

Abstract

Full details of the total synthesis of phostriecin (2), the assignment of its relative and absolute stereochemistry, and the resultant structural reassignment of the natural product previously represented as sultriecin (1), a phosphate versus sulfate monoester, are detailed. Studies with authentic material confirmed that phostriecin, but not sultriecin, is an effective and selective inhibitor of protein phosphatase 2A (PP2A) defining a mechanism of action responsible for its antitumor activity. The extension of the studies to the synthesis and evaluation of a series of key synthetic analogues is disclosed that highlights the importance of the natural product phosphate monoester (vs sulfate or free alcohol, inactive and >250-fold), the α,β-unsaturated lactone (12-fold), and the hydrophobic Z,Z,E-triene tail (C12–C22, ca. 200-fold) including the unique importance of its unsaturation (50-fold, and no longer PP2A selective).
Introduction
Protein phosphatases are key enzymes in the regulation of many biological processes. Their function is to catalyze the dephosphorylation of hydroxyl-containing amino acid side chains of proteins. Protein phosphatase 2A (PP2A) is a major serine/threonine phosphatase, and plays an important role in many cell functions including cell growth, replication, mobility, and signal transduction. Because of the importance of these processes in human diseases and to further understand the pathways by which protein phosphorylation affects cells, there is a need for the development of compounds that selectively inhibit protein phosphatases including PP2A.1
Sultriecin (1)2 was identified as an antitumor antibiotic isolated from Streptomyces roseiscleroticus No. L827-7 and was an early member of a family of natural products3 that now include fostriecin (3),4–6 cytostatin (4),7 phospholine (5, phoslactomycin B),8 the leustroducsins (6),9 and the phoslactomycins (7) (Figure 1).10 Common structural features include an electrophilic α,β-unsaturated lactone and hydrophobic Z,Z,E-triene capping the ends of an extended structure that contains a central functionalized 1,3-diol. Unique to 1 and in contrast to the more recent members of the family that contain phosphate monoesters, sultriecin was assigned as a C9 sulfate ester at the time of its early disclosure. Sultriecin displayed moderate broad spectrum antifungal activity in vitro, moderate in vitro cytotoxic activity against human and murine tumor cell lines, and potent in vivo antitumor activity against P388 leukemia and B16 melanoma.2
FIGURE 1.
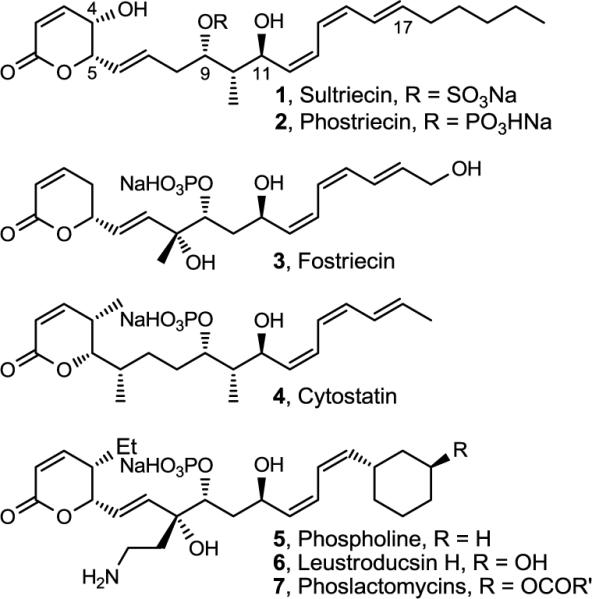
Natural product structures
In efforts on the synthesis and evaluation of members of this class of antitumor agents that have since been shown to act as PP2A inhibitors, we reported total syntheses of 34c and 4,7c the establishment of their relative and absolute configuration,5,7 and the preparation of a series of analogues used to define structural features that are key to their potent and unusually selective inhibition of PP2A.6,7 Based on its functional biological activity and structural similarity to fostriecin (3) and cytostatin (4), we anticipated that sultriecin (1) would also be a selective PP2A inhibitor, albeit with a sulfate versus phosphate interaction with the enzymatic bimetallic catalytic core. Pertinent to the work detailed herein and because of the sultriecin disclosure, we had previously examined and shown that the corresponding sulfate ester of cytostatin (sulfocytostatin) was unexpectedly inactive as a PP2A inhibitor.7c Herein, we report a full account of the first total synthesis and stereochemical determination of sultriecin (1), the unanticipated structural reassignment of the natural product as phosphate monoester 2 (renamed phostriecin), establishment of its biological activity as an inhibitor of PP2A, and the synthesis and biological evaluation of key analogues.11
Stereochemical Assignment
The structure of sultriecin was disclosed without definition of its relative or absolute stereochemistry.2 Based on its biological and structural similarity to fostriecin and cytostatin as well as its reported spectral data, we assigned the (4S,5S,9S,10S,11S)-stereochemistry to sultriecin. The syn C4–C5 stereochemistry assignment was supported by the observed H4–H5 coupling constant in the reported 1H NMR of 1 (J = 2.9 Hz). This coupling constant is similar to that observed for cytostatin (J = 2.7 Hz), and a similar known lactone displays a syn H4–H5 coupling constant (J = 3.1 Hz) distinct from that of the corresponding anti-4,5-disubstituted lactone (J = 8.7 Hz).12 The H10–H11 coupling constant (J = 10.2 Hz) reported for sultriecin is indicative of an anti relationship. It is in particularly good agreement with coupling constants observed for fostriecin (J = 9.6 vs 3.7 Hz) and cytostatin (J = 9.4 Hz), and supports the existence of an intramolecular H-bond between the C11-OH and putative C9 sulfate resulting in a rigid twist-boat cyclic structure as found in 3 and 4 (Figure 2).
FIGURE 2.
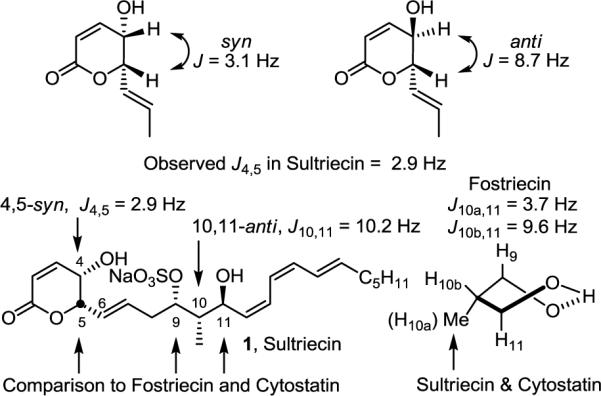
Assignment of relative and absolute stereochemistry
Synthetic Approach
A convergent route to sultriecin was designed that incorporated the flexibility to provide access to analogues and to allow preparation of any diastereomer in the event that the initial stereochemical assignment proved incorrect. The approach relies on a late-stage one-step installation of the Z,Z,E-triene via chelation-controlled addition of the cuprate derived from 9 to aldehyde 8. We envisioned the protected lactol 8 arising from an oxidative ring expansion of an α-hydroxyfuran that would be accessed from the coupling of alkyne 10 with 2-furoyl chloride followed by asymmetric (R)-CBS ketone reduction to set the C5 stereochemistry and subsequent stereoselective alkyne reduction (Figure 3).
FIGURE 3.
