SNP L412F (LRR15) is present in ∼30% of the human population, exhibits a defect for signal transduction in cultured cells, and is correlated with reduced incidence of the dry form of age-related macular degeneration (21, 22). An isoform of TLR3 generated by alternative splicing that lacks 64 amino acids within LRR10 to the end of LRR12 has been reported in human astrocytes; its role in TLR3 function is unknown (23).
How TLR3 subcellular localization relates to function is not well understood. TLR7 and TLR9 are recruited from endoplasmic reticulum through the Golgi to lysosomes upon ligand stimulation in a process that also involves proteolysis (24, 25). TLR3 is present on the cell surface as well as in endosomes, depending on the cell type (26, 27). A cytoplasmic “linker region” between the transmembrane domain and the TIR domain of TLR3 was reported to regulate the TLR3 location in the cell (28, 29). TLR3 trafficking will likely involve Unc93b1, an integral, endoplasmic reticulum-associated protein that regulates TLR7 and TLR9 localization (30, 31). Single amino acid substitutions in Unc93b1 of mice and mutations of UNC93B1 in human fibroblasts were affected for cytokine production mediated by TLR3 (32–34). Murine Unc93b1 can physically interact with TLR3, -7, and -9 through their transmembrane domains (30).
T3ECD could be secreted into the culture medium by both insect and human cells (6, 8, 10). We hypothesize that the requirements for T3ECD secretion mimic that for normal TLR3 trafficking to the cell surface. Herein we documented that T3ECD secretion correlates with the presence of full-length TLR3 on the cell surface and that Unc93b1 is required for secretion as well as affect the cell surface localization of wild-type (WT) TLR3. SNPs associated with human disease susceptibilities decreased T3ECD secretion and the abundance of cell surface-associated TLR3. Interestingly, Loop 1 was found to act to inhibit T3ECD secretion, whereas changes in Loop 2 decreased secretion.
MATERIALS AND METHODS
Plasmid Construction
The WT TLR3 plasmid was previously described in Sun et al. (8). Plasmids containing SNPs were made by site-directed mutagenesis using oligonucleotides and the QuikChange kit (Stratagene). The sequence of the all constructs containing changes from the WT sequence were determined to ascertain that unintended mutations were not introduced. All primers used are shown in supplemental Table 1.
T3ECD was expressed from the pBeth-TLR3ECD-His6 plasmid (8). Briefly, cDNA from TLR3 residues 1–703 to which a C-terminal V5 epitope and a His6 tag was appended to a herpesvirus thymidine kinase promoter in a vector containing the pcDNA3.1 backbone to generate pBeth-TLR3ECD. T3ECDΔL1 was subcloned into the same vector from TLR3ΔL1.
N-L10, N-L11, and N-L12 containing TLR3 residues 1–297, 1–322, and 1–355, respectively, were fused to the sequence coding for residues 660–704 and 6 histidine codons derived from the pBeth-TLR3 plasmid. These three plasmids were made in two steps. First, an XhoI site was created as a silent mutation in codon 694 in pBeth-TLR3ECD to result in pBeth-TLR3ECDx. Fragments encoding N-L10, N-L11, and N-L12 were generated by PCR from T3ECD to contain flanking HindIII and XhoI restriction sites at the 5′ and 3′ ends. The DNA fragments treated with these restriction enzymes were ligated to an acceptor plasmid derived from pBeth-TLR3ECDx and linearized with a HindIII and XhoI restriction digestion. The sequence from 5′ of the TLR3 cDNA to codon 660 was replaced in this manipulation. Plasmid N-L12Δ1 was cloned by the same strategy except that T3ECDΔL1 was used as the template to generate the PCR fragment.
Plasmids expressing N-L10 + 12 and N-L10 + 12ΔL1 were made by chemical synthesis of the two DNA strands that can encode LRR12 or LRR12 lacking the Δ1 loop sequence that were then digested with XhoI before its ligation to the C terminus of LRR10. The TLR3 isoform (TLR3Δ64) was recreated by annealing chemically synthesized DNA molecules designed to remove the sequence missing in Δ64 and extending from the primers using PfuI. The resulting DNA was the digested with flanking restriction sites and ligated in the backbone generated from pBeth-TLR3. pCMV6-Entry-Unc93b1-Myc-FLAG was from Origene Technologies (Rockville, MD). Unc93B1 ΔN36 was PCR-amplified using PCR and cloned into the same vector.
TLR3 Luciferase Reporter Assay
HEK293T cells were plated in CoStar white 96-well plates in DMEM amended with 10% FBS at 4.4 × 104 cells per well and transfected at ∼85–90% confluency with a mixture of the Lipofectamine 2000 and plasmids pNiFty-Luc, pUNO-huTLR3, or TLR3 mutations and phRL-TK as described in Sun et al. (8). For assays using IFN-β-Luc or ISRE-luc reporters, the respective plasmids were substituted for pNifty-luc. The transfected cells were induced 18–22 h later by the addition of poly(I:C) to the culture medium to a final concentration of 2.5 μg/ml. The cells harvested for analysis of luciferase 18–20 h after poly(I:C) addition.
siRNA Knockdown of Unc93b1
Human embryonic kidney cells (HEK293T) were seeded at 1.0 × 105 cells/well in DMEM amended with 10% FBS in a 48-well tissue culture plate or 4.4 × 104 cells/well in a 96-well plate. 6 h later the cells were transfected with siRNA specific to Unc93b1 (Darmacon, Inc.) or ST3GAL4 (Ambion, siRNA ID: s12852) using Lipofectamine 2000 (Invitrogen). The cells were incubated for 24 h before transfection to express TLR3 or TLR3 derivatives. If the cells were examined for TLR3 activation of reporter expression, they were also transfected with the luciferase and Renilla luciferase plasmids as described above.
Secretion Assay
HEK 293T cells and P2.1 cells were plated in 48-well plates in DMEM amended with 10% FBS at 1 × 105 cells/well and transfected with 50 ng of plasmid pUNO-TLR3, pBeth-TLR3ECD, or various plasmids derived from pBeth-TLR3ECD using Lipofectamine 2000. The medium was removed 24 h later and replaced with 150 μl of DMEM medium. The media were harvested after another 24 h, and cells were lysed with the passive lysis buffer (Promega) and sonicated to degrade chromosomal DNA, then subjected to Western blot analysis.
Equal amounts of proteins from the culture medium or cell lysate were separated on NuPAGE 4–12% Bis-Tris gels and blotted onto polyvinylidene difluoride membranes (Invitrogen) for probing with the anti-TLR3 antibody (R&D systems, catalog #AF1487). The anti-human Unc93b1 antibody was from Sigma (catalog #AV49979). Monoclonal anti-FLAG® M2 peroxidase (HRP) antibody was from Sigma (catalog #A8592). The blots were developed with HRP-conjugated secondary antibodies (Santa Cruz Biotechnology) and an ECL-plus Western blotting detection system (Amersham Biosciences).
