

Abstract
Background
We previously reported the association among donor leukocyte chimerism, apoptosis of presumedly IL-2-deficient graft-infiltrating host cells, and the spontaneous donor-specific tolerance induced by liver but not heart allografts in mice. Survival of the rejection-prone heart allografts in the same strain combination is modestly prolonged by the pretransplant infusion of immature, costimulatory molecule (CM) deficient donor dendritic cells (DC), an effect that is markedly potentiated by concomitant CM blockade with anti-CD40L (CD154) monoclonal antibody (mAb). We investigated whether the long survival of the heart allografts in the pretreated mice was associated with donor leukocyte chimerism and apoptosis of graft-infiltrating cells, if these end points were similar to those in the spontaneously tolerant liver transplant model, and whether the pretreatment effect was dependent on sustained inhibition of CM expression of the infused immature donor DC. In addition, apoptosis was assessed in the host spleen and lymph nodes, a critical determination not reported in previous studies of either spontaneous or “treatment-aided” organ tolerance models.
Methods
Seven days before transplantation of hearts from B10 (H-2b) donors, 2 × 106 donor-derived immature DC were infused i.v. into C3H (H-2k) recipient mice with or without a concomitant i.p. injection of anti-CD40L mAb. Donor cells were detected post-transplantation by immunohistochemical staining for major histocompatibility complex class II (I-Ab) in the cells of recipient lymphoid tissue. CM expression was determined by two-color labeling. Host responses to donor alloantigen were quantified by mixed leukocyte reaction, and cytotoxic T lymphocyte (CTL) assays. Apoptotic death in graft-infiltrating cells and in areas of T-dependent lymphoid tissue was visualized by terminal deoxynucleotidyltransferase-catalyzed dUTP-digoxigenin nick-end labeling and quantitative spectrofluorometry. Interleukin-2 production and localization were estimated by immunohistochemistry.
Results
Compared with control heart transplantation or heart transplantation after only DC administration, concomitant pretreatment with immature donor DC and anti-CD40L mAb caused sustained elevation of donor (I-Ab+) cells (microchimerism) in the spleen including T cell areas. More than 80% of the I-Ab+ cells in combined treatment animals also were CD86+, reflecting failure of the mAb to inhibit CD40/CD80/CD86 up-regulation on immature DC in vitro after their interaction with host T cells. Donor-specific CTL activity in graft-infiltrating cells and spleen cell populations of these animals was present on day 8, but decreased strikingly to normal control levels by day 14. The decrease was associated with enhanced apoptosis of graft-infiltrating cells and of cells in the spleen where interleukin-2 production was inhibited. The highest levels of splenic microchimerism were found in mice with long surviving grafts (> 100 days). In contrast, CTL activity was persistently elevated in control heart graft recipients with comparatively low levels of apoptotic activity and high levels of interleukin-2.
Conclusion
The donor-specific acceptance of rejection-prone heart allografts by recipients pretreated with immature donor DC and anti-CD40L mAb is not dependent on sustained inhibition of donor DC CM (CD86) expression. Instead, the pretreatment facilitates a tolerogenic cascade similar to that in spontaneously tolerant liver recipients that involves: (1) chimerism-driven immune activation, succeeded by deletion of host immune responder cells by apoptosis in the spleen and allograft that is linked to interleukin-2 deficiency in both locations and (2) persistence of comparatively large numbers of donor-derived leukocytes. These tolerogenic mechanisms are thought to be generic, explaining the tolerance induced by allografts spontaneously, or with the aid of various kinds of immunosuppression.
Compelling evidence has been summarized elsewhere that organ and bone marrow allograft “acceptance” are related forms of acquired tolerance that are not fundamentally different than the tolerance induced by noncytopathic microorganisms (1). With both varieties of transplantation, coexisting donor and recipient immune cells undergo reciprocal activation followed by various levels of mutual antigen-specific clonal exhaustion-deletion, the maintenance of which in the organ recipient depends on persistence of the donor leukocytes (microchimerism) (2–4). Verification of this concept has been greatly facilitated by the use of rodent models, in which tolerance occurs spontaneously (5), most commonly involving transplantation of the leukocyte-rich liver (6–12).