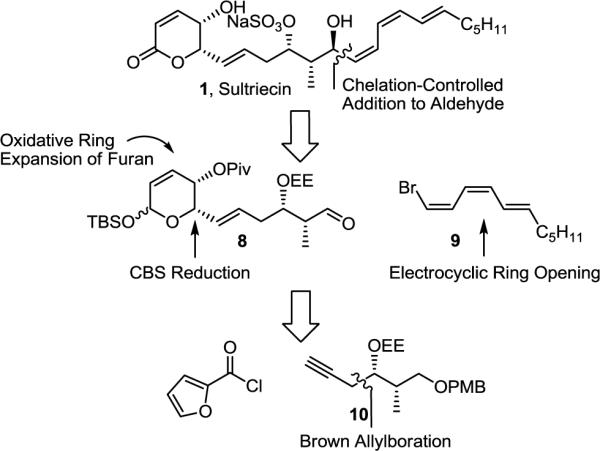
Retrosynthetic plan for sultriecin (1)
Results and Discussion
Synthesis of C1–C11
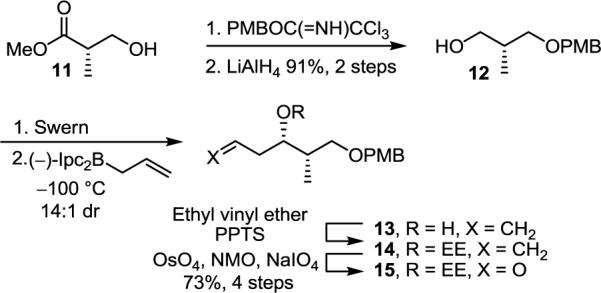
The synthesis began with protection of methyl (S)-3-hydroxy-2-methylpropionate (11) as a PMB ether followed by its reduction to alcohol 12 (PMBOC=(NH)CCl3, camphorsulfonic acid, CH2Cl2, 25°C, 12 h; LiAlH4, Et2O, 0 to 25°C, 12 h, 91%, 2 steps) (Scheme 1). After oxidation of 12 to the corresponding aldehyde (DMSO, oxalyl chloride, Et3N, CH2Cl2, −78 to 0°C, 2 h), asymmetric allylboration (allyldiisopinocampheylborane, −100°C, 4 h; NaOH, H2O2, 25°C, 16 h, 14:1 dr)13 gave alcohol 13 that was protected as the ethoxyethyl acetal (14) (ethyl vinyl ether, PPTS, CH2Cl2, 25°C, 2 h). The ethoxyethyl acetal (EE) protecting group was chosen due to its ability to direct a subsequent chelation-controlled cuprate addition as well as its unique ability to be removed under mildly acidic conditions in the presence of the labile triene and sensitive allylic alcohol despite the complicating diastereomeric mixture it introduces. Oxidative cleavage of the double bond in 14 (OsO4, NaIO4, NMO, THF, H2O, 25°C, 18 h, 73%, 4 steps) provided aldehyde 15.
SCHEME 1.
In the course of our efforts, several routes to the furyl alcohol 19 were investigated. Initially and although not pursued in depth, hydroboration of model alkynes 16 followed by transmetallation to zinc and asymmetric addition to 2-furaldehyde with (−)-MIB [(−)-MIB = (2S)-3-exo-(morpholino)isoborneol] as chiral ligand14 were examined but failed to provide the desired addition products. Similarly, preliminary efforts on diastereoselective alkyne addition of 16 to 2-furaldehyde following established procedures15–17 were also unsuccessful (Scheme 2).
SCHEME 2.
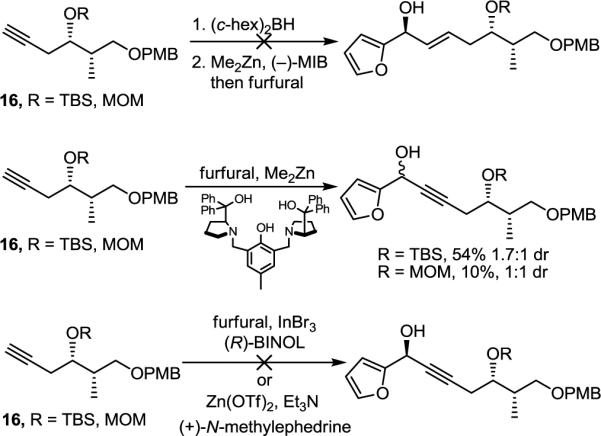
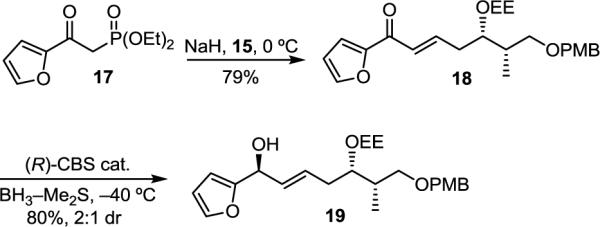
We also examined methods of diastereoselective reduction of ketone 18, synthesized by Horner–Wadsworth–Emmons reaction of phosphonate 17 with aldehyde 15. However, hydrogenation in the presence of ketone selective chiral catalysts (e.g., (S)-BINAP–RuCl2)18 as well as attempted reduction with chiral aluminum hydride reagents (e.g., (S)-BINAL–H)19 reduced the olefin in preference to or along with the ketone. Reduction of 18 with (R)-Me-CBS (1.5 equiv) and BH3–Me2S (4 equiv) gave exclusive ketone reduction in good yield (80%), but with only 2:1 diastereoselectivity (Scheme 3).
SCHEME 3.
With a recognition that higher diastereoselectivity could be obtained by replacing the alkene in 18 with an alkyne providing a smaller substituent and affording a better stereodifferentiation with respect to the furan, aldehyde 15 was subjected to the Corey–Fuchs homologation (CBr4, PPh3, CH2Cl2, 0°C, 10 min, 71%; n-BuLi, THF, −78 to 25°C, 16 h, 93%)20 giving alkyne 10 (Scheme 4). Coupling of 10 with 2-furoyl chloride (Pd(PPh3)2Cl2, CuI, Et3N, 25°C, 24 h, 85%)21 gave ketone 21 that was subjected to asymmetric reduction (methyl-(R)-CBS-oxazaborolidine, BH3–Me2S, THF, −40°C, 3 h) giving the desired alcohol diastereomer 22 with 12.5:1 selectivity and setting the C5 stereochemistry.22 Olefin 19 was subsequently obtained by stereoselective reduction of the alkyne (LiAlH4, THF, 0 to 25°C, 24 h, 84%, 2 steps) (Scheme 4).23
SCHEME 4.