Fluorescence-activated Cell Sorting (FACS) Analysis
Cells were grown in 6-well collagen-coated plates (BD Biosciences) at 2 × 106 cells/well. The cells were transfected with 1 μg of the appropriate plasmids using Lipofectamine 2000. Staining of cell surface-associated TLR3 used cells harvested 24 h after transfection and washed twice with ice-cold FACS buffer (1× phosphate-buffered saline containing 10 mm phosphate, 150 mm NaCl, pH 7.4, 3% fetal bovine serum, 0.04% sodium azide) before suspension at 2.0 × 107 cells/ml in FACS buffer. The cells were stained for 30 min at 4 °C with 1 μg of phycoerythrin-labeled anti-human TLR3 mAb (clone TLR3.7 or a negative control mouse IgG1 control antibody (eBioscience) and incubated for 30 min on ice in the dark. The cells were washed twice with FACS buffer to remove unbound antibody and then resuspended in FACS buffer. Viaprobe (BD Biosciences) was added to the cultures to exclude dead cells. The samples were analyzed using a FACSCalibur machine (BD Biosciences).
RT-PCR Analysis of the Δ64 Isoform Expression
Poly(A)-selected mRNAs extracted from actively growing human astrocytes (NHA) and normal human bronchial epithelial cells (NHBE) were used for cDNA synthesis by reverse transcription and PCR. The primers used are shown in supplemental Table S1. The cDNA product with and without the Δ64 deletion should be ∼700 and 500 base pairs. These were excised from an agarose gel, purified, and subcloned into Invitrogen TOPO pCR4 vector for DNA sequencing.
RESULTS
TLR3 Function, Localization, and Unc93b1
We seek to better understand TLR3 localization in cells and how localization could affect TLR3 function and diseases that could result from improper function. Unc93b1 is likely involved in TLR3 localization as it is required for TLR7 and TLR9 trafficking and the function of TLR3 (31, 33, 34).
We first attempted to confirm that Unc93b1 contributes to TLR3-dependent signaling in a previously validated reporter assay (8, 21). Briefly, HEK293T cells were transfected with three plasmids, one to express TLR3, a second to constitutively express Renilla luciferase, and a third that can express a firefly luciferase reporter under the control of the ISRE promoter. In the presence of poly(I:C), TLR3 will activate signaling leading to an increase in the firefly luciferase activity. The ratio of firefly to Renilla luciferase activities of the poly(I:C)-induced and mock-induced samples is plotted as -fold induction.
The cells to assay TLR3 were also transfected with siRNA specific to Unc93B1 or a control siRNA targeting the sialyltransferase ST3GAL4. The siRNA to Unc93b1 reduced TLR3-dependent expression of the reporters to approximately a third of that of the untreated sample. The control siRNA did not have a comparable effect (Fig. 1A). Furthermore, neither siRNA affected TLR4 signaling with lipopolysaccharide as the agonist (Fig. 1B). These results demonstrate that Unc93b1 can affect TLR3 signaling in HEK293T cells. Furthermore, the siRNA to Unc93b1 did reduce the level of the Unc93b1 protein detected by Western blot analysis, and there were no obvious effects on the morphology of the cells in all siRNAs tested (Fig. 1C and data not shown).
FIGURE 1.

Unc93b1 is required for TLR3 function and T3ECD secretion in HEK293T cells. A, shown is the effects of siRNA to Unc93b1 on TLR3-dependent poly(I:C)-dependent signaling by TLR3. The concentrations of the siRNAs transfected into cells are shown below the bar graph. The control siRNA (Contr.) was designed against ST3GAL4 and was used throughout all experiments. The readouts of the firefly luciferase reporter driven by the ISRE promoter are normalized to the Renilla luciferase driven by the thymidine kinase promoter. The number above each bar represents percentage activity relative to TLR3 activity without siRNA treatment. The error bar denotes 1 S.D. averaged from at least three independent experiments that each had three independent samples. B, shown is the effects of siRNA to Unc93b1 on TLR4-dependent signal transduction. Cells were transfected to express TLR4 and its co-activators and induced with 1 μg/ml lipopolysaccharide as described previously (47). The firefly luciferase reporter is driven by the NF-κB promoter and normalized to the Renilla luciferase driven by the thymidine kinase promoter. The concentration of the siRNA used is shown below the graphs. C, shown is confirmation that the siRNA to Unc93b1 did reduce the level of Unc93b1. The Western blot result contained lysates from cells treated with siRNA for 48 h and probed with a mAb that specifically recognizes Unc93b1. The loading control (L. C.) was a band present on the membrane used for the Western blot. The membrane was stained with Coomassie Blue. D, siRNA knockdown of Unc93b1 decreased TLR3 cell surface expression. siRNA to Unc93b1 or ST3Gal4 was transfected at 50 nm. The cells were either not permeabilized to stain for surface expression of TLR3 or permeabilized to stain for overall expression. The graphs are representative or six individual transfections of each treatment and are shown as an overlap of mock, siRNA-treated, and untreated samples. The numbers show percentage of cells positive for TLR3 and are average of three independent transfections.
To examine whether Unc93b1 can affect TLR3 localization, HEK293T cells expressing TLR3 were transfected with the Unc93b1-specific or the control siRNA and FACS was used to quantify the amount of TLR3 on the surface of intact cells. An aliquot of the cells were permeabilized with detergent, and the total TLR3 was also quantified. HEK293T cells do not express endogenous TLR3, and transfection to express WT TLR3 resulted in ∼18% of the cells exhibiting TLR3 on the cell surface. Cell surface-associated TLR3 was reduced by the siRNA to Unc93b1 to a mean value of 13.4% in six independent samples (Fig. 1D). Given that the cells were transfected with the siRNAs, the observed reduction of TLR3 on the cell surface likely underestimates the potential effect of the Unc93b1 siRNA. Furthermore, the total amount of TLR3-positive cells in the permeabilized samples was not significantly different in the cells knocked down for Unc93b1 or treated with a nonspecific siRNA (Fig. 1D). Finally, a Western blot detecting TLR3 confirmed the FACS analysis that the siRNA to Unc93b1 does not affect the overall level of TLR3 (supplemental Fig. S1). All of these results demonstrate that Unc93b1 is involved in the localization of TLR3 to the cell surface.
Unc93b1 and T3ECD Secretion
We seek to determine whether Unc93b1 can affect the secretion of the TLR3 ectodomain. The effects of the siRNA to Unc93b1 or the control siRNA were examined in HEK293T cells that could secrete T3ECD into the medium. T3ECD secretion was reduced by siRNA to Unc93b1 in a manner dependent on the siRNA concentration (Fig. 2A). The control siRNA did not have this effect. Furthermore, knocking down Unc93b1 did not affect the secretion of matrix metallopeptidase-28 (MMP-28) (Fig. 2B) (35). Finally, we expressed recombinant Unc93b1 in HEK293T cells from a plasmid and observed that T3ECD secretion was not significantly affected, likely because the endogenous Unc93b1 was sufficient for T3ECD secretion. However, expressing a version of Unc93b1 lacking the first 36 residues (ΔN36) reduced T3ECD secretion and increased the level of T3ECD in the cell-associated materials (Fig. 2C). Δ36 was previously reported to down-regulate TLR9 trafficking and up-regulate TLR7 trafficking (36). Altogether, the effects on the localization of TLR3 and T3ECD correspond to the altered Unc93b1 expression profiles showing that Unc93b1 is important in TLR3 localization. Furthermore, the secretion of T3ECD should provide a useful assay to examine the requirements for TLR3 cell surface localization.
