

Abstract
We describe a non-fluorescent, second generation stilbene that very selectively binds to transthyretin in complex biological environments and remains dark until it chemoselectively reacts with the pKa perturbed Lys-15 ε-amino group of transthyretin to form a bright blue fluorescent conjugate. Stilbene A2 is mechanistically unusual in that it remains non-fluorescent in cell lysates lacking transthyretin, even though there is likely some proteome binding. Thus, it is especially useful for cellular imaging, as background fluorescence is undetectable until A2 reacts with transthyretin. The mechanistic basis for the effective lack of environment-sensitive fluorescence of A2 when bound to, but before reacting with, transthyretin is reported. Stilbene A2 exhibits sufficiently rapid transthyretin conjugation kinetics at 37 °C to enable pulse-chase experiments to be performed, in this case demonstrating that transthyretin is secreted from HeLa cells. As the chase compound, we employed C1–a cell permeable, highly selective, non-covalent, transthyretin binding dihydrostilbene that cannot become fluorescent. The progress reported is viewed as a first and necessary step toward our long-term goal of creating a one-chain, one-binding-site transthyretin tag, whose fluorescence can be regulated by adding A2, or an analogous molecule. Fusing proteins of interest to a one-chain, one-binding-site transthyretin tag regulated by A2 should be useful for studying folding, trafficking and degradation in the cellular secretory pathway, utilizing pulse-chase experiments. Immediate applications of A2 include utilizing its conjugate fluorescence to quantify transthyretin concentration in human plasma, reflecting nutritional status, and determining the binding stoichiometry of kinetic stabilizer drugs to transthyretin in plasma.
Introduction
The development of fluorescent tags and fluorescent biosensors to image processes within living cells with spatial resolution has transformed what can be accomplished in biological research.1–3 A very useful macromolecule for fluorescence tagging and a component of many biosensors is the green fluorescent protein (GFP), which first folds and then undergoes an autocatalytic intramolecular chemical reaction to form its chromophore.4–9 Mutants of GFP exhibit emission at a spectrum of wavelengths,10 thus mutagenesis has proven to be a very useful approach to alter the photophysical properties of the chromophore.1 In a complementary approach, a tetra-cysteine tag on a protein of interest can be used to bind the As-containing latent fluorophores FlAsH or ReAsH, rendering the conjugate fluorescent through a sulfide exchange reaction that can be conformation- or quaternary structure-dependent.11–14 Additional methodology to fluorescently tag a protein of interest includes the enzymatic conjugation of fluorophores to proteins,15–18 as well as the snap19–21 and halo tag22,23 approaches.
Wild type (WT) transthyretin is a tetrameric human plasma protein composed of identical 127-amino-acid β-sheet-rich subunits (Figure 1A).24–27 The more labile dimer-dimer interface of transthyretin (TTR) (bisected by the crystallographic 2-fold axis; Figure 1A) creates two identical funnel-shaped thyroxine (T4) binding sites that are interconverted by a C2 axis perpendicular to the crystallographic 2-fold axis.27–29 Previously, we reported stilbenes that bind to the T4 binding sites within TTR in human plasma with very high selectivity over the other 4000+ proteins present.30,31 Active ester analogs of these stilbenes then chemoselectively react with the pKa perturbed Lysine-15 (K15) ε-amino group of TTR to form the amide-linked stilbene conjugate.32 An x-ray crystal structure defined the binding orientation of the stilbene substructure and the apparent cis-amide linkage to TTR.32
Figure 1.

(A) Crystal structure of the TTR-(thyroxine)2 complex with differentially colored subunits. (B) Structures of covalent TTR modifiers A1 and A2 and the fluorescent propylamide stilbene analogue B1 that is not capable of reacting with WT-TTR. (C) Depiction of the structure of the two TTR thyroxine binding sites undergoing a reaction with A1 or A2 to create a WT-TTR–(stilbene)2 conjugate.
Herein, we describe a non-fluorescent, second generation stilbene active ester A2 (Figure 1B), and related stilbenes that selectively bind to and then chemoselectively react with the TTR protein in complex biological environments, creating blue fluorescent conjugates as a consequence of amide bond formation (Figure 1C). It is atypical that stilbenes remain dark when bound to proteins, and in that regard stilbene A2 is especially useful for cellular imaging. Stilbene A2 (Figure 1B) also exhibits highly selective binding to TTR within the secretory pathway of eukaryotic cells and then undergoes a chemoselective amide bond forming reaction that renders the conjugate fluorescent. Stilbene A2 exhibits sufficiently rapid transthyretin conjugation kinetics at 37 °C to enable pulse-chase experiments to be performed–the chase utilizing a highly selective, non-covalent TTR binder that cannot become fluorescent.
The progress reported is viewed as a first and necessary step toward the creation of a one-chain, one-binding-site TTR tag, whose fluorescence can be turned on when a small molecule of the type described herein is added to cells to form a TTR-stilbene conjugate. Folded, monomeric versions of TTR have been published33,34 and were created by introducing sterically demanding mutations into both of TTR’s quaternary structural interfaces.28,35 We have already linked subunits a and c (Figure 1A), employing a low complexity linker using established molecular biological approaches.28 We are currently seeking mutations that prevent the one-chain, one-binding-site TTR molecule from dimerizing to form the equivalent of the tetramer. Fusing proteins of interest to a one-chain, one-binding-site transthyretin tag regulated by A2 should be especially useful for following protein maturation and degradation in the secretory pathway of eukaryotic cells.36–38
Experimental Section
Fluorimetric Assay with Recombinant Wild Type Transthyretin
WT-TTR was expressed in an E. coli expression system and purified as described previously.39 The covalent TTR modifiers (A1 or A2) or the non-covalent TTR ligand B1 (5 μL of a 1.44 mM solution in DMSO, final concentration: 7.2 μM) were added to 1 mL of a solution of WT-TTR homotetramer (0.2 mg/mL, 3.6 μM) in 10 mM phosphate, 100 mM KCl and 1 mM EDTA (pH 7.0 phosphate buffer) in a 2 mL Eppendorf tube. The samples were vortexed, and incubated for 18 h at 25 °C. The fluorescence changes were monitored using a Varian Cary 50 spectrofluorometer at 20 °C in a 1 cm path length quartz cell. The excitation slit was set at 5 nm and the emission slit was set at 5 mm. The samples were excited at 328 nm and the emission spectra were collected from 350 to 550 nm.