Lactol 23 was obtained as a mixture of anomers following oxidative ring expansion of 19 (NBS, NaHCO3, NaOAc, THF/H2O, 0°C, 1 h).24 Installation of a range of lactol protecting groups was examined including Boc, Piv, Bz, and Me groups. However, 23 and its corresponding protected products proved to be especially sensitive to both acidic and basic conditions, and attempts at installation of these protecting groups largely failed. TBS protection of the lactol under mild conditions (TBSCl, AgNO3, pyr, CH2Cl2, 25 °C, 15 min) was most effective,25 providing an inconsequential mixture of inseparable anomers. Due to its instability to silica gel chromatography, diastereoselective 1,2-reduction of the enone was performed on crude 24 (LiAlH4, Et2O, −60°C, 2.5 h, dr >30:1, 51–64% for 3 steps) (Scheme 4). Alternative reducing agents including NaBH4–CeCl3, and DIBAL produced varying amounts of the syn isomer as a minor product, and we were not successful in identifying a reducing agent that would selectively or exclusively provide this syn isomer. The stereochemistry of the C4 alcohol in 25 was necessarily inverted and directly protected as its pivalate ester using the Mitsunobu reaction (DIAD/PPh3, pivalic acid, THF, 0 to 25 °C, 75%).26 Aldehyde 8 was obtained following PMB removal (DDQ, CH2Cl2/H2O, 25 °C, 1 h, 74%) and oxidation of alcohol 27 (DMP, CH2Cl2, 25 °C, 1 h, 92%).27
Synthesis of C12–C22
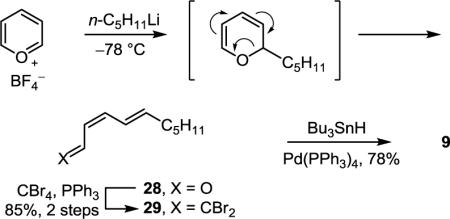
With aldehyde 8 in hand, focus turned toward synthesis of the sensitive triene portion of the molecule. Following an approach adopted in our total synthesis of cytostatin7c and originally introduced by Taylor,28 treatment of pyrilium tetrafluoroborate with n-pentyllithium (THF, −78 °C, 4 h) gave, after room temperature electrocyclic ring opening of the adduct, aldehyde 28 as a single isomer (Eq 1). Because of its instability to storage and its volatility, aldehyde 28 was immediately converted by dibromoolefination (CBr4, PPh3, Et3N, CH2Cl2, 0 °C, 15 min, 85% for 2 steps)20 to 29 which could be stored as a solution in Et2O at 4 °C for several weeks. Unstable Z,Z,E-bromotriene 9 was produced immediately before use by selective E-bromide reduction (Bu3SnH, Pd(PPh3)4, Et2O, 0 °C, 45 min, 78%).29
 |
(1) |
Total Synthesis of Sultriecin and Phostriecin
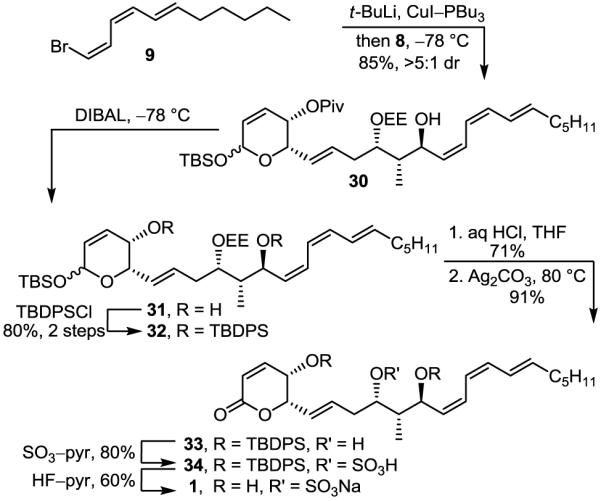
Incorporation of the sensitive Z,Z,E-triene required conversion of 9 to the corresponding cuprate (t-BuLi, Et2O, −78 °C, 1 h; CuI–PBu3, Et2O, −78 °C, 15 min) followed by slow addition of aldehyde 8 (Et2O, −78 °C, 1 h, 85%, >5:1 dr),7c,30 providing 30 derived from a chelation-controlled addition to the aldehyde (Scheme 5). Removal of the pivalate ester (DIBAL, CH2Cl2, −78 °C, 2 h) followed by silylation of the secondary alcohols of 31 (TBDPSCl, AgNO3, pyr/CH2Cl2, 25 °C, 16 h, 80%, 2 steps) afforded 32 that was treated with dilute HCl (THF/H2O, 25 °C, 12 h, 71%) to simultaneously and selectively remove the EE and TBS protecting groups. The resulting lactol was selectively oxidized to give lactone 33 (Ag2CO3–Celite, benzene, 80 °C, 1.5 h, 91%). Sulfate ester introduction (SO3–pyr, THF, 25 °C, 10 min, 80%) followed by desilylation (HF–pyr, pyr/THF, 25 °C, 60%) gave 1, which did not match the spectroscopic (1H NMR, 13C NMR, IR) or physical characteristics (TLC, [α]D, solubility, stability to silica gel) of the natural product.
SCHEME 5.
Synthesis of sultriecin (1)
Several possibilities for this non-correlation with the natural product were envisioned including the accuracy of our stereochemical assignment as well as spectroscopic perturbations derived from the protonation state or salt form of the sulfate. To confirm the C9/C11 anti relationship of synthetic 1, 33 was deprotected (HF–pyr, pyr/THF, 25 °C, 12 h, 90%),31 and the resultant 1,3-diol 35 was converted to acetonide 36 (2,2-dimethoxypropane, TsOH, THF, 25 °C, 45 min, 52%) (Eq 2). The 13C NMR chemical shifts observed for the acetonide carbons of 36 (CD3CN, δ 24.6, 25.0, 101.3) were found to be indicative of the anti-1,3-diol acetonide as desired and anticipated.32
 |
(2) |
To address potential issues of the protonation state and sulfate counterion identity, naturally-derived sultriecin33 was subjected to the conditions of the last step of the synthesis of 1 (HF–pyr, pyr/THF, aq NaHCO3 quench, silica gel chromatography). This action, however, resulted only in decomposition of the natural material indicating that there was a more fundamental distinction in the synthetic (stable) and natural (unstable) materials.
The most apparent difference observed in the 1H NMR of synthetic 1 and the natural product was the chemical shift (CD3OD, δ 4.82 vs 4.64) and multiplicity (ddd, J = 8.4, 6.0, 1.8 Hz vs dddd, J = 9.6, 7.8, 7.2, 1.8 Hz) of C9-H adjacent to the putative sulfate ester (Figure 4). Careful examination of the 1H NMR spectra of synthetic 1 and natural sultriecin revealed that the natural product H9 signal exhibited an additional long range coupling (JP-H9 = 7.8 Hz) that would be characteristic of a phosphate (monoisotopic mass = 492.1889) versus sulfate ester (monoisotopic mass = 492.1794). In addition to the potential ambiguity introduced by the closely related molecular weights, the natural product was reported to provide a negative Hanes test used to characterize a phosphate (vs sulfate) monoester, and exposure of the natural product to a sulfatase resulted in putative sulfate monoester hydrolysis.2 Consequently, phosphate monoester 2 was targeted for synthesis (Scheme 6). Alcohol 33 was phosphorylated (i-Pr2NP(OFm)2, tetrazole, CH2Cl2/CH3CN, 25 °C, 1 h; H2O2, 15 min, 96%)7 to give 37 that was desilylated (HF–pyr, pyr/THF, 25 °C, 4 d). Removal of the fluorenylmethyl groups (Fm) in 38 (Et3N, CH3CN, 25 °C, 16 h; Dowex Na+, 63% for 2 steps) unmasked the phosphate giving 2 (renamed phostriecin), that proved identical to the reported properties of 1 as well as a sample of the natural product (1H NMR, 31P NMR, [α]D, TLC, HPLC, HRMS), the latter of which also displayed a 31P NMR signal like that found with synthetic 2 (δ 3.4, CD3OD). Thus, the total synthesis of 2 and its correlation with authentic natural material requires a structural reassignment of the natural product formerly known as sultriecin.
FIGURE 4.
