


Abstract
The European Large Subunit Ribosomal RNA Database compiles all complete or nearly complete large subunit ribosomal RNA sequences available from public sequence databases. These are provided in aligned format and the secondary structure, as derived by comparative sequence analysis, is included. Additional information about the sequences such as literature references and taxonomic information is also included. The database is available from our WWW server at http://rrna.uia.ac.be/lsu/.
LARGE SUBUNIT RIBOSOMAL RNA
The secondary structure model of the large subunit ribosomal RNA (LSU rRNA) consists of a conserved core that is interspersed with a number of variable areas. Most of the secondary structure of the molecule is known, especially for archaeal, bacterial and plastid sequences. In eukaryotes some areas show considerable variability, both in base composition and sequence length, even when closely related species are compared. This makes alignment more difficult, and as a result the secondary structure annotation, which is based on the primary structure alignment, is less reliable or even absent for these areas. The same problem arises with sequences from animal mitochondria and is even more pronounced with those from kinetoplast mitochondria.
Very often the LSU rRNA is not formed by one continuous molecule, but is fragmented in two or more parts. The most widely known fragments are 5.8S rRNA in eukaryotes and 4.5S rRNA in plastids but some LSU rRNA sequences show more extensive fragmentation. One extreme example is the sequence of Euglena gracilis, which consists of 14 pieces (1). In our alignment these fragments are joined in one continuous sequence but information about the individual fragments is also provided.
Most of our knowledge about the secondary and tertiary structure of LSU rRNA is based on comparative sequence analysis. Recently, the crystal structure of the large ribosomal subunit of the archaebacterium Haloarcula marismortui has been resolved at 2.4 Å resolution (2). This model confirmed the secondary structure model derived by comparative sequence analysis and added a vast amount of information about tertiary interactions. Only a very small number of inaccuracies in the comparative model were noted and have been removed from the alignment. These data again confirm the validity of comparative sequence analysis of RNA molecules and serves as a good indication that secondary structure models of other sequences in our database indeed reflect the true secondary structure of these molecules in the ribosome.
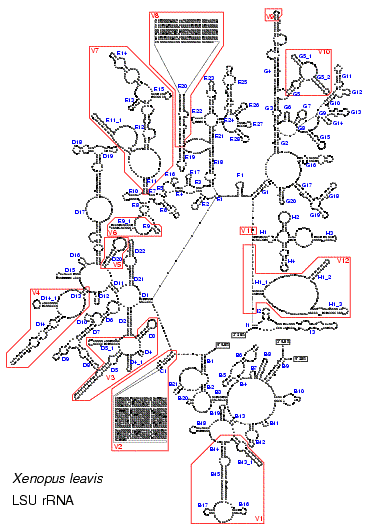
Figure 1 shows the complete secondary structure model of LSU rRNA of the Apicomplexan Toxoplasma gondii. The drawing was made using the software package RnaViz (3). In the drawing, nine substructures branching from the central loop and labeled from A to I are clearly visible. Individual helices are numbered clockwise from the 5′- to the 3′-end in each substructure and are given an individual number if a potential branching point separates them. Substructure A, which is formed by helix A1, is absent in eukaryotes but is present in bacteria, plastids and most archaea.
Figure 1.

Secondary structure model of LSU rRNA from the Apicomplexan T.gondii (EMBL accession number X75453). The sequence is written clockwise from the 5′- to the 3′-terminus.
CONTENTS OF THE DATABASE
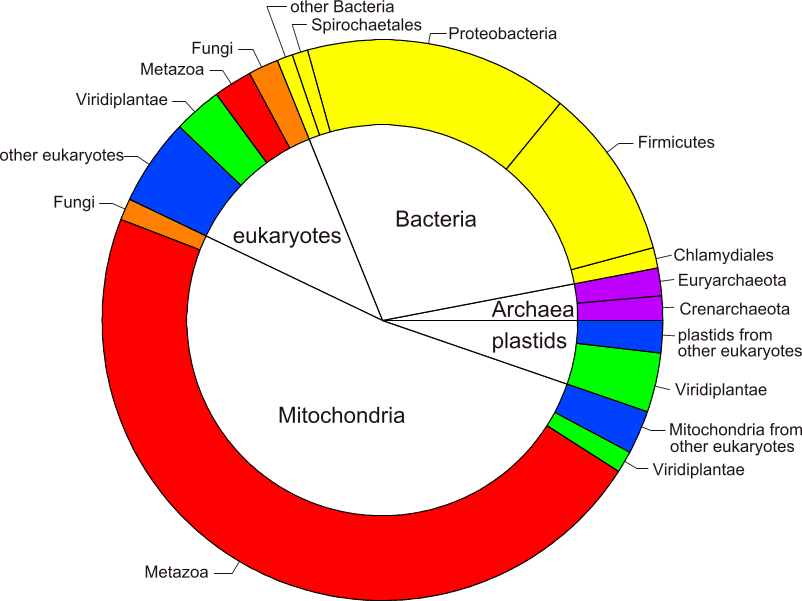
The database contains approximately 1400 sequences. Mitochondria and Bacteria have the largest number of representatives with about 750 and 400 sequences, respectively. There are approximately 150 eukaryote, 50 archaebacterial and 70 plastid sequences. The Supplementary Material includes a pie chart showing a graphical overview of the relative amount of representative sequences for all taxa.