Kinetics of the A2-TTR Conjugation Reaction
The covalent TTR modifier A2 (1 μL of 0.6 mM solution in DMSO, final concentration: 6 μM) was added to 90 μL of HeLa cell lysate (final protein concentration: 2 μg/μL) and 10 μL of WT-TTR (20 μM in pH 7.0 phosphate buffer, final concentration: 2 μM). The time-dependent fluorescence change of the conjugate derived from A2 was monitored utilizing a microplate spectrophotometer reader (Gemini SpectraMax®, Molecular Devices, Sunnyvale, CA) for 3 h at 37 °C. The fluorescence (λex = 328 nm and λem = 430 nm) was measured from the bottom of the plate without shaking.
Binding Stoichiometry of A2 to WT- and K15A-TTR
To 1 mL of WT- or K15A-TTR (5 μM in pH 7.0 phosphate buffer) in a 2 mL Eppendorf tube was added A2 (5 μL of a 2 mM solution in DMSO, final concentration: 10 μM) and then the solution was incubated at 37 °C for 24 h on a rocker plate (30 rpm). A 1:1 (v/v) slurry of unfunctionalized sepharose resin in 10 mM Tris, 140 mM NaCl, pH 8.0 (TSA) buffer was added and the solution was incubated for 1 h at 4 °C on a rocker plate (18 rpm). The solution was then centrifuged and the supernatant was divided into two 400 μL aliquots, which were added to 200 μL of 1:1 (v:v) slurry of anti-TTR antibody conjugated sepharose resin in TSA. The solution was gently rocked (18 rpm) at 4 °C for 20 min, then centrifuged and the supernatant removed. The resin was washed three times by shaking for 1 min with 1 mL of TSA. After centrifugation to remove the supernatant, 155 μL of triethylamine (100 mM, pH 11.5) was added to the sepharose resin to dissociate the TTR and bound test compound from the resin and the suspension was vortexed for 1 min. After centrifugation, the supernatant was analyzed by reverse phase HPLC on a Water 600 E multi-solvent delivery system, using a Waters 486 tunable absorbance detector, a 717 autosampler, and a ThermoHypersil Keystone Betabasic-18 column (150 Å pore size, 3 μm particle size). The “A” mobile phase comprises 0.1% TFA in 94.9% H2O + 5% CH3CN and the “B” mobile phase is made up of 0.1% TFA in 94.9% CH3CN + 5% H2O. Linear gradients were run from 100:0 A:B to 0:100 A:B for 10 min.
Quantum Yield Measurement
The covalent TTR modifier A1 or A2 or the non-covalent TTR ligand B1 (15 μL of a 1.44 mM solution in DMSO, final concentration: 7.2 μM) was added to 3 mL of a solution of WT-TTR homotetramer (0.2 mg/mL, 3.6 μM) in 10 mM phosphate, 100 mM KCl and 1 mM EDTA (pH 7.0 phosphate buffer). The samples were vortexed, and incubated for 18 h at 25 °C. Quantum yields were measured by following the instructions at: www.jobinyvon.com/usadivisions/Fluorescence/applications/quantumyieldstrad.pdf. Quinine bisulfate in 0.5 M H2SO4 was used as a reference for comparison (Φf = 0.546).40
Indirect immunofluorescence detection of TTR
HeLa cells were plated on cover slips in DMEM containing 10 % FBS at 37 °C under 5 % CO2 the day before they were transfected with pcDNA3.1(+)-empty vector or pcDNA3.1(+)-WT-TTR using Fugene 6 (Roche) according to the manufacturer’s instructions. After 48 h, the cells were washed with PBS twice before adding fresh DMEM (without FBS) with A2 (final concentration: 10 μM), and the cells were incubated at 37 °C for 1 h. After two washes with PBS, the cells were fixed at 25 °C for 15 minutes with 3.7 % (w/v) paraformaldehyde in PBS. After two washes with PBS, the cells were permeabilized with 0.2 % saponin (w/v) in PBS at 25 °C for 15 min, followed by blocking at 25 °C for 30 min with blocking buffer (10 % goat serum (v/v), 0.2 % saponin (w/v) in PBS). Cells were incubated for 1 h with anti-TTR (a kind gift from Dr. E. Masliah at UCSD, diluted 1:100) and anti-calnexin (Stressgen, diluted 1:200), washed three times with PBS, and incubated for 1 h with Alexa Fluor 546 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes, A11035 and A11029, respectively) for WT-TTR and calnexin staining. After mounting, microscopic images were acquired by using a fully tunable filter-based emission collection system (Bio-Rad (Zeiss) Radiance 2100 Rainbow laser scanning confocal microscope) that enables the precise collection of 420–460 nm for compound detection (from a 405 nm laser line). Image analyses were performed using either Zen 2008 Light Edition software (Carl Zeiss MicroImaging) or ImageJ software (NIH).
Plasmid Constructs Harboring TTR
The pcDNA3.1(+)-WT-TTR plasmid was produced by ligating the WT-TTR cDNA (excised from the pCMV5-WT-TTR plasmid36 using HindIII and XhoI restriction sites) into the pcDNA3.1(+) plasmid (Invitrogen). A point mutation in the WT-TTR at position 133 (A>G) was corrected using Quikchange II site-directed mutagenesis (Stratagene) using the following primers (5′ to 3′): CCGAGGCAGTCCTGCCATCAATGTGGC and GCCACATTGATGGCAGGACTGCCTCGG. The proper construction of all plasmids was confirmed by DNA sequencing. Restriction endonucleases and Pfu Turbo DNA polymerase were purchased from New England Biolabs and Stratagene, respectively. TTR plasmid DNA was isolated with a QIAprep Spin Miniprep kit (Qiagen Inc.).