FIGURE 2.
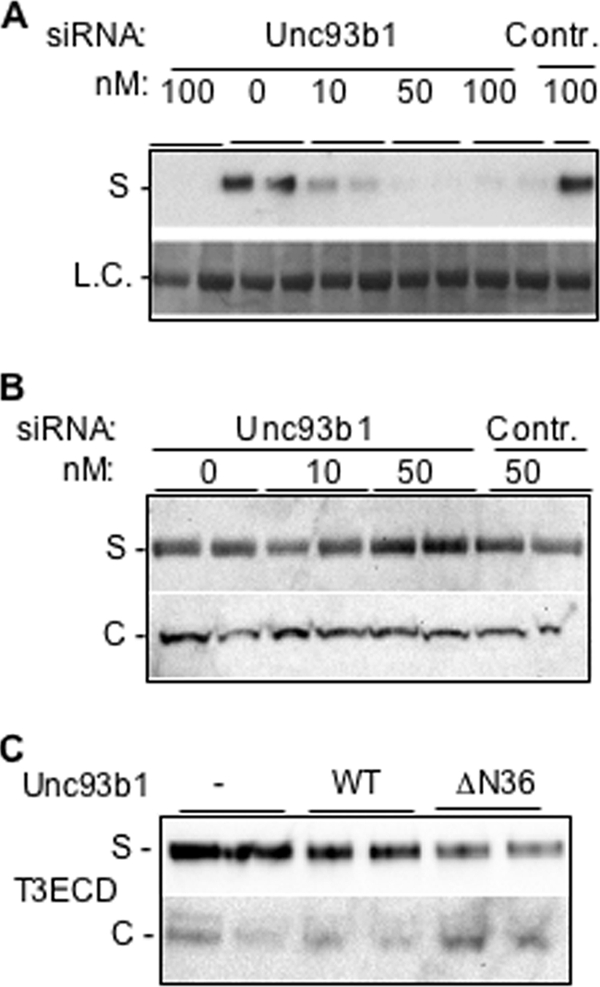
Unc93b1 and T3ECD secretion. A, shown are the effects of siRNA to Unc93b1 on the amount of T3ECD present in the culture medium. An empty vector or plasmid expressing T3ECD was transfected into HEK293T cells. The concentrations and types of siRNA transfected are shown above the blot image. The medium was harvested and subjected to Western blot analysis. Each construct was tested in two independent samples. Contr. denotes a control siRNA. The result is representative of three independent experiments. S, supernatant; L.C., loading control. B, siRNA to Unc93b1 did not affect the secretion of matrix metalloproteinase, MMP-28. The identities and the concentrations of the siRNAs used are shown above the image of the results from the Western blots. The images are from Western blots of the cell culture supernatant (S) and the cell lysate (C) probed with a MMP-28-specific polyclonal antibody. C, shown is overexpression of Unc93b1 and T3ECD secretion. A truncated Unc93b1 is affected for the secretion of T3ECD. HEK293T cells were either mock-transfected or transfected with plasmids expressing WT or an N-terminal-truncated Unc93b1, ΔN36. The cells were also transfected to express T3ECD. The amount of T3ECD present either in the cell lysate (C) or the supernatant (S) was detected by Western blots. The reduced secretion of T3ECD with ΔN36 Unc93b1 relative to wild-type Unc93b1 was observed in four independent samples.
Glycosylation and T3ECD Secretion
Post-translational modifications including glycosylation should be prerequisite to the secretion of T3ECD. In fact, the T3ECD secreted in the medium, and that remained in the cell had distinct electrophoretic mobilities when they were combined and electrophoresed in the same lane (Fig. 3A).
FIGURE 3.

Glycosylation and T3ECD secretion. A, the secreted T3ECD has a distinct electrophoretic mobility in comparison to that of the cell-associated T3ECD. Samples from the culture medium transfected to express T3ECD (S) or the cell lysate (C) were separately processed for Western blot analysis in the two upper panels or mixed together in equal proportions for the lower Western blot. B, shown are the effects of substitutions that affected TLR3 ECD glycosylation and ligand binding on T3ECD secretion. The results show a Western blot of the amount of T3ECD present in the medium and the cells when they harbor amino acid substitutions. The medium and cell lysate were combined with each sample as the secreted form of T3ECD (designated S) and the cell-associated form (designated C) are easily distinguishable. All samples were from independently transfected cells processed in parallel. The N413A mutation was characterized to be an N-linked glycosylation site (7, 11). The H539E substitution was characterized to be defective in ligand binding (7, 9). C, effects of tunicamycin and swainsonine on T3ECD secretion. The Western blot image shows the T3ECD present in the medium (S) or the cell lysate (C) of HEK293T cells transfected to express T3ECD. The medium and cell lysates from the transiently transfected HEK293T cells were analyzed separately, and the final concentrations of the glycosylation inhibitor (glyc. Inh.) added to the samples are shown above the Western blot images. S denotes the secreted T3ECD, whereas C denotes the cell-associated T3ECD. The loading controls in these blots are GAPDH proteins detected by Western blots.
Residue Asn-413 was shown to be glycosylated in T3ECD (5, 6, 8, 10). A N413A substitution reduced the secreted T3ECD when compared with WT (Fig. 3B). In contrast, the H539E substitution, which affected ligand binding by T3ECD and abolished TLR3-dependent activation of reporters (6, 9), allowed significantly high levels of T3ECD secretion (Fig. 3B). The latter observation is consistent with a model in which ligand binding by TLR3 is not required for the trafficking of TLR3 (9, 37). L412F, a substitution linked to macular degeneration and defective TLR3 signal transduction (21, 22), also affected the secretion of the T3ECD, possibly through impacting the glycosylation of the neighboring residue, Asn-413.
Tunicamycin, which inhibits the first step in the addition of N-linked glycosyl groups, was previously shown to inhibit the activation of reporters by TLR3 (8). We treated HEK293T cells with tunicamycin up to 80 ng/ml and did not observe any obvious cytotoxicity (8). When we tested T3ECD secretion by these cells, secreted T3ECD was usually at a 2–4-fold higher abundance in cells treated with tunicamycin lower than 5 ng/ml. However, tunicamycin concentrations higher than 5 ng/ml resulted in a reduction in T3ECD expression, and the cell-associated T3ECD migrated as lower molecular mass bands, likely due to an inhibition of glycosylation (Fig. 3C). There was also a relative decrease in the amount of the secreted T3ECD when compared with the levels associated with cells. These results show that tunicamycin can affect both T3ECD expression as well as secretion. In contrast, swainsonine, an inhibitor of mannosidase that did not previously affect TLR3 activation of reporters (8), did not have an obvious effect on T3ECD secretion. All of the results in Fig. 3 suggest that proper glycosylation will influence T3ECD secretion.