The alignment is regularly updated from the EMBL sequence database (4). The LSU rRNA features are extracted from new and updated entries. Using the alignment editor DCSE (5), each new sequence is automatically aligned to the complete sequence of its closest relative and retained only if it shares at least 70% of the homologous positions. Next, the secondary structure pattern is applied to the new sequence. Where necessary, manual corrections are made.
The secondary structure of a sequence is indicated by the insertion of special characters in the alignment. Square brackets are used to indicate helix segments. The number of this helix can be found by looking at the corresponding position in a special sequence called ‘Helix numbering’. Internal loops and bulge loops are annotated by braces. Non-standard base pairs (base pairs other than A·U, G·C or G·U) are indicated by round brackets.
In addition to primary and secondary structure, other information such as literature references, accession numbers and taxonomy are also available. The taxonomic descriptions are the same as those adopted by the NCBI (6).
AVAILABILITY
The European LSU rRNA Database is accessible at http://rrna.uia.ac.be/lsu/. Sequence alignments can be downloaded in different formats. The ‘DCSE alignment’, ‘Distribution format’ and ‘Printable alignment’ formats incorporate both primary and secondary structure information. TREECON, NBRF/PIR and EMBL formats only include the primary structure in aligned format.
Three interfaces are available to select the desired sequences. Using the list interface one can select individual sequences that can be downloaded in the distribution format. The forms interface allows the selection of groups of sequences. The query interface allows one to search for sequences by species name, accession number and literature data. It is possible to do searches on the entire database or to limit the search to certain taxa.