Labeling of endogenous TTR in cells
Huh-7 cells or Gaucher’s patient-derived fibroblasts (Coriell Cell Repositories, GM08760) were maintained in DMEM containing 10 % FBS at 37 °C under 5 % CO2. The cells were plated the day before compound treatment and imaging. Cells were washed twice with PBS and then treated with A2 (final concentration: 10 μM) for 30 min at 37 °C in a CO2 incubator. After two washes, cells were fixed at 25 °C for 15 min with 3.7 % paraformaldehyde (w/v) in PBS and microscopic images were acquired as described above.
Monitoring secretion of WT-TTR from HeLa Cells Utilizing a Pulse-Chase Experiment
HeLa cells were transfected with pcDNA3.1(+)-WT-TTR using Fugene 6 (Roche). After 48 h post-transfection, the cells were washed with PBS twice before adding fresh DMEM (without FBS) with A2 (final concentration: 20 μM). The cells were incubated at 37 °C for 30 min (pulse) to label the synthesized WT-TTR, followed by the chase using 10 μM C1 over a time course of 2 h. After two washes, cells were fixed at 25 °C for 15 min with 3.7 % paraformaldehyde (w/v) in PBS at the indicated times and microscopic images were acquired as described above. The cell culture media was collected at the indicated time points during the pulse-chase experiment and the proteins were precipitated using a methanol/chloroform mixture (4:1 v/v). The protein pellet was solubilized in 8 M urea (pH 8) and half of the mixture was analyzed for the presence of TTR in the media using western blot analysis.
Results
Chemoselective Conjugation of A2 to WT-TTR in buffer creating Blue Fluorescence
The ability of compounds A1 and A2 (Figure 1B), auxochromic analogs of previously reported stilbenes,24,27,30–32,41 to form a chemoselective amide bond with the K15 residue of TTR was assessed by reverse-phase-HPLC and liquid chromatography-mass spectrometry analysis. Incubating WT-TTR (3.6 μM) with A1 or A2 (7.2 μM, due to the two thyroxine binding sites per TTR homotetramer) in phosphate buffer (pH 7) exhibits two peaks of nearly equal intensity (Figure S1A and S1B, left panels). Since only 2 of the 4 subunits comprising the tetramer can be modified by A2 (Figure 1C), a ~ 1:1 peak ratio is the expected result for binding and chemoselective conjugation (the change in molar absorbtivity of the conjugate was accounted for, note the slightly increased intensity of the conjugate). Analogous incubation of A1 or A2 with the K15A-TTR homotetramer followed by HPLC analysis revealed no conjugation, consistent with the K15 TTR residue being essential for chemoselective conjugation (Figure S1A and S1B, right panels). The binding of the covalent modifier A2 to K15A-TTR (in the absence of amide bond conjugation) was confirmed by incubating A2 with K15A-TTR in phosphate buffer (pH 7) followed by K15A immunoisolation using a Sepharose resin-conjugated anti-TTR polyclonal antibody.42 After the dissociation of K15A-TTR and bound A2 at high pH, HPLC analysis showed that 0.88 equivalents out of maximum 2 equivalents of A2 were non-covalently bound to K15A-TTR (Figure S2), an underestimation of bound A2 owing to the washing steps preceding dissociation.
Furthermore, compound A2 was evaluated to determine its ability to bind selectively to and then react chemoselectively with TTR over the 4000+ other proteins in human blood plasma. Compound A2 (10.8 μM) was incubated with human blood plasma (TTR concentration is 3.6 – 5.4 μM) for 24 h at 37 °C before immunoisolating human TTR using a Sepharose resin-conjugated anti-TTR polyclonal antibody.42 After dissociation of TTR from the resin-linked antibody at high pH, HPLC analysis revealed the ratio of TTR monomer and TTR monomer covalently attached to the benzoyl portion of A2 via amide bond formation is effectively 1:1 (taking into account the differences in molar absorbtivity) (Figure S3A). Compound A2 must exhibit very high binding selectivity to TTR in human plasma in order to chemoselectively label 48% of the TTR subunits through amide bond formation, as only 2 of the 4 subunits comprising the tetramer can be modified (Figure 1C), corresponding to maximal subunit labeling of 50%. Mass spectrometric analysis of the conjugate showed the expected masses (Figure S3B).32 It is clear that these second generation auxochromic stilbenes exhibit exceptional binding selectivity and amide bond forming chemoselectivity to TTR, comparable to the first generation stilbenes. This will be important for determination of plasma TTR concentration and kinetic stabilizer binding stoichiometry to TTR (see discussion).32
While incubation of recombinant WT-TTR with A1 or A2 in phosphate buffer (pH 7) for 18 h produced bright blue fluorescence resulting from amide bond conjugation (Figures S4 and 2A, respectively, blue traces), barely detectable fluorescence resulted from A1 or A2 alone in buffer (Figure S4 and 2A, respectively, red traces). Notably, no significant fluorescence increase was observed when A2 was incubated with the recombinant K15A-TTR homotetramer, which binds A2 (Figure S2), but cannot amide bond conjugate (Figure 2A, black trace). A red shift in the weak, but detectable fluorescence of A1 was observed with K15A-TTR, providing evidence that it is binding to the T4 binding sites within K15A-TTR (Figure S4, black trace), but not reacting (Figure S1A, right panel).
Figure 2.

(A) Fluorescence observed after an 18 h incubation of the reactive stilbene-based TTR modifier A2 with WT- and K15A-TTR. (B) Fluorescence observed after a 10 min incubation of non-reactive stilbene B1 with WT- and K15A-TTR (λex = 328 nm).