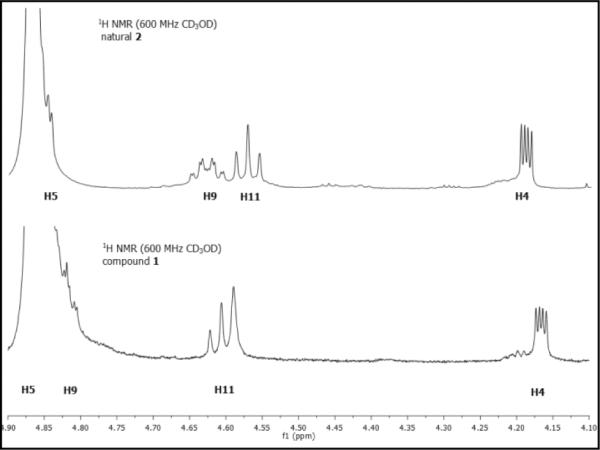
Diagnostic region of 1H NMR of 1 and 2
SCHEME 6.
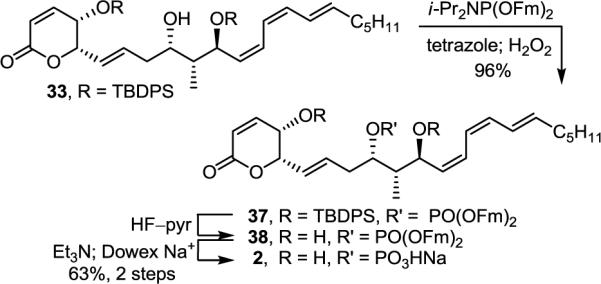
Synthesis of phostriecin (2)
Synthesis of Key Analogues
With a synthesis and structural reassignment of the natural product in hand, we extended the efforts to the preparation of a series of analogues designed to probe and identify key features necessary for potent and selective PP2A inhibition. To address the importance of the α,β-unsaturated lactone and the presence of a phosphate rather than sulfate ester for PP2A inhibition, truncated analogues 45 and 48 were targeted (Scheme 7).
SCHEME 7.
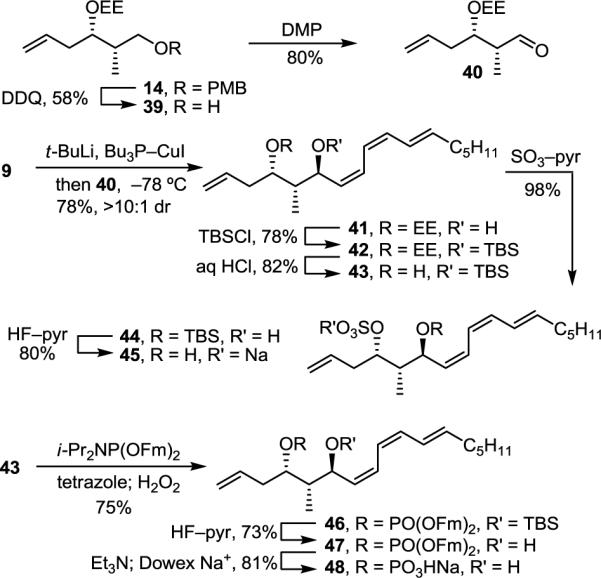
Synthesis of analogues 45 and 48
Deprotection of PMB ether 14 (DDQ, CH2Cl2/H2O, 25 °C, 1 h, 58%) gave alcohol 39 that was oxidized to aldehyde 40 (DMP, CH2Cl2, 1 h, 80%) (Scheme 7). Addition of the cuprate derived from 9 (Et2O, −78 °C, 1 h, 78%, >10:1 dr) to 40 provided alcohol 41 that was subsequently protected (TBSCl, AgNO3, pyr/CH2Cl2, 25 °C, 30 min, 78%). The EE acetal of 42 was selectively removed with dilute HCl (THF/H2O, 25 °C, 2.5 h, 82%) to yield alcohol 43. Sulfate installation (SO3–pyr, THF, 25 °C, 10 min, 98%) and desilylation (HF–pyr, pyr/THF, 25 °C, 3 h, 80%) provided sulfate 45, whereas phosphorylation of 43 (i-Pr2NP(OFm)2, tetrazole, CH2Cl2/CH3CN, 25 °C, 1 h; H2O2, 15 min, 75%) followed by desilylation (HF–pyr, pyr/THF, 25 °C, 18 h, 73%), and fluorenylmethyl removal (Et3N, CH3CN, 25 °C, 16 h; Dowex Na+, 81%) provided the corresponding phosphate monoester 48 lacking the unsaturated lactone unit.
The importance of the Z,Z,E-triene tail as well as the C11 hydroxyl group was investigated with the synthesis of 55 and 58 (Scheme 8). Replacement of the pivalate ester of 26 with a TBDPS protecting group (LiAlH4, THF, 0 °C, 2 h; TBDPSCl, AgNO3, pyr/CH2Cl2, 16 h) followed by EE and TBS deprotection (aq HCl, THF, 12 h, 52% for 3 steps) gave lactol 51 that was oxidized to lactone 52 (Ag2CO3–Celite, benzene, 80 °C, 1.5 h, 59%). Compound 52 was phosphorylated (i-Pr2NP(OFm)2, tetrazole, CH2Cl2/CH3CN, 25 °C, 1 h; H2O2, 15 min, 95%) to give protected phosphate 53. In the course of these studies, we found that reversing the final deprotection steps conducting first the fluorenylmethyl removal followed by desilylation of the crude phosphate with TAS–F34 often gave higher yields and cleaner reactions than the order and final HF–pyridine conditions typically employed. By using this method, 55 was synthesized (Et3N, CH3CN, 25 °C, 16 h; TAS–F, CH3CN, 25 °C, 16 h, 67% for two steps). The PMB group of 53 was removed (DDQ, CH2Cl2/H2O, 25 °C, 1.5 h, 70%), and 56 was fully deprotected under the same conditions (Et3N, CH3CN, 25 °C, 16 h; TAS–F, CH3CN, 25 °C, 16 h, 84% for two steps) to give 58.
SCHEME 8.

Synthesis of analogues 55 and 58
The storage instability of fostriecin led to a premature discontinuation of its clinical trials,35 and a possible source of this instability is the sensitive Z,Z,E-triene. Thus, analogues that replace the triene with more stable hydrophobic groups are of special interest. The synthesis of one such key analogue 65, bearing a fully saturated variant of the Z,Z,E-triene, began with chelation-controlled addition of the cuprate derived from 1-bromoundecane to aldehyde 8 (Et2O, −78 °C, 1 h, 71%, >19:1 dr) (Scheme 9). Notably, the diastereoselectivity of the addition and the remarkable overall clean conversion with this substrate are the best we have observed among the substrates that we have examined. Pivalate removal (LiAlH4, THF, 0 °C, 3 h) was followed by silylation of the secondary alcohols of 60 (TBDPSCl, AgNO3, pyr/CH2Cl2, 25 °C, 16 h) to give 61. Selective TBS and EE removal (aq HCl, THF, 25 °C, 12 h, 48% for 3 steps) resulted in lactol 62 that was oxidized to lactone 63 (Ag2CO3–Celite, benzene, 80 °C, 1.5 h, 84%). The alcohol of 63 was phosphorylated as before (i-Pr2NP(OFm)2, tetrazole, CH2Cl2/CH3CN, 25 °C, 1 h; H2O2, 15 min, 90%). At this time, we also found that TAS–F is sufficiently basic to remove the fluorenylmethyl groups on the phosphate, and that 64 could be fully deprotected in one step using this reagent (TAS–F, CH3CN, 25 °C, 3 d, 73%).
SCHEME 9.