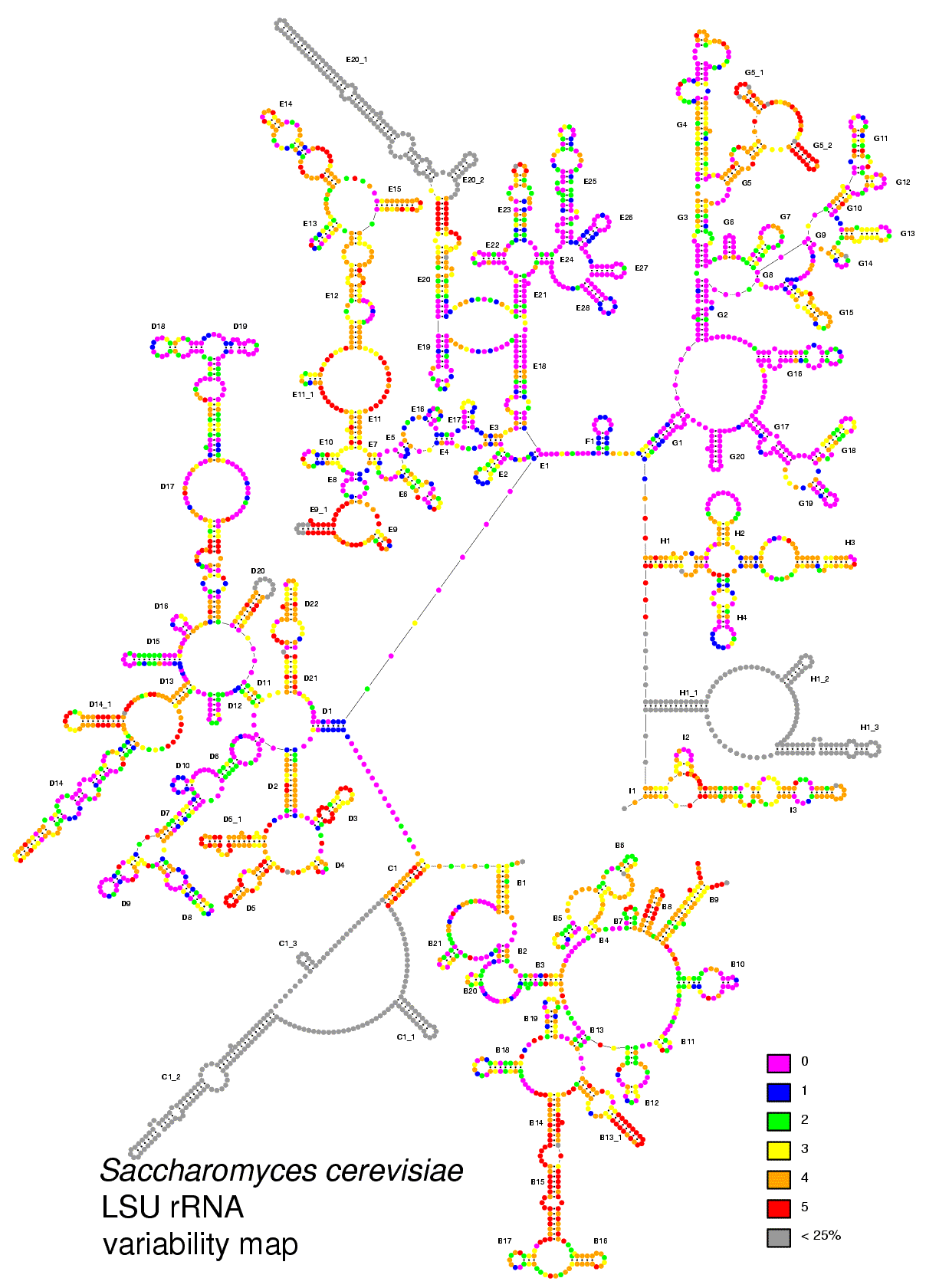
Additional information available on the RNA server includes a database with information on PCR and sequencing primers used in our laboratory, detailed variability maps based on substitution rate calibration (7) and links to other interesting sites. Various software packages for molecular biology are also available. These include DCSE (5), an alignment editor that supports secondary structure annotation, RnaViz (3), a program to make RNA secondary structure drawings, and Forcon (8), a program for the conversion between various alignment formats.
From our anonymous ftp server at ftp://rrna.uia.ac.be/pub/lsu/ individual sequences in the distribution format are available. A compressed version of the complete DCSE alignment file and the DCSE reference file is also available.
SUPPLEMENTARY MATERIAL
The following additional information can be found at NAR Online:
• pie chart showing the distribution of different taxa in the database;
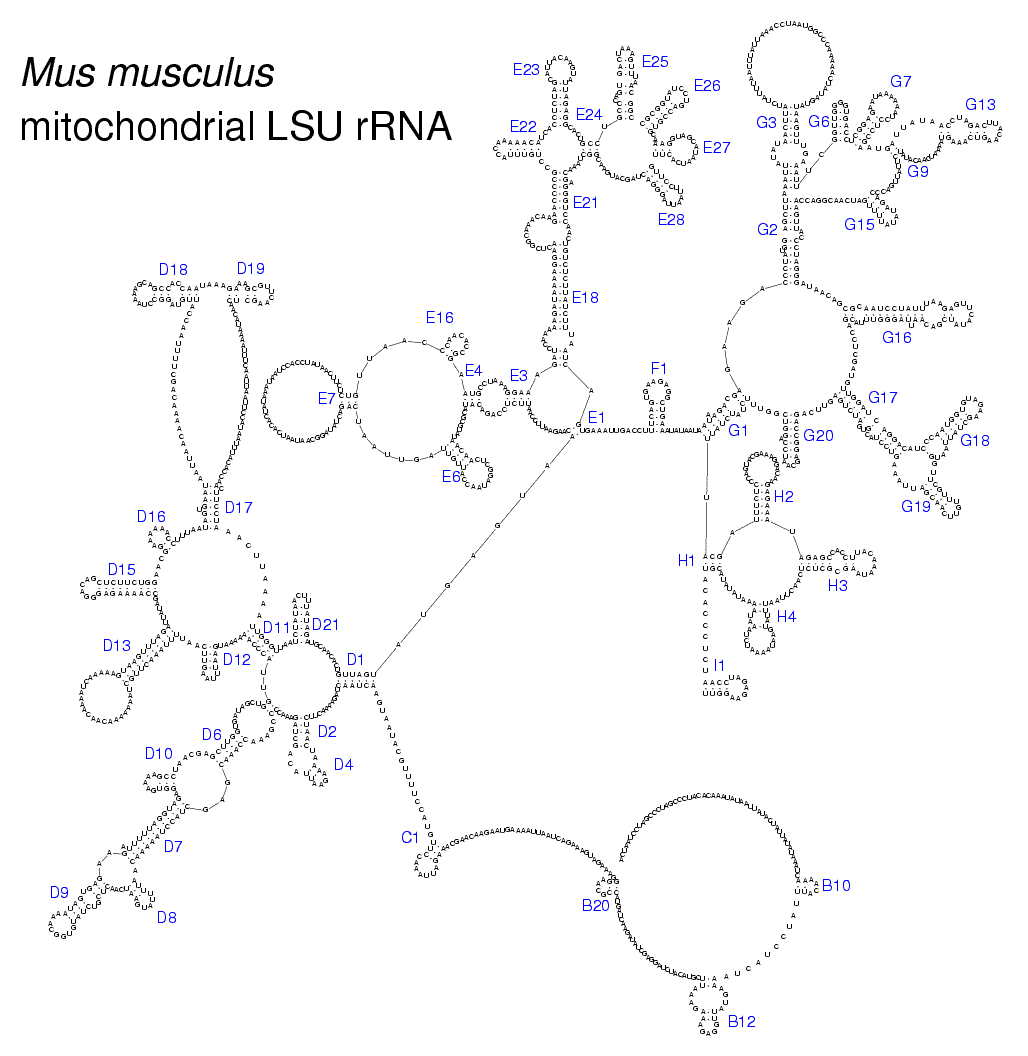
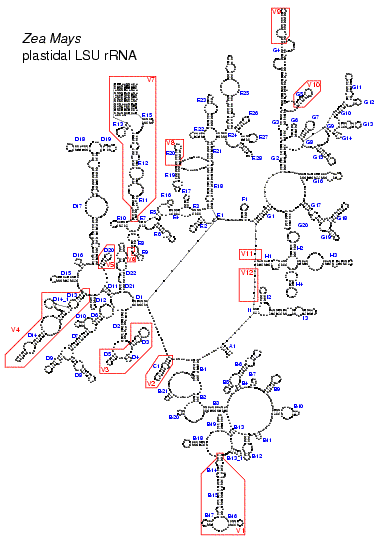
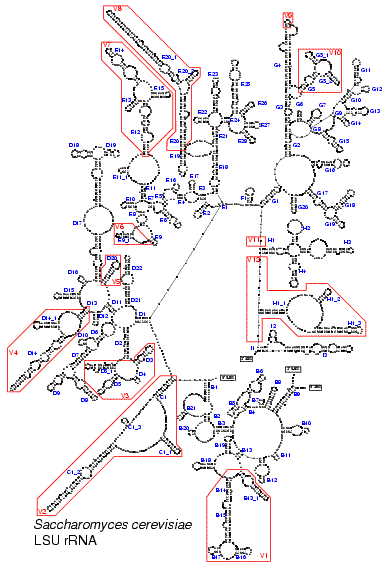
• detailed secondary structure models of several LSU rRNA species;
• nucleotide variability maps of eukaryotic and bacterial LSU rRNA molecules.
Acknowledgments
ACKNOWLEDGEMENTS
Our research is supported by the Special Research Fund of the University of Antwerp (Belgium). P.D.R. and Y.V.d.P. are Research Fellows of the Fund for Scientific Research (Flanders).
References