Incubation of unreactive B1 (7.2 μM; Figure 1B), comprising a built-in amide bond, with WT-TTR (3.6 μM) for 10 min reveals a significant increase in fluorescence intensity (Figure 2B, blue trace, cf. B1 alone in buffer, red trace). Collectively, these data support the hypothesis that amide bond linkage of the benzoyl portion of A1 or A2 to TTR enhances WT-TTR–(stilbene)2 conjugate fluorescence. In stark contrast to A2 which remains non-fluorescent upon binding to K15A-TTR, B1 exhibited a ~ 4-fold increase in fluorescence intensity upon binding to K15A-TTR (Figure 2B, black trace), providing additional evidence that this family of stilbenes binds to K15A-TTR. The fluorescence intensity was diminished relative to WT-TTR binding, presumably due to changes in the T4 binding site environment or dynamics associated with the K15A mutant.43
We also incubated A2 with recombinant WT- or K15A-TTR and the resulting solution fluorescence was detected using a hand-held UV lamp (365 nm). While intense blue fluorescence resulted from the incubation of WT-TTR with A2 (Figure S5, vial 1), no fluorescence was observed when K15A-TTR was incubated with A2 (vial 2). In contrast, intense blue fluorescence was produced when amide B1 was incubated with either WT-TTR or K15A-TTR (Figure S5, cf. vials 3 and 4, respectively). The reaction chemoselectivity of A2 for WT-TTR was further demonstrated by the observation that WT-TTR, but not K15A-TTR, was detected by conjugate fluorescence on an SDS-PAGE gel (Figure S6).
Investigating the Mechanism by Which Stilbene Conjugation to TTR creates Blue Fluorescence
The data outlined above and presented below suggest that amide bond conjugation is required for the observation of TTR–(stilbene)n≤2 conjugate fluorescence. As mentioned above, it is unusual that stilbenes remain dark when bound to proteins, including TTR, and in this regard we hypothesize that stilbene A2 could be especially useful for cellular imaging. What makes A2 atypical in this regard? To answer this question, it is useful to consider the excited state relaxation mechanism available to most stilbenes.44–49
Upon promotion of an electron from the stilbene HOMO to the LUMO, a singlet excited state is formed that repopulates the ground state in solution primarily by isomerization to a cis-stilbene, and to a lesser extent by stilbene fluorescence.44–49 The trans form of the singlet excited state can twist about the central C-C bond orienting the planes of the two aryl rings perpendicular to each other (Figure 3A), affording a mixture of cis and trans isomers after internal conversion and conformational relaxation.44–49 While TTR can bind to small molecules composed of two aromatic rings when the tethered rings are up to 40° out of plane (based on TTR-stilbene crystal structures24,27,30–32,50), it would be energetically unfavorable to bind the excited singlet state in a conformation where the two aryl rings are oriented perpendicular to one another. Thus, we propose that the increase in fluorescence quantum yield of B1 from 0.09 in buffer to 0.55 in complex with TTR results from the resculpted excited singlet state energy surface of the stilbene in complex with TTR, such that the trans-conformer is the only state of relatively low potential energy (Figure 3B),44–49 preventing photoisomerization-based relaxation of B1. That B1 binding to TTR prevents B1 photoisomerization is consistent with the observation that the irradiation of B1 in buffer (Life Technologies, TFX-35M, 312 nm) for 10 sec affords 60% of the cis isomer, along with other minor photoreaction products (Figure S7A), whereas irradiation of B1 bound to TTR yields only 20% cis isomer (Figure S7B).
Figure 3.

Structures and energetics associated with stilbene photochemistry and photophysics. (A) Perpendicular singlet excited state (1P*) conformation required for the trans to cis photoisomerization of stilbenes. (B) Relative energies of the ground (S0) and excited singlet states (S1) of stilbenes as a function of their conformation (c, cis isoform; t, trans isoform) in solution (black S1 curve) or when the stilbenes are bound to TTR (red S1 curve), wherein the perpendicular singlet excited state (1P*) and the cis singlet excited state (S1) are strongly destabilized by the constraints imposed by the TTR binding site.
It appears that the increase in fluorescence quantum yield of A2 from 0.00 in buffer to 0.27 in the TTR–(stilbene)n≤2 conjugate is only partly explained by the mechanism proposed for B1.44–49 That A2 is different from B1 is supported by observation that A2 does not readily form the cis isomer or undergo photoreactions upon irradiation in buffer for 10 sec (Figure S7C). Nor does A2 fluoresce upon binding to K15A-TTR (Figure 2A). It is also notable that the fluorescence intensity of A2 does not increase appreciably in dichloromethane (Figure S8A), unlike B1, which is an environmentally sensitive fluorophore (Figure S8B). The very low fluorescence intensity of A2 combined with its resistance to photoisomerization suggests that the thioester comprising A2 quenches its fluorescence, both in solution and when bound to K15A-TTR. However, upon amide bond formation with TTR, the conjugate derived from A2 becomes fluorescent because the stilbene is no longer quenched by the thioester functionality and because the perpendicular excited singlet state conformation cannot be accommodated in the TTR–(stilbene)n≤2 conjugate structure (Figure 3B), preventing the photoisomerization-based relaxation. The formation of the perpendicular excited singlet state conformation required for photoisomerization (Figure 3A) is envisioned to be even more energetically inaccessible in the amide bond conjugate, as the aryl ring occupying the inner T4 binding subsite is rigidly held through complementary non-covalent interactions24,27,30–32,41,50–63 and the ring occupying the outer T4 binding subsite is covalently tethered to the protein through a m-amide linkage.32 The thioester stilbene quenching hypothesis is supported by observations that A1 and B1 are quenched, dose dependently, by the addition of mM concentrations of thiophenol (Figure S9).
Antibodies that bind to stilbenes that have aromatic side chains in their stilbene binding sites can form either fluorescence exciplexes or charge-transfer-based luminescent antibody-stilbene complexes.64,65 However, the TTR stilbene binding sites do not comprise any aromatic residues, making an exciplex fluorescence or charge-transfer luminescence explanation unlikely.64,65
Summing up the mechanistic details; A2 remains dark, even if bound to proteins, including TTR, because of a thioester quenching mechanism. Upon amide bond conjugation to TTR, the fluorescence quantum yield of the blue conjugate is reasonably high because the trans-to-cis photoisomerization mechanism is energetically disfavored owing to TTR binding and photobleaching is minimized by the lower energy excitation and emission resulting from the dimethylamino auxochromic substituent on one of the aromatic rings.