We recently showed in such a mouse liver transplant tolerance model that the clonal deletion associated with immune activation was largely due to apoptosis of cytotoxic T lymphocytes (CTL)* (13). The apoptosis in graft-infiltrating cells (GIC) reached a peak between days 7 and 14, effectively forestalling rejection and eventually eliminating most of the antigraft CTL. Although low but significant CTL activity could still be detected in the allografts and host spleens at 150 days, such liver recipients are stable thereafter (7). The programed cell death of CTL was inhibited by administration of interleukin-(IL)2 (13), with consequent increased hepatocyte destruction. Similar findings have been reported in a spontaneously tolerant rat model of liver (or composite liver/intestinal) transplantation (14). In our investigation (13) and in that of Meyer et al. (14), apoptosis was not determined in the host lymphoid organs where “high dose” clonal exhaustion-deletion has been described (11).
In our investigation using a mouse strain combination in which livers but not hearts reliably induce tolerance (13), the mechanisms of engraftment of the less tolerogenic heart allografts were studied after the preoperative administration of an immunosuppressant with a known molecular target, in combination with adjunct infusions of well-characterized donor leukocytes. The heart recipients were pretreated with a monoclonal antibody (mAb) directed against the CD40 ligand (CD40L, CD154) that is expressed by activated T cells. The adjunct cells given concomitantly were donor-derived immature DC that have a phenotype resembling interstitial DC resident within nonlymphoid tissue (e.g., heart or liver) (15) and are known to have low immunogenicity that is associated with poor B7 expression (16). Given alone, a single pretransplant i.v. infusion of these immature donor cells (propagated either from liver or bone marrow) on day –7 modestly prolongs the survival of pancreatic islet (17) and cardiac allografts (18). This minor therapeutic effect is dramatically enhanced with the concomitant i.p. administration on day –7 of anti-CD40L mAb, allowing consistently long or indefinite survival of normally rejection-prone cardiac allografts (16).
The principal objective of our studies was to determine if the heart allograft acceptance under these therapeutically altered conditions occurred by the same chimerism and apoptosis-associated mechanisms as with spontaneous liver allograft acceptance. This appeared to be true. In addition, apoptosis in the host lymphoid tissues was determined using the spleen as the prototype lymphoid organ. This revealed programed cell death in the spleen at least equal to that in the heart grafts.
MATERIALS AND METHODS
Mice
Male C57BL/10J (B10; H-2b), C3H/HeJ (C3H; H-2k), and BALB/c (H-2d) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). They were maintained in the specific pathogen-free facility of the University of Pittsburgh Medical Center, provided with Purina Rodent Chow (Ralston Purina, St. Louis, MO) and tap water ad libitum, and used at 8–12 wk old.
Anti-CD40L mAb
M158, a rat IgG mAb specific for mouse CD40L (CD154) was provided by the Immunex Corporation, Seattle WA. It was administered i.p. (200 μg) as a single injection.
Heterotopic cardiac transplantation
Fully allogeneic intraab-dominal vascularized heart transplantation was performed from normal B10 donors to size-matched C3H recipients, as described (18). Graft survival was assessed by daily transabdominal palpation of the hearts. Rejection was determined as cessation of heartbeat, and confirmed histologically.
Experimental organ transplantation protocol
Normal C3H mice received donor (B10) or control syngeneic or third-party (BALB/c)-derived immature DC, either alone or in combination with anti-CD40L mAb (or control rat IgG). Cells (2 × 106) were administered i.v. with or without anti-CD40L mAb, 7 days before B10 heart transplantation on day 0. On days 0, 7, and 14 posttransplant, groups of mice were killed. Spleens and heart grafts were examined by immunohistochemical staining for donor major histocompatibility complex (MHC) class II, and B7 molecule expression, and by terminal deoxynucleotidyl transferase-catalyzed dUTP-digoxigenin nick-end labeling (TUNEL) staining for apoptotic cells. At the same times (days 7 and 14), antidonor CTL activity of both GIC and spleen cells was determined.