SNP P554S and Loop 2
We seek to examine whether SNPs of TLR3, in addition to L412F, could affect the secretion of T3ECD and the localization of TLR3 in cells. SNP P554S is associated with increased susceptibility to herpes simplex virus-1 encephalitis in children (19). To examine whether P554S affected TLR3 trafficking, this substitution was constructed in full-length TLR3 and in T3ECD. Both proteins with the P554S substitution were produced at a level comparable with that of WT in transiently transfected HEK293T cells. The same result was also observed in the fibrosarcoma cell line P2.1 (Fig. 4A). The P554S T3ECD was found in the culture medium at a level severalfold lower than WT T3ECD in both HEK293T and P2.1 cells, indicating that the defect in secretion is not specific to HEK23T cells.
FIGURE 4.

Effects of P554S on TLR3 localization and T3ECD secretion. A, shown is Western blot analysis of the effects of the P554S SNP on full-length TLR3 expression and T3ECD expression and secretion. The upper two Western blot results labeled with TLR3 show the total level of full-length TLR3 or TLR3-P554S proteins produced. The lower two panels show the secretion (S) and cell-associated (C) forms of WT T3ECD and T3ECD-P554S. All samples were tested in two independently transfected samples. B, FACS analysis of the TLR3 and TLR3-P554S expressed on the surface of HEK293T cells and in detergent-permeabilized cells is shown. The fluorescence due to binding of the antibody to TLR3 is in log scale (horizontal axis) and plotted against the number of events (vertical axis). The blue line shows the level of staining with a control antibody (anti-mouse IgG), and the black line shows the staining with anti-hTLR3 conjugated with phycoerythrin. C, quantitative results from the FACS experiment are shown. MFI, mean fluorescence intensity. S.E. were from a minimum of three independently assessed samples. The numbers that lacked a S.E. are the average of two independent samples. D, TLR3-dependent and poly(I:C)-dependent signaling of TLR3-P554S is shown. Activities of the reporter driven by promoters responsive to NFκB, ISRE, or IFNβ were normalized to the Renilla luciferase driven by the thymidine kinase promoter as a ratio. The number above each bar represents the ratio of poly(I:C)-induced samples over the ratio of uninduced samples.
FACS analysis showed that TLR3 with the P554S substitution was reduced in the amount of TLR3 on the cell surface relative to WT (Fig. 4, B and C). In permeabilized cells, however, comparable levels of both proteins were observed, demonstrating that the defect is not in protein expression (Fig. 4, B and C). Together, the secretion and FACS results show that P554S was altered for trafficking to the cell surface.
Zhang et al. (19) reported that P554S was produced as a truncated product of TLR3 and reduced TLR3 activation of reporter expression. We did not find an obvious defect for TLR3-P554S induced reporter activity in the presence of poly(I:C) (Fig. 4D), and we did not observe a truncated form of TLR3 in Western blots (data not shown). These results suggest that sufficient amounts of the TLR3-P554S are present to activate signal transduction. Our results are consistent with those of Wang et al. (38) who examined the effects of P554S on hepatitis C virus infection and did not observe a defect associated with the P554S SNP. The disparate results could be due to the cell lines used.
Loop 2 and T3ECD Secretion
P554 resides in Loop 2. Deletion of Loop 2 was previously found to decrease signal transduction by TLR3 (7). We engineered versions of T3ECD that lacked the entire Loop 2 of nine residues (ΔL2a) or five of the residues (ΔL2b) and found that both decreased T3ECD secretion (Fig. 5, A and B). Replacing Loop 2 with the murine sequence in the context of full-length TLR3 (L2m) restored secretion (Fig. 5B). These results along with those from the analysis of P554S suggest that Loop 2 of the T3ECD is required for T3ECD secretion.
FIGURE 5.

Loop 2 of the TLR3 ectodomain contributes to secretion. A, shown are sequences in Loop 2 affected in various mutants. The WT Loop 2 sequence is in bold, and the deleted residues are shown as dashes. Amino acid substitutions are underlined. B, shown are the effects of mutations in Loop 2 of T3ECD on secretion. Each construct was tested in two independently transfected cultures. In this assay, the medium and the cell lysates were loaded in the same well of the SDS-PAGE because the two bands can be clearly distinguished. The secreted (S) and cell-associated (C) T3ECD are identified to the right of the gel images. M denotes the molecular mass marker, and Vec. denotes that the cells were transfected with an empty vector. FL, full-length.
The Δ64 Isoform and the F303S SNP
Yang et al (23) identified an alternative splice variant of TLR3 (Δ64) expressed in primary human astrocytes (NHA) and glioblastoma cell lines. Ménager et al. (39) subsequently reported that the isoform was also expressed in neuroblastoma cells. Δ64 lacks residues 289–352, which span LRR10 to LRR12 (Fig. 6A). We wanted to examine whether the Δ64 mutation would affect the secretion of T3ECD and TLR3 cell surface expression.
FIGURE 6.

The TLR3 isoform Δ64. A, shown is the sequence affected in the Δ64 isoform. The sequences in LRR 10–12 are shown, and the sequence deleted in Δ64 is in bold. The Phe-303 residue that is substituted in the F303S SNP and the Loop 1 sequence are in grey and underlined. B, expression of Δ64 isoform in NHA cells and NHBE cells is shown. The gel shows the results of an RT-PCR reaction separated by agarose gel electrophoresis and visualized by staining with ethidium bromide. The HEK293T cells transfected with constructs expressing WT TLR3 or Δ64 served as positive controls. The cDNAs were generated from polyadenylated RNAs primed with oligo(dT) and random hexamer primers (supplemental Table S1) as described by Yang et al. (23) for PCR. C, the overexpression of Δ64 protein in HEK293T cells is shown. Plasmids encoding the wild-type TLR3 and Δ64 were transiently transfected to HEK293T cells. The Western blots were probed with a mixture of two mAbs, one to detect TLR3 and a second to detect α-Actin. The latter signal was intended as the internal loading controls. A nonspecific band recognized in Western blots is identified with an asterisk. D, the Δ64 isoform is defective for activation of reporter gene expression. Three independent luciferase reporters were used, as driven by the INFβ, ISRE, and NF-κB promoter elements. Whether the isoform was able to act as a dominant negative mutant to the wild-type TLR3 was also examined by transfecting into cells a 3 m excess of the isoform expression plasmid relative to the wild-type TLR3. In these assays the bona fide dominant negative mutant, ΔTIR, was used as a control. E, FACS analysis of the cell surface (Nonpermea.) and total (Permeab.) TLR3, TLR3 Δ64. The data forTLR3 and Δ64 are identified. The black lines show the sample stained with a control antibody to mouse IgG conjugated with phycoerythrin. F, Western blots to detect the secretion of T3ECDΔ64 are shown. S denotes the secreted form of T3ECD. The loading controls (L.C.) were taken from the same membrane used for Western analysis.