Kinetics of TTR Fluorescent Conjugate Formation at 37 °C in Human Cell Lysate
Since the one-chain, one-binding-site TTR tag being created is envisioned to be useful for doing pulse-chase experiments in the secretory pathway of eukaryotic cells, the kinetics of WT-TTR modification by A2 in concentrated cell lysate was investigated (Figure 4A). HeLa cell lysate was employed because these cells do not make transthyretin. We added WT-TTR at a final concentration of 2 μM (tetramer) to the HeLa cell lysate (2 μg/μL total protein concentration) to which A2 (6 μM) was added. At 37 °C, the conjugate formed with a t50 of 18 min, within experimental error of the t50 (19 min) of TTR (2 μM) conjugate formation with A2 (6 μM) in phosphate buffer (pH 7), Figure 4A. That A2 remains dark, even if bound to other proteins in the absence of TTR, is demonstrated by the HeLa cell lysate (2 μg/μL total protein concentration) incubated with A2 (2 μM) for 180 min, Figure 4A (red trace).
Figure 4.

Kinetics and chemoselectivity of the A2 conjugation reaction with WT-TTR in HeLa cell lysate vs. buffer. (A) Time-dependence of fluorescent conjugate formation, created by reacting A2 and WT-TTR in phosphate buffered saline (pH 7; green trace). Time-dependent fluorescence changes or lack thereof occurring when HeLa cell lysate (lacking TTR) is incubated with A2 alone (red trace) or when HeLa cell lysate is incubated alone with added TTR (without A2, blue trace) or when HeLa cell lysate with added WT-TTR is treated with A2 at 37 °C (black trace). (B) RP-HPLC analysis of TTR immunoisolated from HeLa cell lysate utilizing an anti-TTR antibody conjugated to resin. The nearly equal peak areas resulting from the unmodified subunits and the conjugated subunits (the molar absorptivity changes associated with benzoylation were accounted for) demonstrate high binding selectivity and a highly chemoselective amide bond forming reaction with TTR (49 % out of a maximum of 50 % of the subunits were modified).
Selectivity of the A2 TTR Conjugation Reaction at 37 °C in Human Cell Lysate
The ability of A2 to form a chemoselective amide bond with the K15 residue of TTR in cell lysate was assessed by RP-HPLC and LC-MS analysis. Incubating WT-TTR (5 μM) with A2 (15 μM) in 900 μL of HeLa cell lysate (2 μg/μL total protein concentration, excluding TTR) exhibits peaks of nearly equal intensity (Figure 4B; the molar absorptivity changes associated with benzoylation were accounted for) demonstrating high binding selectivity and a highly chemoselective amide bond forming reaction with TTR (49 % out of a maximum of 50 % of the subunits were modified, Figure 1C). Immunoisolation of TTR affords the expected mass resulting from conjugate formation (14,185 m/z [M+H]+ for benzoylation by A2). The equal rates of TTR conjugate formation in HeLa cell lysate and in buffer (Figure 4A and 4B) demonstrate that the binding selectivity of A2 to TTR and the chemoselectivity of amide bond conjugation to TTR are excellent in complicated biological environments.
Detection of WT-TTR in Transfected HeLa cells and in Huh-7 Human Liver Cells
Having demonstrated that A2 is a non-fluorescent stilbene in solution (Figure 2A) and in HeLa cell lysate not containing TTR (Figure 4A, red trace), we next investigated whether specific labeling of TTR could be achieved in the complex environment of the eukaryotic cellular secretory pathway. Secreted WT-TTR was produced in HeLa cells by transient transfection and the cells were treated with A2 for 1 h prior to imaging using confocal fluorescence microscopy. The expression of WT-TTR in the HeLa cells after transient transfection was confirmed using western blot analysis (Figure S10A), as was the absence of TTR in the empty vector control. Only HeLa cells expressing WT-TTR, and not cells transfected with empty vector (control), afforded strong conjugate fluorescence upon A2 treatment, Figure 5A, first column. Indirect TTR immunofluorescence (Figure 5A, second column) afforded an analogous image, revealing that TTR was being imaged by A2 treatment. TTR conjugate fluorescence co-localized with calnexin indirect immunofluorescence, a marker for the endoplasmic reticulum (ER), confirming that WT-TTR within the secretory pathway is being observed (Figure 5A, right most column, colocalization artificially colored white). TTR conjugate fluorescence is also expected in the Golgi and in secretory vesicles, explaining why TTR conjugate fluorescence is observed in organelles in addition to the ER.
Figure 5.
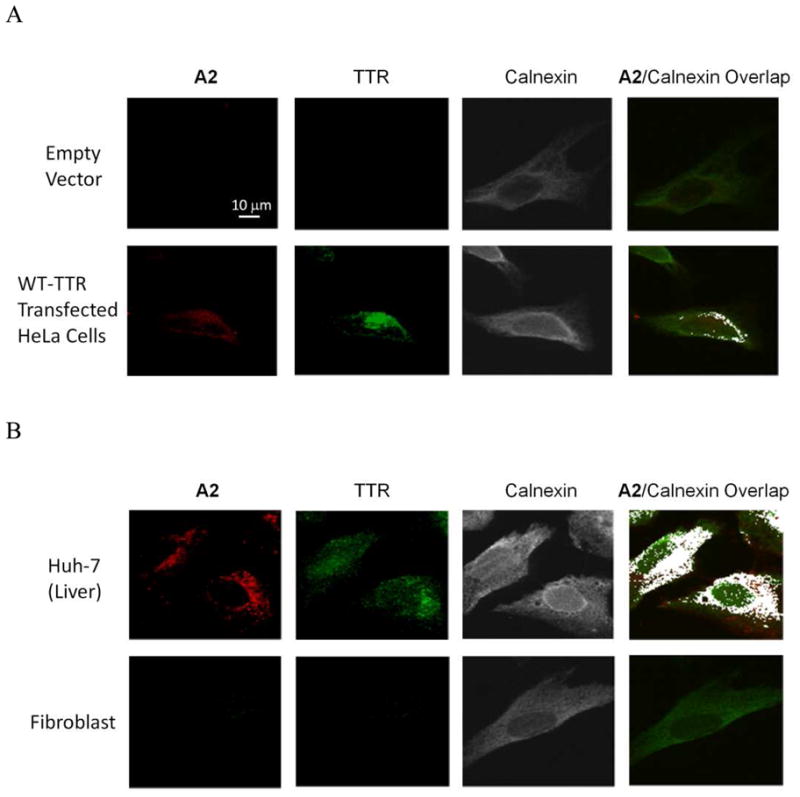
(A) Confocal fluorescence microscopy images of A2 (10 μM) treated HeLa cells transfected with empty vector (top panels) or WT-TTR (bottom panels). (B) Confocal fluorescence miscroscopy images of Huh-7 cells (liver) (top panels) and fibroblasts 30 min after 10 μM A2 treatment (bottom panels). In (A) and (B), the fluorescence from A2 is artificially colored red, WT-TTR was detected by indirect immunofluorescence (green staining) and the endoplasmic reticulum was labeled using indirect calnexin immunofluorescence (artificially colored grey). The extreme right column shows the colocalization between the A2-derived fluorescence and calnexin (artificially colored white).