Propagation and purification of BM-derived myeloid dendritic cells (DC)
BM cells harvested from femurs of normal B10 or control strain mice were cultured in 24-well plates (2 × 106/well) in 2 ml of RPMI-1640 (Life Technologies, Gaithersburg, MD) supplemented with antibiotics, and 10% v/v fetal bovine serum (referred to subsequently as complete medium), 4 ng/ml recombinant (r) mouse granulocyte-macrophage colony-stimulating factor (GM-CSF), and either 1000 U/ml r mouse IL-4 (both cytokines from Schering Plough, Kenilworth, NJ), or r human transforming growth factor β (TGFβ)1 (0.2 ng/ml; R & D Systems Inc., Minneapolis, MN). The culture and selection procedures used to generate and purify the DC were similar to those reported initially by Inaba et al. (19) and modified as described (16, 20).
Flow cytometry of cultured cells
Cell surface molecule expression was analyzed by cytofluorography, using an EPICS ELITE flow cytometer (Coulter Corporation, Hialeah, FL). Staining with primary rat or hamster mAbs, including antibodies (Abs) directed against mouse dentric cell-(DC) restricted (CD11c and DEC205) and myeloid lineage markers (CD11b and F4/80), and rat anti-mouse CD40, CD80 (B7-1), or CD86 (B7-2) (PharMingen, San Diego, CA) was followed by fluorescein isothiocyanate-(FITC) conjugated goat anti-rat IgG2a, or goat anti-hamster Ig, as described (20), MHC class II (I-Ab) staining was performed using biotin-conjugated mouse anti-mouse mAbs, with FITC streptavidin as the secondary reagent (8).
T cell allostimulatory activity
One-way mixed leukocyte reaction (MLR) cultures were established in 96-well, round-bottom microculture plates (Corning, Corning, NY). Graded doses of γ-irradiated syngeneic (C3H) or allogeneic (B10) stimulator cells were added to 1 or 2×105 nylon wool-eluted C3H spleen cells used as responders (8). Cultures were maintained for 72 hr; [3H]TdR (1 μCi/well) was added for the final 18 hr, and incorporation of [3H]TdR into DNA assessed by liquid scintillation counting. Results were expressed as mean counts per minute (cpm) ± 1 SD.
Isolation of GIC
Animals were perfused in situ via the left ventricle with 30 ml collagenase type IV (0.1 mg/ml; Sigma Chemical Co., St. Louis, MO). Heart grafts from experimental or control groups (three mice per group) were pooled, diced, and further digested in collagenase (1 mg/ml) at 37°C for 30 min. This was followed by Percoll (Sigma) centrifugation, as described previously for the isolation of nonparenchymal cells (18). The cells between the top (digested parenchymal/myocardial cells) and the lower (erythrocyte) layers were collected, washed, and the number adjusted to 5 × 106 ml in RPMI-1640 complete medium.
Cell-mediated lymphocytotoxicity (CML) assay
Freshly isolated normal heart nonparenchymal cells, heart graft-infiltrating cells, or spleen cells were used as effectors. The EL4 (H-2b) lymphoma cell line [TIB39; American Type Culture Collection (ATCC), Rockville. MDI was used as a source of specific target cells. P815 (H-2d) mouse mastocytoma cells (TIB64, ATCC) were used as specificity controls, and as lymphokine-activated killer cell targets. The target cells were labeled with 100 μCi Na251Cr O4 (NEN, Life Science Products, Boston, MA), washed, and plated at 5 × 103 cells/well in 96-well, round-bottomed culture plates (Corning). Serial, 2-fold dilutions of effector cells were added to give effector:target ratios of 100:1, 50:1. and 25:1, in a total volume of 200 μl/well. The percentage of specific 51Cr release was determined after incubating the plates for 4 hr at 37°C in 5% CO2 in air. An aliquot (100 μl) of supernatant was recovered from each well after centrifugation at 500×g for 1 min. Maximum 51Cr release was determined by osmotic lysis of the target cells. The percent specific cytotoxicity was calculated using the following formula: % cytotoxicity = 100 × [experimental (cpm) − spontaneous (cpm)/maximum (cpm) − spontaneous (cpm)]. The results are expressed as means± 1 SD of percent specific 51Cr release in triplicate cultures.