We first determined using RT-PCR that a message corresponding to Δ64 was expressed in NHA cells and also in normal human bronchial epithelial cells (NHBE), albeit at low levels relative to that of full-length TLR3 (Fig. 6B). Sequencing of the shorter cDNA from both cell types revealed the exact sequence reported by Yang et al. (23).
The Δ64 deletion was engineered into DNA constructs of full-length TLR3 to examine for effects on TLR3 activation of reporter expression and TLR3 trafficking. Western blot analysis showed that the resulting protein accumulated in HEK293T cells (Fig. 6C). However, the Δ64 mutant was defective for activation of reporter gene expression in the presence of poly(I:C), and it did not act as a dominant negative mutant to WT TLR3 (Fig. 6D). In FACS analysis, the Δ64 protein was decreased for cell surface expression when compared with WT TLR3, although it was expressed at a similar level as WT in permeabilized cells (Fig. 6E). T3ECD containing the Δ64 deletion also failed to secrete into the medium (Fig. 6F). These results demonstrate that the Δ64 isoform of TLR3 is defective for both signaling and cell surface expression in HEK293T cells.
SNP F303S of TLR3, found in a patient with influenza-associated encephalopathy, resides in the region deleted in Δ64. TLR3 with the F303S SNP was thoroughly characterized by Hidaka et al. (20), and we confirmed that it exhibited decreased ability to activate reporter gene expression in comparison to the WT TLR3 (supplemental Fig. S2A). In addition, it was not dominant negative to the WT TLR3 (supplemental Fig. S2A). T3ECD with the F303S substitution was also defective for secretion (supplemental Fig. S2B).
TLR3 Mutations Interact with Unc93b1
Unc93b1 has been reported to co-immunoprecipitate with TLR3 (30). We seek to examine whether the SNPs and mutant versions of TLR3 are affected for interaction with Unc93B1. A C-terminal myc and FLAG-tagged Unc93b1 was co-expressed with TLR3 and subjected to immunoprecipitation with the antibody to the myc tag. The presence of Unc93b1 and/or TLR3 in the precipitate was examined by Western blots. TLR3 co-precipitated with Unc93b1, consistent with the report of Brinkman et al. (30). The P554S, ΔL2, and F303S variants expressed to different levels. However, the amounts of the TLR3 variant that co-precipitated with Unc93b1 can be compared with the amount in the input (Fig. 7). The relative amounts of WT TLR3, P554S, ΔL2, and F303S that co-precipitated were comparable to the input (Fig. 7). These results were observed in four independent experiments. Importantly, the Δ64 did not co-immunoprecipitate as well with Unc93b1 when compared with the other TLR3 variants or to WT TLR3. When quantified, ∼30% of the input WT TLR3 co-immunoprecipitated with Unc93b1, whereas only 6% of the input Δ64 protein did so. Although these results do not inform us as to whether the region missing from Δ64 contacts Unc93b1, it shows that the ectodomain of TLR3 can affect interaction with Unc93b1.
FIGURE 7.

The interaction of Unc93b1 with TLR3 and variants. Plasmid expressing Unc93b1-Myc-FLAG was co-transfected with either an empty vector of plasmids expressing TLR3 or TLR3 mutants. 10% of the cell lysates were saved as input, and the rest 90% were immunoprecipitated (IP) with anti-Myc. Both input and immunoprecipitates were subjected to Western blots (IB) to detect TLR3. Immunoprecipitates were also blotted to detect Unc93b1. The images are representative of three independent experiments.
Truncated TLR3 ECD Can Secrete
Examination of the SNPs suggests that motifs within the TLR3 ectodomain are required for secretion. To better understand whether the motif(s) deleted in Δ64 is related to secretion, we made three truncations containing the N terminus of TLR3 through LRR10 (named N-L10), LRR11 (N-L11) or LRR12 (N-L12) (Fig. 8A). Each truncation contained a partial C-terminal cap (residues 660–704), and all three proteins were expressed at high levels (Fig. 8B, bottom panel). Unexpectedly, the secreted level of N-L10 was far higher than that of T3ECD, whereas N-L11 was comparable to T3ECD, and N-L12 was not detected in the medium (Fig. 8B, top panel). Consistent with the secretion defect of N-L12, a construct containing the first 15 LRRs of the TLR3 also failed to secrete (supplemental Fig. S3). These results suggest that LRR11 and LRR12 in the T3ECD contain motifs that negatively regulated T3ECD secretion.
FIGURE 8.
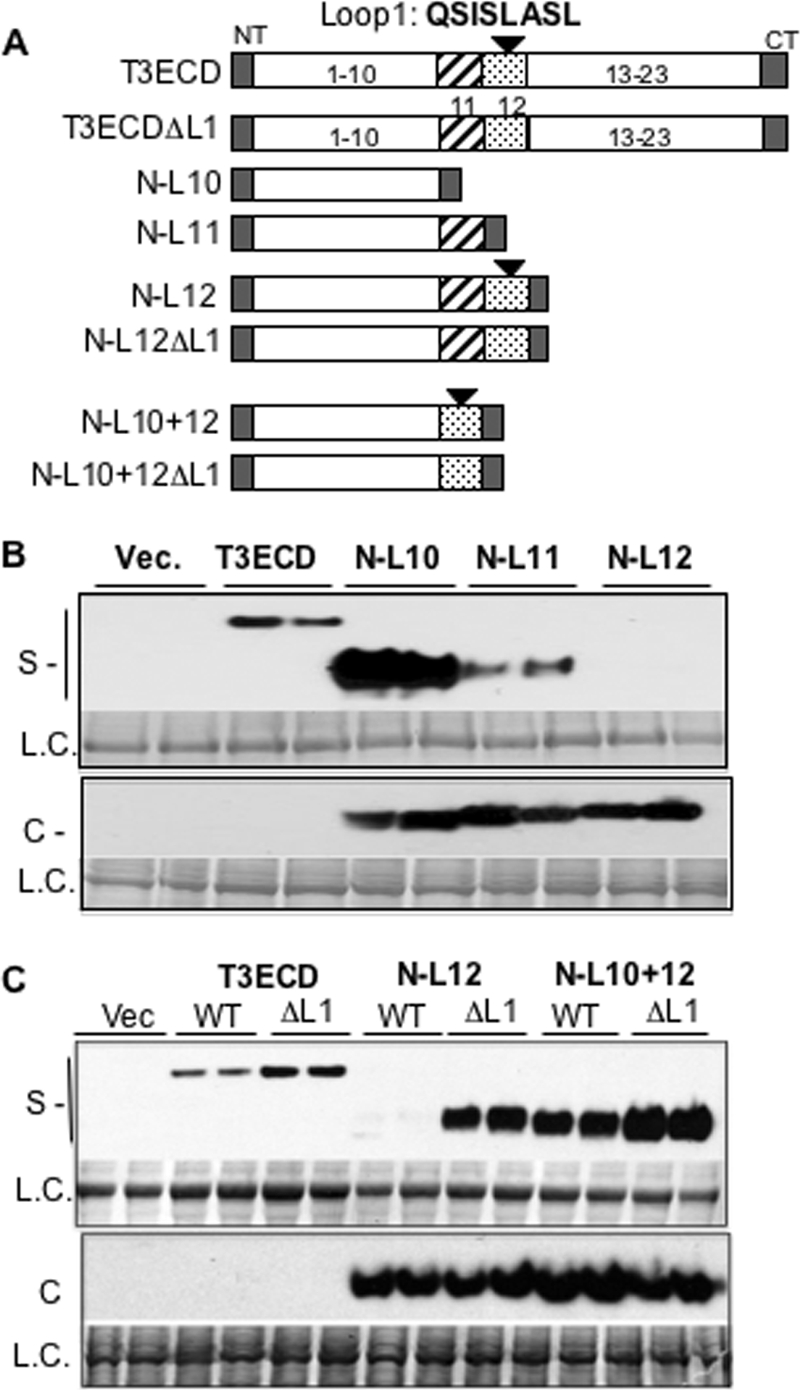
Loop 1 acts as a negative regulator of T3ECD secretion. A, shown is a schematic of the constructs used. NT and CT represent the N-terminal and C-terminal caps (residues 661–704). The numbers within or above the bars denote the number of LRRs in T3ECD. Loop 1 is denoted by a solid triangle above LRR12. B, LRR 12 can inhibit secretion. The WT T3ECD is expressed at a lower level in comparison to the truncations and requires a longer exposure for visualization. The loading controls were taken from the same membrane used for Western analysis. S, supernatant; L.C., loading control; C, cell lysate. C, deletion of Loop 1 rescues secretion of N-L12 and increases secretion of WT ECD and N-L10 + 12. The cell-associated level was unchanged when Loop 1 is deleted from N-L12 or N-L10 + 12.