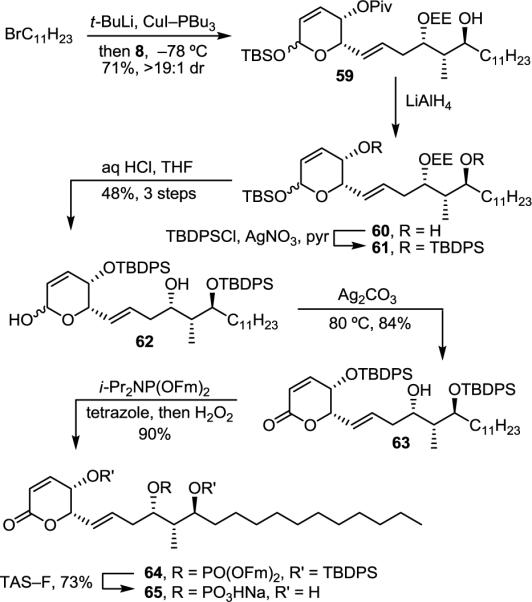
Synthesis of analogue 65
Biological Evaluation
The natural products and their key analogues were examined for phosphatase inhibition against native PP2Ac (Crassostrea virginica), rhPP1cα, and rhPP5c using phosphohistone1 as substrate (Figure 5). Sultriecin (1) was devoid of PP2A inhibitory activity indicating that a sulfate is a poor phosphate substitute for coordination to the bimetallic core of the enzyme. This is in good agreement with our prior observation that the sulfate derivative of cytostatin (sulfocytostatin) showed no activity against all three phosphatases as well.7c Although this latter observation was viewed as unexpected and unusual at the time it was made, the result now is in full agreement with the sultriecin observations. The actual natural product, phostriecin (2), exhibited PP2A inhibitory activity as anticipated, albeit at a level that is less potent and selective than either fostriecin (3, 170-fold) or cytostatin (4, 15-fold). Dephosphophostriecin (35) was inactive against all three phosphatases confirming the importance of the phosphate for inhibitory activity. Compound 48, lacking the α,β-unsaturated lactone, was significantly less potent than the natural product, but still showed moderate inhibition of PP2A (4.5 μM) and good selectivity. Notably, this compound lacking the electrophile responsible for reversible conjugate addition of the PP2A active site Cys2696 proved to be only 12-fold less active than phostriecin itself and still selective for PP2A. Although its potency is 1000-fold lower than fostriecin and 100-fold lower than cytostatin, it maintains PP2A selectivity indicating that this portion of the natural products productively contributes to their properties. This is consistent with a prior observation made with fostriecin and a related set of key analogues6 reinforcing a generality to this observation. Compound 45, identical to 48 lacking the unsaturated lactone, but bearing a sulfate versus phosphate monoester was inactive providing a result also in line with present expectations. Also consistent with the importance of this portion of the molecule, removing the Z,Z,E-triene tail (C12–C22) providing 58 led to a 200-fold loss in activity, and its replacement with a PMB ether (55), albeit with removal of the important C11 secondary alcohol,7c resulted in a 100-fold loss in activity. Important in these comparisons is the observation that 48 is 15-fold more potent than 58 emphasizing the key role the C12–C22 tail plays in contributing to the properties of 2. Most surprising and most interesting, simply replacing the Z,Z,E-triene tail (C12–C22) with its saturated counterpart providing 65 lacking only the π-unsaturation not only lost potency (50-fold), but also lost selectivity for PP2A. Although the activity is modest, potentially masking more subtle distinctions, 65 was essentially equally active against PP2A, PP1, and PP5 indicating that the unsaturation in the C12–C22 tail contributes in a significant manner to the PP2A selectivity of this class of natural products.
FIGURE 5.

Protein phosphatase activity (IC50,μM)
Conclusion
Total syntheses of 1 and 2 unequivocally established the structural composition and stereochemical configuration of the natural product previously known as sultriecin (renamed phostriecin). Key steps include a CBS reduction to establish the lactone stereochemistry, oxidative ring expansion of an α-hydroxyfuran to access a pyran lactol precursor, and one-step installation of the sensitive Z,Z,E-triene unit using a chelation-controlled cuprate addition to an intermediate aldehyde. Studies with authentic material established that phostriecin, but not sultriecin, is an effective and selective inhibitor of protein phosphatase 2A (PP2A) defining a mechanism of action responsible for its antitumor activity. Examination of a series of synthetic analogues prepared by extending the route developed for 1 and 2 defined key structural features of the natural product contributing to the potency and selectivity of the PP2A inhibition.
Experimental Section
(2S,3S)-6-((tert-Butyldimethylsilyl)oxy)-2-((4S,5S,6S,E)-4-(1-ethoxyethoxy)-6-hydroxy-5-methylheptadec-1-en-1-yl)-3,6-dihydro-2H-pyran-3-yl Pivalate (59)
t-BuLi (1.7 M in pentane, 0.97 mL, 1.7 mmol) was slowly added to a degassed solution of 1-bromoundecane (0.22 g, 0.92 mmol) in Et2O (6.3 mL) under Ar with stirring at −78 °C under N2. After 1.5 h, a solution of CuI–PBu3 (0.14 g, 0.37 mmol) in 2.2 mL of Et2O was added at −78 °C and the mixture was stirred for 15 min. A solution of 8 (0.095 g, 0.19 mmol) in 5.3 mL of anhydrous Et2O was added slowly to the mixture down the side of the flask over 15 min at −78 °C. After 1 h, the reaction was quenched by addition of saturated aqueous NH4Cl/NH4OH (pH 8, 2 mL) and warmed to room temperature. The mixture was extracted with Et2O and the combined organic phases were washed with H2O, saturated aqueous NaCl, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography (SiO2 pretreated with 2% Et3N/hexanes, 10–20% EtOAc/hexanes gradient) to give 0.088 g (71%) of 59 as a yellow oil: [α]25D +65 (c 0.47, CHCl3); 1H NMR (C6D6, 400 MHz)δ5.87 (dd, J = 9.6, 5.6 Hz, 1H), 5.85–5.65 (m, 3H), 5.35 (d, J = 2.8 Hz, 1H), 5.02 (dd, J = 5.6, 2.4 Hz, 1H), 4.81 (q, J = 5.2 Hz, 1H), 4.64 (dd, J = 5.6, 2.4 Hz, 1H), 3.97 (td, J = 7.6, 2.8 Hz, 1H), 3.66–3.57 (m, 1H), 3.57–3.48 (m, 1H), 3.42–3.32 (m, 1H), 2.85 (d, J = 5.6 Hz, 1H), 2.60–2.50 (m, 1H), 2.42–2.32 (m, 1H), 1.80–1.66 (m, 2H), 1.67–1.57 (m, 1H), 1.50–1.24 (m, 18H), 1.27 (d, J = 5.2 Hz, 3H), 1.21 (s, 9H), 1.13 (t, J = 7.2 Hz, 3H), 0.96 (s, 9H), 0.90 (d, J = 6.8 Hz, 3H), 0.90 (t, J = 6.8 Hz, 3H), 0.17 (s, 3H), 0.11 (s, 3H); 13C NMR (C6D6, 150 MHz)δ177.7, 133.0, 130.4, 128.9, 124.1, 100.0, 89.4, 77.6, 73.0, 70.4, 65.2, 62.7, 60.1, 41.2, 39.1, 35.8, 35.5, 33.3, 32.3, 30.5, 30.3, 30.21, 30.19, 30.1, 29.8, 27.3, 26.2, 26.00, 25.9, 23.1, 20.4, 18.2, 15.6, 14.4, 11.3, −4.0, −5.3; IR (film) νmax 35.07, 1727, 1024 cm−1; HRMS (ESI-TOF) calcd for C38H72O7Si + Na+ 691.4939; found 691.4938.