In situ nick-end labeling
DNA strand breaks were identified in cryostat sections (13) or in cell suspensions allowed to settle on glass slides (21) by terminal deoxynucieotidyl transferase-catalyzed dUTP-digoxigenin nick-end labeling (TUNEL). Slides were immersed in 2% H2O2 for 5 min at room temperature (RT) to quench endogenous peroxidase activity. They were then incubated with 20 μg/ml proteinase K (Sigma) for 15 min at RT, washed in phosphate-buffered saline, then immersed in 100 μl reaction buffer [0.2 M potassium cacodylate, 25 mM Tris-HCl (pH 6.6), 0.25 mg/ml bovine serum albumin, and 2.5 mM cobalt chloride] supplemented with TdT (0.3 enzyme units/ml and digoxigenin-conjugated-dUTP. Each experiment was performed with a negative control (without dUTP) and a positive control (10-min pretreatment of slides with 1 mg/ml DNase in reaction buffer). Peroxidase-labeled Ab to digoxigenin (Oncor Technical Assistance, Gaithersburg, MD) was added (30 min RT), and the reaction developed with 3-amino-5-ethycarbizol. The slides were counterstained with Harris’ hematoxylin, and mounted with Crystal mount (Biomeda Corp., Foster City, CA).
DNA fragmentation assay
DNA fragmentation in cell lysates was determined by spectrofluorometric assay, as described (21).
lmmunohistochemistry
Immunohistochemical staining of tissue sections for donor MHC class II antigen and CM, including two-color labeling, was performed as described (18, 22).
Statistical analyses
Results are expressed as means ± SD. Statistical analyses were performed using the Student’s t test, or the Mann Whitney U test, as appropriate.
RESULTS
Surface immunophenotype and allostimulatory activity of BM-derived cells propagated in GM-CSF+TGFβ1 (immature DC) or GM-CSF+IL-4 (mature DC)
Low buoyant density cells were propagated from normal B10 mouse BM in GM-CSF plus either TGFβ1 or IL-4 to obtain immature or mature myeloid DC, respectively. The cells were harvested at day 5, stained for surface expression of lineage-restricted and other antigens using an extensive panel of mAbs, and analyzed by flow cytometry. As reported previously (16), both immature and mature DC were negative for lymphoid markers, but expressed the mouse DC-restricted antigens DEC 205 and CD11c, and MHC class I and II. The IL-4-induced mature DC, that strongly stimulated naïve allogeneic T cell proliferation in MLR, also expressed high levels of CM (CD40, CD80, and CD86), but very low levels of the macrophage-associated markers F4/80, CD11b, and CD32. By contrast, TGFβ-stimulated immature DC that exhibited weak allostimulatory activity, expressed only low levels of CD40, CD80, and CD86 (data not shown).
The capacity of donor immature DC to prolong heart graft survival is markedly enhanced by anti-CD40L mAb, and dependent on donor MHC class II expression
As shown in Figure 1a, the capacity of donor immature DC to prolong heart allograft survival when infused 7 days before transplant, was strikingly enhanced by coadministration of anti-CD40L mAb on day –7. Infusion of mature donor DC (propagated in GM-CSF+ IL-4) 7 days before heart transplant resulted in accelerated graft rejection compared with untreated controls. Coadministration of anti-CD40L mAb reversed this effect partially, but not significantly (Fig. 1b). Neither the mAb alone (Fig. 1A) nor third-party immature DC (BALB/c; H-2d) [median graft survival time 17 days (n=9), compared with 13 days in syngeneic immature DC-pretreated controls (n=5)] affected graft survival significantly. Control IgG reversed the effect of immature donor DC infusion (Fig. 1A). The mechanism underlying this effect is unknown. Conceivably, the Ig may have opsonized or activated the donor DC leading to their enhanced elimination/host sensitization.