LRR12 contains Loop 1 (Fig. 8A). We examined whether a deletion of the eight residues of Loop 1 from T3ECD named ΔL1 would affect secretion. ΔL1 consistently secreted better than T3ECD by 2-fold (Fig. 8C). Furthermore, N-L12 that lacked Loop 1 (in construct N-L12ΔL1) was dramatically improved for secretion relative to N-L12 (Fig. 8C).
To address whether the inhibitory activity of Loop 1 functions in a position-dependent manner, a construct that fused LRR12 to the C terminus of N-L10 (named N-L10 + 12) was tested. We also made a version of N-L10 + 12 that lacked Loop 1 named N-L10 + 12ΔL1 (Fig. 8A). N-L10 + 12 was competent for secretion, whereas the removal of Loop1 increased secretion of N-L10 + 12ΔL1 slightly (Fig. 8C). These results suggest that Loop 1 needs to act in a position-dependent manner or that the fusion of noncontiguous LRRs disrupted the Loop 1 ability to suppress secretion.
Finally, we determined whether the secretion of ΔL1 or the N-terminal portion of T3ECD retains the requirement for Unc93b1 (Fig. 9). siRNAs targeting Unc93b1 or the control siRNA were transiently transfected into HEK293T cells that can express ΔL1, N-L10, or N-L11 (Fig. 9A). The secretion of ΔL1 was decreased by siRNA targeting Unc93b1 (Fig. 9A). Similar results were observed for N-L10 and N-L11 (Fig. 9B). These results demonstrate that Unc93b1 can directly or indirectly participate in the secretion of the N-terminal portion of T3ECD but that the removal of Loop 1 within the ectodomain did not affect interaction with Unc93b1.
FIGURE 9.

siRNA knockdown of Unc93b1 can decrease secretion of the T3ECD lacking Loop 1(ΔL1) and two truncations require Unc93b1. A, shown is secretion of the ΔL1. The Western blot results show the amounts of the ΔL1 protein secreted in the absence or presence of transfected siRNA targeting Unc93b1 or a control, ST3GAL4. 50 ng of an appropriate plasmid expressing T3ECDΔL1 was transfected per well of cells. The format of the results is as described previously, where S and L.C. stand for secreted and loading control, respectively. B, shown is secretion of truncated TLR3ECD, N-L10, and N-L11 in response to siRNA targeting the Unc93b1 gene containing the first 10 or 11 LRRs of TLR3. L. C. denotes a loading control. S denotes the secreted proteins present in the cell culture medium. Con, control.
DISCUSSION
Although endosomes are likely the sites of ligand binding and signaling by TLR3 (40), the role for the cell surface-associated TLR3 is not clear. However, monoclonal antibodies targeting cell surface TLR3 can inhibit cytokine production by cultured human fibroblasts, lung epithelial BEAS-2B cells, and NHBE cells (41, 42), indicating that cell surface-associated TLR3 can affect signal transduction. These results motivated additional analysis of TLR3 trafficking.
In this work we observed that the location of TLR3 on the cell surface and the secretion of TLR3 ectodomain required Unc93b1, whereas the secretion of another innate immune protein, MMP-28, did not. The secretion assay was, therefore, useful for identifying features in the TLR3 ECD required for transport out of the cell. The secreted T3ECD was differentially glycosylated from the cellular T3ECD (Fig. 2A). Cells treated with tunicamycin decreased both T3ECD glycosylation and secretion (Fig. 2C). The role of glycosylation in secretion was supported by the mutant N413A, which negatively affected secretion when introduced in to T3ECD. However, full-length TLR3 harboring the N413A mutation did not show significant difference in surface expression compared with WT TLR3 (9), suggesting that there may be compensatory glycosylation sites for cell surface localization. The secretion assay allowed us to determine that Loop 1 and Loop 2 in the TLR3 ectodomain have opposing regulatory effects on the secretion of T3ECD and that three SNPs and a human TLR3 isoform are also affected for T3ECD secretion (Figs. 3 and 5 and supplemental Fig. S1).
The relationship between TLR3 secretion/cell surface expression, signal transduction, and disease pathologies will be a complex one. Although SNPs P554S, F303S, and L412F decreased TLR3 ECD secretion and TLR3 cell surface levels, they had different outcomes on human diseases; P554S and F303S increased pathologies to viral infection, whereas the L412F SNP is associated with a reduction in the incidence of the dry form of macular degeneration (21, 22). These apparently contradictory outcomes likely reflect TLR3 dual roles in pathogen-induced and non-pathogen-associated immune responses. In addition, TLR3 can contribute to increased pathology of rabies virus infection (39). It is also important to note that the ligand could be recognized by functionally overlapping innate immune responses. For example, the L412F SNP was not linked to infection by the hepatitis C virus (43), perhaps because HCV RNA is also recognized by the cytoplasmic RNA sensor, RIG-I (44). All of these complexities highlight the need for cells to carefully regulate both the expression as well as the activity of TLR3. In this vein it is likely that the amount of secreted protein in transiently transfected cells may not be linearly related to the levels needed to detect TLR3 activation of reporter production.
The cell-associated T3ECD or truncations derived from T3ECD accumulated to a lower level than their secreted counterparts. The levels of the variants of full-length TLR3 also accumulated to different levels in highly reproducible manner. These results suggest that TLR3 is actively degraded within the cell and that a defect in cell surface localization is one factor in TLR3 turnover. TLR3 has been localized to pericentriolar cytoplasmic bodies called aggresomes that are linked to proteolytic degradation (45). Because the N-terminal 10–11 LRRs of the TLR3 ectodomain accumulated better than the WT T3ECD, the signal for degradation exists between LRR12 and the C terminus of the TLR3 ectodomain. Because all of the truncations contained N- and C-terminal caps, the degradation signal exists within the LRRs of the ectodomain.