We also demonstrated that endogenous TTR could be detected with A2-derived fluorescence in a common cell line secreting TTR, which is challenging, as the low quantities of TTR being detected are undergoing folding and tetramerization in the endoplasmic reticulum, vesicular trafficking to the Golgi, followed by vesicular trafficking to the plasma membrane and release into the media.36 While A2 treatment gave no significant fluorescence in human fibroblasts, consistent with reports that these cells do not make TTR (Figure 5B, bottom panels, and S10B),66 the A2-treated Huh-7 human liver (hepatoma) cells afforded strong fluorescence that colocalized with calnexin immunofluorescence (Figure 5B, top panels). The confocal fluorescence observations are consistent with substantial evidence that the liver secretes the vast majority, if not all, of the TTR into the human bloodstream.67 That TTR could be visualized in the secretory pathway addresses the sensitivity of A2-derived TTR fluorescent conjugate-based detection.36
Pulse-Chase Experiment Demonstrating WT-TTR Secretion from HeLa cells
Since imaging WT-TTR in the secretory pathway of HeLa cells post-transfection employing A2 is feasible (Figure 5A) and A2 conjugation kinetics in HeLa cell lysate (Figure 4A) is sufficiently rapid, we next utilized A2-based fluorescence to label WT-TTR in a pulse-chase experiment in living, transiently transfected HeLa cells. WT-TTR in HeLa cells (48 h post-transfection) was labeled using a 20 μM A2 pulse for 30 min, followed by a chase employing C1 (10 μM) over a 2 h time course (Figure 6). C1 is a non-fluorescent, highly selective, non-covalent, dihydrostilbene-based TTR binder31 (see Figure 6A for a line drawing of its structure). While notable TTR conjugate fluorescence was observed 30 and 60 min into the chase period, no conjugate fluorescence was detected after 120 min (Figure 6C, first column), indicating that the WT-TTR has been secreted out of the cells over that period (Figure 6B). The movement of TTR through the secretory pathway in our pulse-chase experiment (Figure 6C) is consistent with previous data from BHK and MMH cells transiently overexpressing WT-TTR demonstrating that WT-TTR secretion takes about 90 min.36 It appears that the 20 min t50 for TTR fluorescent conjugate formation observed in HeLa cell lysate (Figure 4A) is a reasonable facsimile for what happens in a transiently transfected living HeLa cell, as a 30 min pulse afforded a strong signal in the pulse-chase experiment (Figure 6C) that disappeared over time as a consequence of the C1 chase and secretion on the expected 2 h scale, with TTR secretion into the media being confirmed (Figure 6B). The reaction kinetics of A2 would become problematic if higher time resolution than tens of minutes were required for a particular application.
Figure 6.

The secretion time course of WT-TTR from HeLa cells monitored utilizing a pulse-chase experiment. HeLa cells were transfected with WT-TTR and after 48 h were treated with 20 μM A2 for a 30 min pulse. The secretion time course of labeled WT-TTR was followed by chasing the cells with the selective, non-covalent, non-fluorescent TTR kinetic stabilizer C131 (10 μM) over a chase period of 2 h. (A) Structure of the chase compound, C1. (B) Western blot demonstration that WT-TTR was secreted into the cell culture media over the same time course that TTR left the secretory pathway. (C) Confocal fluorescence microscopy images wherein the fluorescence from A2 is artificially colored red (column 1). The expression of WT-TTR in the HeLa cells only after transfection was confirmed by indirect immunofluorescence (green staining, column 2) as a function of time and the endoplasmic reticulum was labeled using indirect calnexin immunofluorescence (artificially colored grey, column 3). The extreme right column shows the colocalization between the A2 fluorescence and calnexin (artificially colored white) as a function of time.
Discussion
We describe a non-fluorescent, second generation, auxochromic stilbene A2 that very selectively binds to transthyretin in the eukaryotic cellular secretory pathway (remains dark in mammalian cells lacking TTR) and then chemoselectively reacts with a pKa perturbed Lys-15 ε-amino group in TTR–creating a bright blue fluorescent conjugate. Stilbene A2 exhibits sufficiently rapid TTR conjugation kinetics at 37 °C to enable pulse-chase experiments to be performed within the secretory pathway of HeLa cells, demonstrating that transthyretin can be imaged as it transits through the secretory pathway. The progress reported is viewed as a first and necessary step toward our long-term goal of creating a one-chain, one-binding-site transthyretin tag that can be used in the eukaryotic secretory pathway to follow protein secretion and turnover utilizing A2, or an analogous molecule. While the reaction kinetics of A2 with TTR are well-suited for studying the folding, trafficking, secretion and degradation of TTR and TTR-tagged proteins, other thiol containing functional groups (required to keep the latent fluorophores dark) with faster reaction kinetics are being explored to carry out pulse-chase experiments with higher time resolution.