- 1.Schnare M.N., Cook,J.R. and Gray,M.W. (1994) Fourteen internal transcribed spacers in the circular ribosomal DNA of Euglena gracilis. J. Mol. Biol., 215, 85–91. [DOI] [PubMed] [Google Scholar]
- 2.Ban N., Nissen,P., Hansen,J., Moore,P.B. and Steitz,T.A. (2000) The complete atomic structure of the large ribosomal subunit at 2.4Å resolution. Science, 289, 905–920. [DOI] [PubMed] [Google Scholar]
- 3.De Rijk P. and De Wachter,R. (1997) RnaViz, a program for the visualisation of RNA secondary structure. Nucleic Acids Res., 25, 4679–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker W., van den Broek,A., Camon,E., Hingamp,P., Sterk,P., Stoesser,G. and Tuli,M.A. (2000) The EMBL Nucleotide Sequence Database. Nucleic Acids Res., 28, 19–23. Updated article in this issue: Nucleic Acids Res. (2001), 29, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Rijk P. and De Wachter,R. (1993) DCSE, an interactive tool for sequence alignment and secondary structure research. Comput. Appl. Biosci., 9, 735–740. [DOI] [PubMed] [Google Scholar]
- 6.Wheeler D.L., Chappey,C., Lash,A.E., Leipe,D.D., Madden,T.L., Schuler,G.D., Tatusova,T.A. and Rapp,B.A. (2000) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 28, 10–14. Updated article in this issue: Nucleic Acids Res. (2001), 29, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van de Peer Y., Van der Auwera,G. and De Wachter,R. (1996) The evolution of stramenopiles and alveolates as derived by “substitution rate calibration” of small ribosomal subunit RNA. J. Mol. Evol., 42, 201–210. [DOI] [PubMed] [Google Scholar]
- 8.Raes J. and Van de Peer,Y. (1999) ForCon: a software tool for the conversion of sequence alignments. EMBnet.news, 6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}