Bell et al. (46) proposed that local modifications within the LRRs could confer specialized functions. We determined that the nine-amino acid Loop 2 is required for TLR3 secretion, and the location of P554S within Loop 2 suggests that improper localization may be a factor in herpes simplex virus-1 pathologies. The eight-amino acid Loop 1 acts as a part of a larger element within LRR11 and 12 that negatively regulates secretion (Fig. 8). However, because T3ECD can secrete and N-L12 cannot, some portion of the T3ECD can overcome the negative effect of Loop 1.
Interestingly, the Δ64 isoform may have removed multiple signals linked to cell surface expression of TLR3. The sequence contains Loop 1 and the F303S SNP. Although the removal of Loop 1 increased the secretion of truncated TLR3 ectodomain, the lack of the sequence in Δ64 had the puzzling effect of decreasing secretion of the ectodomain. Finally the absence of these 64 residues decreased its ability to co-immunoprecipitate with Unc93b1 (Fig. 7). This region of the TLR3 ectodomain deserves more detailed analysis to elucidate whether all of the effects are linked or function independently.
Unc93b1 has been reported to interact with the transmembrane domain of TLRs to regulate the trafficking of TLR7 and TLR9 (30, 31). Given that Unc93b1 regulates TLR3 activity and can co-immunoprecipitation with TLR3, we expect a similar interaction between the transmembrane domain of TLR3 and Unc93b1. The interaction between Unc93b1 and the TLR3 ectodomain was unexpected. At this time, we cannot discern whether this interaction is direct or indirect. We have attempted to co-immunoprecipitate T3ECD with myc-tagged Unc93b1 but failed to detect T3ECD.3 This may be due to the interaction not being robust in the absence of the transmembrane domain or that appropriate conditions need to be identified. Nevertheless, the information in this work should be relevant to understanding the defects in several human SNPs, a human TLR3 isoform, and potentially the coordinated regulation of TLR3, -7, and -9 by Unc93b1.
Supplementary Material
Acknowledgments
We are grateful for the helpful discussions with Linda Wu, R. Goldschmidt at Centocor, and members of the Kao laboratory at Indiana University. We thank L. Kao for editing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant RAI075015A (to R. Q. and C. T. R.-K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S3.
R. Qi, unpublished results.
- TLR
- Toll-like receptor
- TIR
- Toll-interleukin-1 receptor
- ECD
- ectodomain
- T3ECD
- TLR3 ectodomain
- SNP
- single nucleotide polymorphism
- LRR
- leucine-rich repeat
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MMP
- matrix metallopeptidase.
REFERENCES
- 1.Kumar H., Kawai T., Akira S. (2009) Biochem. Biophys. Res. Commun. 388, 621–625 [DOI] [PubMed] [Google Scholar]
- 2.Barton G. M., Kagan J. C. (2009) Nat. Rev. Immunol. 9, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaturvedi A., Pierce S. K. (2009) Traffic 10, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGettrick A. F., O'Neill L. A. (2010) Curr. Opin. Immunol. 22, 20–27 [DOI] [PubMed] [Google Scholar]
- 5.Bell J. K., Botos I., Hall P. R., Askins J., Shiloach J., Segal D. M., Davies D. R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10976–10980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choe J., Kelker M. S., Wilson I. A. (2005) Science 309, 581–585 [DOI] [PubMed] [Google Scholar]
- 7.Ranjith-Kumar C. T., Miller W., Xiong J., Russell W. K., Lamb R., Santos J., Duffy K. E., Cleveland L., Park M., Bhardwaj K., Wu Z., Russell D. H., Sarisky R. T., Mbow M. L., Kao C. C. (2007) J. Biol. Chem. 282, 7668–7678 [DOI] [PubMed] [Google Scholar]
- 8.Sun J., Duffy K. E., Ranjith-Kumar C. T., Xiong J., Lamb R. J., Santos J., Masarapu H., Cunningham M., Holzenburg A., Sarisky R. T., Mbow M. L., Kao C. (2006) J. Biol. Chem. 281, 11144–11151 [DOI] [PubMed] [Google Scholar]
- 9.Bell J. K., Askins J., Hall P. R., Davies D. R., Segal D. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 8792–8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu L., Botos I., Wang Y., Leonard J. N., Shiloach J., Segal D. M., Davies D. R. (2008) Science 320, 379–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schröder N. W., Diterich I., Zinke A., Eckert J., Draing C., von Baehr V., Hassler D., Priem S., Hahn K., Michelsen K. S., Hartung T., Burmester G. R., Göbel U. B., Hermann C., Schumann R. R. (2005) J. Immunol. 175, 2534–2540 [DOI] [PubMed] [Google Scholar]
- 12.Schröder N. W., Meister D., Wolff V., Christan C., Kaner D., Haban V., Purucker P., Hermann C., Moter A., Göbel U. B., Schumann R. R. (2005) Genes Immun. 6, 448–451 [DOI] [PubMed] [Google Scholar]
- 13.Lorenz E., Mira J. P., Cornish K. L., Arbour N. C., Schwartz D. A. (2000) Infect. Immun. 68, 6398–6401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tantisira K., Klimecki W. T., Lazarus R., Palmer L. J., Raby B. A., Kwiatkowski D. J., Silverman E., Vercelli D., Martinez F. D., Weiss S. T. (2004) Genes Immun. 5, 343–346 [DOI] [PubMed] [Google Scholar]
- 15.Redecke V., Häcker H., Datta S. K., Fermin A., Pitha P. M., Broide D. H., Raz E. (2004) J. Immunol. 172, 2739–2743 [DOI] [PubMed] [Google Scholar]
- 16.Cook D. N., Pisetsky D. S., Schwartz D. A. (2004) Nat. Immunol. 5, 975–979 [DOI] [PubMed] [Google Scholar]
- 17.Misch E. A., Hawn T. R. (2008) Clin. Sci. 114, 347–360 [DOI] [PubMed] [Google Scholar]
- 18.Dhiman N., Ovsyannikova I. G., Cunningham J. M., Vierkant R. A., Kennedy R. B., Pankratz V. S., Poland G. A., Jacobson R. M. (2007) J. Infect. Dis. 195, 21–29 [DOI] [PubMed] [Google Scholar]