An immediate application of A2 is to utilize the conjugate fluorescence to quantify TTR concentration in human plasma. The TTR concentration in plasma is an established metric of nutritional status and is an often used clinical laboratory test.68 A2 demonstrates sufficient binding selectivity to TTR in human plasma and amide bond reaction chemoselectivity with Lys-15 in TTR (Figure S3A, B) to create a new method that would replace the error-prone immunoprecipitation–turbidity assay widely used in clinical laboratories today to quantify TTR plasma levels.
Another immediate application of A2 is the quantification of kinetic stabilizer binding stoichiometry to rare variants of TTR in human plasma.42 Tafamidis, a TTR kinetic stabilizer that we discovered,52 has recently been shown in a phase II/III placebo-controlled clinical trial to halt the progression of familial amyloid polyneuropathy (FAP). Negatively cooperative binding of Tafamidis to one of the two T4 binding sites in the TTR tetramer is sufficient to increase the kinetic barrier for tetramer dissociation (through selective ground state stabilization) such that it becomes insurmountable under physiological conditions–preventing amyloidogenesis that causes FAP.29,69–71 While the human oral dose required for occupancy of more than one TTR binding site is known for the most common TTR FAP-associated mutations, this has not been established for all mutations. It is now feasible to utilize A2-derived conjugate fluorescence to measure the stoichiometry of kinetic stabilizers, such as Tafamidis, bound to TTR in patient plasma using an A2-kinetic stabilizer competition assay. After assay optimization, physicians could use such an approach to titrate the dose of kinetic stabilizer to be used in patients with rare mutations to be sure that the kinetic stabilizer occupies at least one TTR binding site per tetramer.57 Since there are over one hundred TTR mutations that cause FAP, developing the A2 method for determining kinetic stabilizer TTR binding stoichiometry is important and especially timely, since Tafamidis, the first kinetic stabilizer to be shown to be efficacious, is expected to become a regulatory agency approved drug during the coming year.52,57,69,70
Supplementary Material
Acknowledgments
We thank the NIH (DK46335) as well as the Skaggs Institute for Chemical Biology and the Lita Annenberg Hazen Foundation for financial support and Professors David Millar at Scripps, Alice Ting at MIT and the reviewers for helpful comments. Technical support from Dr. Tingwei Mu, Dr. Steve Bourgault and Mike Saure are also greatly appreciated.
Footnotes
Supporting Information Available: Procedures for the reverse phase-HPLC analyses, the binding stoichiometry measurement, photoisomerization, the detection of TTR labeling by SDS-PAGE, the western blot analyses, the synthesis of A1, A2, and B1, and additional data referred to in the body of the manuscript are available free of charge via Internet at http://pubs.acs.org.
References
- 1.Tsien RY. Angew Chem Int Ed Engl. 2009;48:5612–5626. doi: 10.1002/anie.200901916. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Suarez M, Ting AY. Nat Rev Mol Cell Biol. 2008;9:929–43. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 3.VanEngelenburg SB, Palmer AE. Curr Opin Chem Biol. 2008;12:60–65. doi: 10.1016/j.cbpa.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 5.Heim R, Cubitt AB, Tsien RY. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 6.Heim R, Prasher DC, Tsien RY. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 8.Shimomura O, Johnson FH, Saiga Y. J Cell Comp Physiol. 1962;59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- 9.Tsien RY. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 10.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Eisman MH, Lohse MJ. Nat Methods. 2005;2:171–176. doi: 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- 12.Adams SR, Tsien RY. Nat Protoc. 2008;3:1527–34. doi: 10.1038/nprot.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luedtke NW, Dexter RJ, Fried DB, Schepartz A. Nat Chem Biol. 2007;3:779–84. doi: 10.1038/nchembio.2007.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan B, Gierasch LM. Chem Biol. 2008;15:1104–15. doi: 10.1016/j.chembiol.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen I, Howarth M, Lin W, Ting AY. Nat Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 16.Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Proc Natl Acad Sci USA. 2004;101:9955–9959. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guignet EG, Hovius R, Vogel H. Nat Biotechnol. 2004;22:440–4. doi: 10.1038/nbt954. [DOI] [PubMed] [Google Scholar]
- 18.Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. Proc Natl Acad Sci USA. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer AE. ACS Chem Biol. 2009;4:157–159. doi: 10.1021/cb9000525. [DOI] [PubMed] [Google Scholar]
- 20.Gautier A, Juillerat A, Heinis C, Correa IR, Kindermann M, Beaufils F, Johnsson K. Chem Biol (Cambridge, MA, U S) 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Regoes A, Hehl AB. BioTechniques. 2005;39:809–810. 812. doi: 10.2144/000112054. [DOI] [PubMed] [Google Scholar]
- 22.Brecht A, Gibbs T. BIOforum Eur. 2005;9:50–51. [Google Scholar]
- 23.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 24.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 25.Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C. J Mol Biol. 1978;121:339–56. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 26.Hornberg A, Eneqvist T, Olofsson A, Lundgren E, Sauer-Eriksson AE. J Mol Biol. 2000;302:649–69. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]