(5S,6S)-5-((tert-Butyldiphenylsilyl)oxy)-6-((4S,5R,6S,E)-6-((tert-butyldiphenylsilyl)oxy)-4-hydroxy-5-methylheptadec-1-en-1-yl)-5,6-dihydro-2H-pyran-2-one (63)
LiAlH4 (1.0 M in THF, 0.26 mL, 0.26 mmol) was added to a solution of 59 in 1.8 mL of anhydrous THF at 0 °C under N2 with stirring. After 3 h, the reaction was quenched at 0 °C by careful addition of H2O, and the mixture was extracted with EtOAc. The combined organic extracts were dried (Na2SO4) and concentrated. The crude 60 was dissolved in CH2Cl2/pyridine (1:1, 2.6 mL). AgNO3 (0.21 g, 1.2 mmol) was added followed by TBDPSCl (0.36 g, 1.3 mmol), and the mixture was stirred at room temperature for 24 h in the dark. The mixture was diluted with Et2O and filtered through Celite. The filtrate was concentrated and the residue purified by flash chromatography (SiO2 pretreated with 2% Et3N/hexanes, 3% EtOAc/hexanes) to give 61 as a colorless oil which was taken to the next step without further characterization. A solution of 0.5 M aqueous HCl (2.6 mL) was added to a solution of 61 in THF (54 mL) at room temperature, and the mixture was stirred overnight. The reaction was quenched by addition of saturated aqueous NaHCO3, and the aqueous phase was extracted with EtOAc. The combined organic phases were washed with saturated aqueous NaCl, dried (Na2SO4), concentrated, and the residue was purified by flash chromatography (SiO2, 10–30% EtOAc/hexanes gradient) to give 0.055 g (48% overall for 3 steps) of 62 as a mixture of anomers as a colorless oil that was immediately taken to the next step without further characterization. Ag2CO3 (50% on Celite, 2.52 g, 4.6 mmol) was added in four portions over the course of 1 h to a solution of 62 (0.055 g, 0.063 mmol) in 6.3 mL of benzene at reflux. After the final addition, the mixture was stirred for 30 min, cooled to room temperature, and filtered through a plug of Celite while washing with EtOAc. The filtrate was concentrated and the residue was purified by flash chromatography (SiO2, 10% EtOAc/hexanes) to give 0.046 g (84%) of 63 as a colorless oil: [α]25D +44 (c 0.75, CHCl3); 1H NMR (C6D6, 600 MHz)δ7.85–7.77 (m, 4H), 7.27–7.15 (m, 12H), 5.98 (dd, J = 15.0, 6.6 Hz, 1H), 5.90 (dt, J = 15.0, 7.2 Hz, 1H), 5.86 (dd, J = 9.6, 4.8 Hz, 1H), 5.66 (d, J = 9.6 Hz, 1H), 4.29 (t, J = 6.6 Hz, 1H), 4.26 (dd, J = 7.2, 3.0 Hz, 1H), 3.86 (dd, J = 4.8, 3.6 Hz, 1H), 3.86–3.82 (m, 1H), 3.34 (brs, 1H), 2.51–2.43 (m, 1H), 2.18–2.10 (m, 1H), 1.80–1.72 (m, 1H), 1.64–1.55 (m, 2H), 1.39–0.94 (m, 18H), 1.17 (s, 9H), 1.15 (d, J = 6.6 Hz, 3H), 1.08 (s, 9H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (C6D6, 150 MHz)δ162.3, 144.2, 136.5, 136.4, 136.22, 136.18, 134.5, 133.8, 133.5, 133.23, 133.17, 130.4, 130.32, 130.31, 130.1, 128.0, 127.1, 122.5, 81.2, 80.0, 70.1, 65.2, 39.5, 38.8, 35.1, 32.3, 30.09, 30.05, 30.0, 29.9, 29.81, 29.78, 27.3, 27.0, 25.8, 23.1, 19.7, 19.5, 14.4, 11.3; IR (film) νmax 3504, 1731, 1106, 701 cm−1; HRMS (ESI-TOF) calcd for C55H76O5Si2 + H+ 873.5304; found 873.5302.
Bis((9H-fluoren-9-yl)methyl) ((4S,5S,6S,E)-6-((tert-butyldiphenylsilyl)oxy)-1-((2S,3S)-3-((tert-butyldiphenylsilyl)oxy)-6-oxo-3,6-dihydro-2H-pyran-2-yl)-5-methylheptadec-1-en-4-yl) Phosphate (64)
i-Pr2NP(OFm)2 (0.044 g, 0.084 mmol) in CH2Cl2 (0.64 mL) was added at room temperature to a stirred solution of 63 (0.020 g, 0.023 mmol) and tetrazole (0.45 M in MeCN, 0.14 mL, 0.064 mmol) in anhydrous MeCN (0.33 mL) under N2. After 1 h, aqueous H2O2 (35%, 0.12 mL) was added, and the mixture was stirred vigorously for 15 min. Saturated aqueous NaHCO3 (4.6 mL) was added, and the mixture was extracted with CH2Cl2. The combined organic extracts were dried (Na2SO4), concentrated, and the residue was purified by flash chromatography (SiO2, 20–25% EtOAc/hexanes gradient) to give 0.027 g (90%) of 64 as a colorless oil: [α]25D +34 (c 0.20, CHCl3); 1H NMR (C6D6, 600 MHz)δ7.86–7.77 (m, 3H), 7.67–7.57 (m, 4H), 7.55–7.45 (m, 6H), 7.39–7.35 (m, 1H), 7.34–7.07 (m, 22H), 5.92 (dd, J = 10.2, 4.8 Hz, 1H), 5.79 (dd, J = 15.6, 6.6 Hz, 1H), 5.71 (d, 10.2 Hz, 1H), 5.67 (dt, J = 15.6, 7.8 Hz, 1H), 4.49 (p, 6.0 Hz, 1H), 4.30–4.23 (m, 2H), 4.23–4.17 (m, 2H), 4.14 (dd, J = 6.6, 3.0 Hz, 1H), 4.00–3.96 (m, 1H), 3.95 (t, J = 6.6 Hz, 1H), 3.88 (t, J = 6.6 Hz, 1H), 3.82 (dd, J = 4.8, 3.0 Hz, 1H), 2.48–2.40 (m, 1H), 2.11–2.04 (m, 1H), 1.71–1.44 (m, 3H), 1.42–1.07 (m, 18H), 1.21 (s, 9H), 1.10 (d, J = 3H), 1.03 (s, 9H), 0.92 (t, J = 7.2 Hz, 1H); 13C NMR (C6D6, 150 MHz)δ162.2, 144.0, 143.84, 143.82, 143.81, 143.76, 141.80, 141.79, 141.77, 136.54, 136.47, 136.2, 134.9, 134.5, 133.7, 133.1, 130.5, 130.4, 130.3, 130.0, 129.9, 128.5, 127.5, 127.40, 127.39, 127.3, 125.65, 125.57, 125.5, 122.7, 120.23, 120.21, 120.16, 80.7, 80.6, 80.5, 75.0, 69.14, 69.10, 69.07, 65.0, 48.44, 48.42, 48.39, 48.37, 43.24, 43.21, 37.2, 33.1, 32.4, 30.3, 30.26, 30.19, 30.15, 30.1, 29.8, 27.4, 27.0, 26.6, 23.1, 22.5, 19.7, 19.5, 14.4, 10.0; 31P NMR (CD3OD 160 MHz)δ–0.6; IR (film) νmax 1733, 1106, 988 cm−1; HRMS (ESI-TOF) calcd for C83H97O8PSi2 + H+ 1309.6532; found 1309.6513.