- 19.Zhang S. Y., Jouanguy E., Ugolini S., Smahi A., Elain G., Romero P., Segal D., Sancho-Shimizu V., Lorenzo L., Puel A., Picard C., Chapgier A., Plancoulaine S., Titeux M., Cognet C., von Bernuth H., Ku C. L., Casrouge A., Zhang X. X., Barreiro L., Leonard J., Hamilton C., Lebon P., Héron B., Vallée L., Quintana-Murci L., Hovnanian A., Rozenberg F., Vivier E., Geissmann F., Tardieu M., Abel L., Casanova J. L. (2007) Science 317, 1522–1527 [DOI] [PubMed] [Google Scholar]
- 20.Hidaka F., Matsuo S., Muta T., Takeshige K., Mizukami T., Nunoi H. (2006) Clin. Immunol. 119, 188–194 [DOI] [PubMed] [Google Scholar]
- 21.Ranjith-Kumar C. T., Miller W., Sun J., Xiong J., Santos J., Yarbrough I., Lamb R. J., Mills J., Duffy K. E., Hoose S., Cunningham M., Holzenburg A., Mbow M. L., Sarisky R. T., Kao C. C. (2007) J. Biol. Chem. 282, 17696–17705 [DOI] [PubMed] [Google Scholar]
- 22.Yang Z., Stratton C., Francis P. J., Kleinman M. E., Tan P. L., Gibbs D., Tong Z., Chen H., Constantine R., Yang X., Chen Y., Zeng J., Davey L., Ma X., Hau V. S., Wang C., Harmon J., Buehler J., Pearson E., Patel S., Kaminoh Y., Watkins S., Luo L., Zabriskie N. A., Bernstein P. S., Cho W., Schwager A., Hinton D. R., Klein M. L., Hamon S. C., Simmons E., Yu B., Campochiaro B., Sunness J. S., Campochiaro P., Jorde L., Parmigiani G., Zack D. J., Katsanis N., Ambati J., Zhang K. (2008) N. Engl. J. Med. 359, 1456–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang E., Shin J. S., Kim H., Park H. W., Kim M. H., Kim S. J., Choi I. H. (2004) Yonsei Med. J. 45, 359–361 [DOI] [PubMed] [Google Scholar]
- 24.Barton G. M., Kagan J. C., Medzhitov R. (2006) Nat. Immunol. 7, 49–56 [DOI] [PubMed] [Google Scholar]
- 25.Park B., Brinkmann M. M., Spooner E., Lee C. C., Kim Y. M., Ploegh H. L. (2008) Nat. Immunol. 9, 1407–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto M., Funami K., Tanabe M., Oshiumi H., Shingai M., Seto Y., Yamamoto A., Seya T. (2003) J. Immunol. 171, 3154–3162 [DOI] [PubMed] [Google Scholar]
- 27.Blasius A. L., Beutler B. (2010) Immunity 32, 305–315 [DOI] [PubMed] [Google Scholar]
- 28.Funami K., Matsumoto M., Oshiumi H., Akazawa T., Yamamoto A., Seya T. (2004) Int. Immunol. 16, 1143–1154 [DOI] [PubMed] [Google Scholar]
- 29.Nishiya T., Kajita E., Miwa S., Defranco A. L. (2005) J. Biol. Chem. 280, 37107–37117 [DOI] [PubMed] [Google Scholar]
- 30.Brinkmann M. M., Spooner E., Hoebe K., Beutler B., Ploegh H. L., Kim Y. M. (2007) J. Cell Biol. 177, 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y. M., Brinkmann M. M., Paquet M. E., Ploegh H. L. (2008) Nature 452, 234–238 [DOI] [PubMed] [Google Scholar]
- 32.Tabeta K., Hoebe K., Janssen E. M., Du X., Georgel P., Crozat K., Mudd S., Mann N., Sovath S., Goode J., Shamel L., Herskovits A. A., Portnoy D. A., Cooke M., Tarantino L. M., Wiltshire T., Steinberg B. E., Grinstein S., Beutler B. (2006) Nat. Immunol. 7, 156–164 [DOI] [PubMed] [Google Scholar]
- 33.Casrouge A., Zhang S. Y., Eidenschenk C., Jouanguy E., Puel A., Yang K., Alcais A., Picard C., Mahfoufi N., Nicolas N., Lorenzo L., Plancoulaine S., Sénéchal B., Geissmann F., Tabeta K., Hoebe K., Du X., Miller R. L., Héron B., Mignot C., de Villemeur T. B., Lebon P., Dulac O., Rozenberg F., Beutler B., Tardieu M., Abel L., Casanova J. L. (2006) Science 314, 308–312 [DOI] [PubMed] [Google Scholar]
- 34.Koehn J., Huesken D., Jaritz M., Rot A., Zurini M., Dwertmann A., Beutler B., Korthäuer U. (2007) Hum. Immunol. 68, 871–878 [DOI] [PubMed] [Google Scholar]
- 35.Rodgers U. R., Kevorkian L., Surridge A. K., Waters J. G., Swingler T. E., Culley K., Illman S., Lohi J., Parker A. E., Clark I. M. (2009) Matrix Biol. 28, 263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukui R., Saitoh S., Matsumoto F., Kozuka-Hata H., Oyama M., Tabeta K., Beutler B., Miyake K. (2009) J. Exp. Med. 206, 1339–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Limmon G. V., Arredouani M., McCann K. L., Corn Minor R. A., Kobzik L., Imani F. (2008) FASEB J. 22, 159–167 [DOI] [PubMed] [Google Scholar]
- 38.Wang N., Liang Y., Devaraj S., Wang J., Lemon S. M., Li K. (2009) J. Virol. 83, 9824–9834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ménager P., Roux P., Mégret F., Bourgeois J. P., Le Sourd A. M., Danckaert A., Lafage M., Préhaud C., Lafon M. (2009) PLoS Pathog. 5, e1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonard J. N., Ghirlando R., Askins J., Bell J. K., Margulies D. H., Davies D. R., Segal D. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duffy K. E., Lamb R. J., San Mateo L. R., Jordan J. L., Canziani G., Brigham-Burke M., Korteweg J., Cunningham M., Beck H. S., Carton J., Giles-Komar J., Duchala C., Sarisky R. T., Mbow M. L. (2007) Cell. Immunol. 248, 103–114 [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto M., Kikkawa S., Kohase M., Miyake K., Seya T. (2002) Biochem. Biophys. Res. Commun. 293, 1364–1369 [DOI] [PubMed] [Google Scholar]
- 43.Askar E., Bregadze R., Mertens J., Schweyer S., Rosenberger A., Ramadori G., Mihm S. (2009) J. Med. Virol. 81, 1204–1211 [DOI] [PubMed] [Google Scholar]
- 44.Sumpter R., Jr., Loo Y. M., Foy E., Li K., Yoneyama M., Fujita T., Lemon S. M., Gale M., Jr. (2005) J. Virol. 79, 2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wileman T. (2007) Annu. Rev. Microbiol. 61, 149–167 [DOI] [PubMed] [Google Scholar]
- 46.Bell J. K., Botos I., Hall P. R., Askins J., Shiloach J., Davies D. R., Segal D. M. (2006) J. Endotoxin Res. 12, 375–378 [DOI] [PubMed] [Google Scholar]
- 47.Ranjith-Kumar C. T., Duffy K. E., Jordan J. L., Eaton-Bassiri A., Vaughan R., Hoose S. A., Lamb R. J., Sarisky R. T., Kao C. C. (2008) Mol. Cell. Biol. 28, 4507–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.