- 27.Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Curr Opin Struc Biol. 2010;20:54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foss TR, Wiseman RL, Kelly JW. Biochemistry. 2005;44:15525–33. doi: 10.1021/bi051608t. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 30.Johnson SM, Connelly S, Wilson IA, Kelly JW. J Med Chem. 2008;51:6348–6358. doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi S, Reixach N, Connelly S, Johnson SM, Wilson IA, Kelly JW. J Am Chem Soc. 2010;132:1359–1370. doi: 10.1021/ja908562q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi S, Connelly S, Reixach N, Wilson IA, Kelly JW. Nat Chem Biol. 2010;6:133–139. doi: 10.1038/nchembio.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hurshman AR, White JT, Powers ET, Kelly JW. Biochemistry. 2004;43:7365–81. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 34.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. Biochemistry. 2001;40:11442–52. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 35.Foss TR, Kelker MS, Wiseman RL, Wilson IA, Kelly JW. J Mol Biol. 2005;347:841–54. doi: 10.1016/j.jmb.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 36.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 38.Mu TW, Ong DST, Wang YJ, Balch WE, Yates JR, Segatori L, Kelly JW. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lashuel HA, Wurth C, Woo L, Kelly JW. Biochemistry. 1999;38:13560–73. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- 40.Crosby GA, Demas JN. J Phys Chem. 1971;75:991–1024. [Google Scholar]
- 41.Baures PW, Peterson SA, Kelly JW. Bioorg Med Chem. 1998;6:1389–401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 42.Purkey HE, Dorrell MI, Kelly JW. Proc Natl Acad Sci USA. 2001;98:5566–5571. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammarstrom P, Jiang X, Deechongkit S, Kelly JW. Biochemistry. 2001;40:11453–9. doi: 10.1021/bi010673+. [DOI] [PubMed] [Google Scholar]
- 44.Gorner H, Kuhn HJ. Adv, Photochem. 1995;19:1. [Google Scholar]
- 45.Malkin S, Fischer E. J Phys Chem. 1964;68:1153. [Google Scholar]
- 46.Saltiel J. J Am Chem Soc. 1967;89:1036–1037. [Google Scholar]
- 47.Saltiel J, Dagostin Jt. J Am Chem Soc. 1972;94:6445. [Google Scholar]
- 48.Saltiel J, Marinari A, Chang DWL, Mitchener JC, Megarity ED. J Am Chem Soc. 1979;101:2982–2996. [Google Scholar]
- 49.Saltiel J, Zafiriou OC, Megarity ED, Lamola AA. J Am Chem Soc. 1968;90:4759. [Google Scholar]
- 50.Baures PW, Oza VB, Peterson SA, Kelly JW. Bioorg Med Chem. 1999;7:1339–47. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 51.Petrassi HM, Klabunde T, Sacchettini JC, Kelly JW. J Am Chem Soc. 2000;122:2178–2192. [Google Scholar]
- 52.Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN, Deechongkit S, Chiang KP, Dendle MT, Sacchettini JC, Kelly JW. Angew Chem Int Ed Engl. 2003;42:2758–2761. doi: 10.1002/anie.200351179. [DOI] [PubMed] [Google Scholar]
- 53.Purkey HE, Palaninathan SK, Kent KC, Smith C, Safe SH, Sacchettini JC, Kelly JW. Chem Biol. 2004;11:1719–1728. doi: 10.1016/j.chembiol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Johnson SM, Connelly S, Wilson IA, Kelly JW. J Med Chem. 2008;51:260–270. doi: 10.1021/jm0708735. [DOI] [PubMed] [Google Scholar]
- 55.Johnson SM, Connelly S, Wilson IA, Kelly JW. J Med Chem. 2009;52:1115–1125. doi: 10.1021/jm801347s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrassi HM, Johnson SM, Purkey H, Chiang KP, Walkup T, Jiang X, Powers ET, Kelly JW. J Am Chem Soc. 2005;127:6662–6671. doi: 10.1021/ja044351f. [DOI] [PubMed] [Google Scholar]
- 57.Wiseman RL, Johnson SM, Kelker MS, Foss T, Wilson IA, Kelly JW. J Am Chem Soc. 2005;127:5540–51. doi: 10.1021/ja042929f. [DOI] [PubMed] [Google Scholar]
- 58.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. J Med Chem. 2004;47:355–74. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 59.Green NS, Palaninathan SK, Sacchettini JC, Kelly JW. J Am Chem Soc. 2003;125:13404–14. doi: 10.1021/ja030294z. [DOI] [PubMed] [Google Scholar]
- 60.Oza VB, Petrassi HM, Purkey HE, Kelly JW. Bioorg Med Chem Lett. 1999;9:1–6. doi: 10.1016/s0960-894x(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 61.Oza VB, Smith C, Raman P, Koepf EK, Lashuel HA, Petrassi HM, Chiang KP, Powers ET, Sachettinni J, Kelly JW. J Med Chem. 2002;45:321–32. doi: 10.1021/jm010257n. [DOI] [PubMed] [Google Scholar]
- 62.Miller SR, Sekijima Y, Kelly JW. Lab Invest. 2004;84:545–52. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 63.Johnson SM, Petrassi HM, Palaninathan SK, Mohamedmohaideen NN, Purkey H, Nichols C, Chiang KP, Walkup T, Sacchettini JC, Sharpless KB, Kelly JW. J Med Chem. 2005;48:1576–1587. doi: 10.1021/jm049274d. [DOI] [PubMed] [Google Scholar]
- 64.Simeonov A, Matsushita M, Juban EA, Thompson EH, Hoffman TZ, Beuscher AE, Taylor MJ, Wirsching P, Rettig W, McCusker JK, Stevens RC, Millar DP, Schultz PG, Lerner RA, Janda KD. Science. 2000;290:307–13. doi: 10.1126/science.290.5490.307. [DOI] [PubMed] [Google Scholar]
- 65.Debler EW, Kaufmann GF, Meijler MM, Heine A, Mee JM, Pljevaljcic G, Di Bilio AJ, Schultz PG, Millar DP, Janda KD, Wilson IA, Gray HB, Lerner RA. Science. 2008;319:1232–5. doi: 10.1126/science.1153445. [DOI] [PubMed] [Google Scholar]
- 66.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Felding P, Fex G. Biochimica et Biophysica Acta. 1982;716:446–9. doi: 10.1016/0304-4165(82)90040-x. [DOI] [PubMed] [Google Scholar]
- 68.Ingenbleek YYV. Ann Rev Nutrition. 1994;14:495–533. doi: 10.1146/annurev.nu.14.070194.002431. [DOI] [PubMed] [Google Scholar]
- 69.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Science. 2003;299:713–6. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 70.Hammarstrom P, Schneider F, Kelly JW. Science. 2001;293:2459–62. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 71.Lai Z, McCulloch J, Lashuel HA, Kelly JW. Biochemistry. 1997;36:10230–9. doi: 10.1021/bi963195p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.