Sodium (4S,5S,6S,E)-6-hydroxy-1-((2S,3S)-3-hydroxy-6-oxo-3,6-dihydro-2H-pyran-2-yl)-5-methylheptadec-1-en-4-yl Hydrogenphosphate (65)
TAS–F (1.0 M in DMF, 0.037 mL, 0.037 mmol) was added to a solution of 64 (4.8 mg, 0.0037 mmol) in MeCN (0.23 mL), and the mixture was stirred for 72 h at room temperature after which time it was concentrated. The residue was dissolved in 0.1 M sodium phosphate buffer (pH 7) and was purified by flash chromatography (C18 reverse phase SiO2, 0–50% MeCN/H2O gradient) giving 1.34 mg (73%) of 65 as a white solid: [α]25D +38 (c 0.03, MeOH); 1H NMR (CD3OD, 600 MHz)δ7.04 (dd, J = 9.6, 5.4 Hz, 1H), 6.06 (d, J = 9.6 Hz, 1H), 5.92 (dt, J = 15.3, 7.2 Hz, 1H), 5.81 (dd, J = 15.3, 7.8 Hz, 1H), 4.85 (dd, J = 8.4, 2.7 Hz, 1H), 4.61 (m, 1H), 4.20 (dd, J = 5.4, 2.7 Hz, 1H), 3.53 (t, J = 8.4 Hz, 1H), 2.66–2.59 (m, 1H), 2.39–2.32 (m, 1H), 1.66–1.41 (m, 3H), 1.41–1.24 (m, 18H), 0.90 (t, J = 7.2 Hz, 3H), 0.89 (d, J = 7.2 Hz, 3H); 13C NMR (CD3OD, 150 MHz)δ166.3, 147.4, 133.9, 127.8, 122.7, 83.4, 74.65, 74.61, 73.3, 63.7, 44.01, 43.98, 38.2, 35.5, 33.1, 30.97, 30.93, 30.89, 30.86, 30.84, 30.5, 27.2, 23.8, 14.5, 9.8; 31P NMR (CD3OD 160 MHz)δ3.4; IR (film) νmax 3386, 1720, 1084, 945 cm−1; HRMS (ESI-TOF) calcd for C23H41O8P + H+ 477.2612; found 477.2593.
Supplementary Material
Acknowledgements
We gratefully acknowledge the financial support of the National Institutes of Health (DLB: CA042056, REH: CA060750), and the studies in Alabama were conducted in a facility constructed with support from the Research Facilities Improvement Program (Grant CD6-RR11174) from the NIH National Center for Research Resources. We thank Nadia Haq and Danielle Soenen for early studies targeting sultriecin in which many of the strategic elements of the total synthesis were explored.
Footnotes
Supporting Information Available: Full experimental details and 1H, 13C, and 31P NMR spectra for 1, 2, 35, 36, and 39–58 are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Swingle M, Ni L, Honkanen RE. Methods Mol. Biol. 2007;365:23. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Virshup DM, Shenolikar S. Mol. Cell. 2009;33:537. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]; (c) McConnell JL, Wadzinski BE. Mol. Pharmacol. 2009;75:1249. doi: 10.1124/mol.108.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ohkuma H, Naruse N, Nishiyama Y, Tsuno T, Hoshino Y, Sawada Y, Konishi M, Oki T. J. Antibiot. 1992;45:1239. doi: 10.7164/antibiotics.45.1239. [DOI] [PubMed] [Google Scholar]
- (3).Lewy DS, Gauss C-M, Soenen DR, Boger DL. Curr. Med. Chem. 2002;9:2005. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]
- (4).Isolation: Tunac JB, Graham BD, Dobson WE. J. Antibiot. 1983;36:1595. doi: 10.7164/antibiotics.36.1595. Stampwala SS, Bunge RH, Hurley TR, Willmer NE, Brankiewicz AJ, Steinman CE, Smitka TA, French JC. J. Antibiot. 1983;36:1601. doi: 10.7164/antibiotics.36.1601. Total syntheses: Boger DL, Ichikawa S, Zhong W. J. Am. Chem. Soc. 2001;123:4161. doi: 10.1021/ja010195q. Chavez DE, Jacobsen EN. Angew. Chem. Int. Ed. 2001;40:3667. doi: 10.1002/1521-3773(20011001)40:19<3667::aid-anie3667>3.0.co;2-6. Reddy YK, Falck JR. Org. Lett. 2002;4:969. doi: 10.1021/ol025537r. Miyashita K, Ikejiri M, Kawasaki H, Maemura S, Imanishi T. Chem. Commun. 2002:742. doi: 10.1039/b201302a. Miyashita K, Ikejiri M, Kawasaki H, Maemura S, Imanishi T. J. Am. Chem. Soc. 2003;125:8238. doi: 10.1021/ja030133v. Esumi T, Okamoto N, Hatakeyama S. Chem. Commun. 2002:3042. doi: 10.1039/b209742g. Fujii K, Maki K, Kanai M, Shibasaki M. Org. Lett. 2003;5:733. doi: 10.1021/ol027528o. Trost BM, Frederiksen MU, Papillon JP, Harrington PE, Shin S, Shireman BT. J. Am. Chem. Soc. 2005;127:3666. doi: 10.1021/ja042435i. Maki K, Motoki R, Fujii K, Kanai M, Kobayashi T, Tamura S, Shibasaki M. J. Am. Chem. Soc. 2005;127:17111. doi: 10.1021/ja0562043. Yadav JS, Prathap I, Tadi BP. Tetrahedron Lett. 2006;47:3773. Hayashi Y, Yamaguchi H, Toyoshima M, Okado K, Toyo T, Shoji M. Org. Lett. 2008;10:1405. doi: 10.1021/ol800195g. Sarkar SM, Wanzala EN, Shibahara S, Takahashi K, Ishihara J, Hatakeyama S. Chem. Commun. 2009:5907. doi: 10.1039/b912267b. Robles O, McDonald FE. Org. Lett. 2009;11:5498. doi: 10.1021/ol902365n.
- (5).(a) Boger DL, Hikota M, Lewis BM. J. Org. Chem. 1997;62:1748. [Google Scholar]; (b) Hokanson GC, French JC. J. Org. Chem. 1985;50:462. [Google Scholar]
- (6).(a) Buck SB, Hardouin C, Ichikawa S, Soenen DR, Gauss C-M, Hwang I, Swingle MR, Bonness KM, Honkanen RE, Boger DL. J. Am. Chem. Soc. 2003;125:15694. doi: 10.1021/ja038672n. [DOI] [PubMed] [Google Scholar]; (b) Swingle MB, Amable L, Lawhorn BG, Buck SB, Burke CP, Ratti P, Fisher KL, Boger DL, Honkanen RE. J. Pharmacol. Exp. Ther. 2009;331:45. doi: 10.1124/jpet.109.155630. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takeuchi T, Takahashi N, Ishi K, Kusayanagi T, Kuramochi K, Sugawara F. Bioorg. Med. Chem. 2009;17:8113. doi: 10.1016/j.bmc.2009.09.050. [DOI] [PubMed] [Google Scholar]
- (7).Isolation: Amemiya M, Someno T, Sawa R, Naganawa H, Ishizuka M, Takeuchi T. J. Antibiot. 1994;47:541. doi: 10.7164/antibiotics.47.541. Amemiya M, Ueno M, Osono M, Masuda T, Kinoshita N, Nishida C, Hamada M, Ishizuka M, Takeuchi T. J. Antibiot. 1994;47:536. doi: 10.7164/antibiotics.47.536. Total syntheses: Lawhorn BG, Boga SB, Wolkenberg SE, Colby DA, Gauss C-M, Swingle MR, Amable L, Honkanen RE, Boger DL. J. Am. Chem. Soc. 2006;128:16720. doi: 10.1021/ja066477d. Lawhorn BG, Boga SB, Wolkenberg SE, Boger DL. Heterocycles. 2006;70:65. Bialy L, Waldmann H. Chem. Eur. J. 2004;10:2759. doi: 10.1002/chem.200305543. Bialy L, Waldmann H. Angew. Chem. Int. Ed. 2002;41:1748. doi: 10.1002/1521-3773(20020517)41:10<1748::aid-anie1748>3.0.co;2-x. Jung W-H, Guyenne S, Riesco-Fagundo C, Mancuso J, Nakamura S, Curran DP. Angew. Chem. Int. Ed. 2008;47:1130. doi: 10.1002/anie.200704893.
- (8).Isolation: Ozasa T, Tanaka K, Sasamata M, Kaniwa H, Shimizu M, Matsumoto H, Iwanami M. J. Antibiot. 1989;42:1339. doi: 10.7164/antibiotics.42.1339. Total syntheses: Wang Y-G, Takeyama R, Kobayashi Y. Angew. Chem. Int. Ed. 2006;45:3320. doi: 10.1002/anie.200600458. Shibahara S, Fujino M, Tashiro Y, Takahashi K, Ishihara J, Hatakeyama S. Org. Lett. 2008;10:2139. doi: 10.1021/ol8004672.
- (9).Isolation: Kohama T, Enokita R, Okazaki T, Miyaoka H, Torikata A, Inukai M, Kaneko I, Kagasaki T, Sakaida Y, Satoh A, Shiraishi A. J. Antibiot. 1993;46:1503. doi: 10.7164/antibiotics.46.1503. Kohama T, Nakamura T, Kinoshita T, Kaneko I, Shiraishi A. J. Antibiot. 1993;46:1512. doi: 10.7164/antibiotics.46.1512. Total syntheses: Matsuhashi H, Shimada K. Tetrahedron. 2002;58:5619. Shimada K, Kaburagi Y, Fukuyama T. J. Am. Chem. Soc. 2003;125:4048. doi: 10.1021/ja0340679. Miyashita K, Tsunemi T, Hosokawa T, Ikejiri M, Imanishi T. J. Org. Chem. 2008;73:5360. doi: 10.1021/jo8005599.
- (10).Isolation: Fushimi S, Nishikawa S, Shimazu A, Seto H. J. Antibiot. 1989;42:1019. doi: 10.7164/antibiotics.42.1019. Total synthesis: König CM, Gebhardt B, Schleth C, Dauber M, Koert U. Org. Lett. 2009;11:2728. doi: 10.1021/ol900757k.
- (11).Burke CP, Haq N, Boger DL. J. Am. Chem. Soc. 2010;132:2157. doi: 10.1021/ja9097252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yang Z-C, Jiang X-B, Wang Z-M, Zhou W-S. J. Chem. Soc., Perkin Trans.1. 1997:317. [Google Scholar]
- (13).(a) Racherla US, Brown HC. J. Org. Chem. 1991;56:401. [Google Scholar]; (b) Nicolaou KC, Patron AP, Ajito K, Richter PK, Khatuya H, Bertinato P, Miller RA, Tomaszewski MJ. Chem. Eur. J. 1996;2:847. [Google Scholar]
- (14).Oppolzer W, Radinov RN. Helv. Chim. Acta. 1992;75:170. [Google Scholar]
- (15).Trost BM, Weiss AH, von Wangelin AJ. J. Am. Chem. Soc. 2006;128:8. doi: 10.1021/ja054871q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Takita R, Yakura K, Oshima T, Shibasaki M. J. Am. Chem. Soc. 2005;127:13760. doi: 10.1021/ja053946n. [DOI] [PubMed] [Google Scholar]
- (17).Frantz DE, Fässler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806. [Google Scholar]
- (18).(a) Noyori R, Ohta M, Hsiao Y, Kitamura M, Ohta T, Takaya H. J. Am. Chem. Soc. 1986;108:7117. [Google Scholar]; (b) Noyori R, Ohkuma T, Kitamura H, Takaya H, Sayo H, Kumobayashi S, Akutagawa S. J. Am. Chem. Soc. 1987;109:5856. [Google Scholar]
- (19).(a) Noyori R, Tomino I, Tanimoto Y, Nishizawa M. J. Am. Chem. Soc. 1984;106:6709. [Google Scholar]; (b) Noyori R, Tomino I, Yamada M, Nishizawa M. J. Am. Chem. Soc. 1984;106:6717. [Google Scholar]
- (20).(a) Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]; (b) Ramirez F, Desai NB, McKelvie N. J. Am. Chem. Soc. 1962;84:1745. [Google Scholar]
- (21).Tohda Y, Sonogashira K, Hagihara N. Synthesis. 1977:777. [Google Scholar]
- (22).Corey EJ, Bakshi RK, Shibata S. J. Am. Chem. Soc. 1987;109:5551. [Google Scholar]
- (23).Corey EJ, Katzenellenbogen JA, Posner GH. J. Am. Chem. Soc. 1967;89:4245. [Google Scholar]
- (24).(a) Georgiadis MP, Couladouros EA. J. Org. Chem. 1986;51:2725. [Google Scholar]; (b) Babu RS, Zhou M, O'Doherty GA. J. Am. Chem. Soc. 2004;126:3428. doi: 10.1021/ja039400n. [DOI] [PubMed] [Google Scholar]
- (25).Hakimelahi GH, Proba ZA, Ogilvie KK. Tetrahedron Lett. 1981;22:4775. [Google Scholar]
- (26).Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- (27).Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- (28).Belosludtsev YY, Borer BC, Taylor RJK. Synthesis. 1991:320. [Google Scholar]
- (29).Uenishi J, Kawahama R, Yonemitsu O, Tsuji J. J. Org. Chem. 1998;63:8965. [Google Scholar]
- (30).Still WC, Schneider JA. Tetrahedron Lett. 1980;21:1035. [Google Scholar]
- (31).1H NMR data for “desulfated” 1 were reported, however this data alone was not sufficient to confirm that it and triol 35 share the same structure. See ref 2.
- (32).(a) Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945. [Google Scholar]; (b) Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;31:7099. [Google Scholar]
- (33).Commercially available from Bioaustralis.
- (34).Sheidt KA, Chen H, Follows BC, Chemler SR, Coffey DS, Roush WR. J. Org. Chem. 1998;63:6436. [Google Scholar]
- (35).De Jong RS, Mulder NH, Uges DRA, Sleijfer D. Th., Hoppener FJP, Groen HJM, Willemse PHB, Van der Graf WTA, De Vries EGE. Br. J. Cancer. 1999;79:882. doi: 10.1038/sj.bjc.6690141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.