Figure 1.

A, Anti-CD40L mAb enhances the capacity of immature donor-derived DC to prolong vascularized cardiac allograft survival. Immature or mature DC were propagated from B10 (H-2b) mouse bone marrow, as described in Materials and Methods, and injected (2 × 106) i.v. either alone, or together with 200 μg anti-CD40L mAb, i.p. into C3H (H-2k) recipients, 7 days before B10 heart transplantation (HTX) on day 0. Control groups received either no treatment (PBS), anti-CD40L mAb, or normal rat IgG (isotype control) alone on day –7. B, Pretreatment with mature DC resulted in accelerated graft rejection, that was not reversed by mAb administration. Anti-CD40L mAb pretreatment alone did not significantly affect allograft survival. Results obtained from groups of 6–19 mice. Immature DC+anti-CD40L versus immature DC+ IgG, P<0.001; immature DC+anti-CD40L versus anti-CD40L, P<0.01; immature DC+anti-CD40L versus immature DC, P<0.01; immature DC versus PBS, P<0.01; mature DC versus PBS, P<0.01; mature DC+anti-CD40L versus PBS, P<0.05.
Donor immature DC+anti-CD40L mAb inhibit anti-donor CTL activity of GIC and host spleen cells
We next examined the influence of immature donor DC+mAb treatment 7 days before cardiac transplantation on ex vivo indices of antidonor immune reactivity. Approximately equivalent numbers of cells, exhibiting similar phenotypic distribution were isolated from heart grafts of both experimental and control groups on days 8 and 14 posttransplant. Evaluation of specific anti-donor CTL activity of freshly isolated GIC from the group pretreated with immature DC plus anti-CD40L on day 8 posttransplant revealed a reduction of 30–50% in CTL activity in GIC compared with untreated, heart-grafted controls. By day 14, CTL activity in GIC was almost completely suppressed in the experimental group, whereas it was maintained in controls (Fig. 2).
Figure 2.
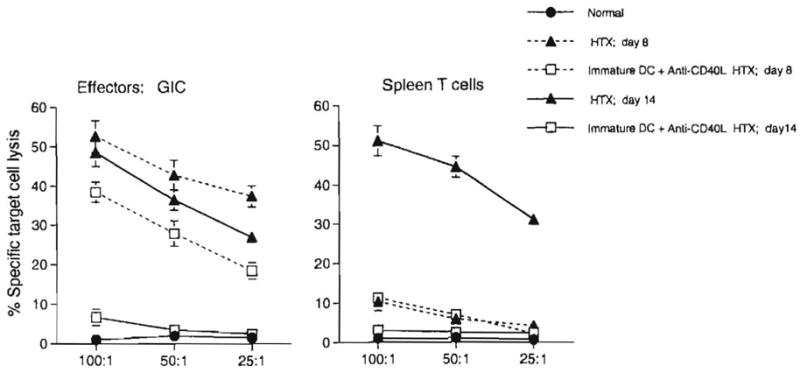
Cytotoxic activity of freshly isolated normal (C3H) heart nonparenchymal cells, heart graft-infiltrating cells (GIC), and recipient (C3H) spleen cells against target cells (EL4) expressing donor (B10) alloantigen (H-2b). A 4-hr 51Cr-release assay was used to determine cytotoxicity 8 or 14 days after organ transplantation. Treatment of heart graft recipients with immature DC + anti-CD40L mAb 7 days before heart transplantation (HTX) inhibited the anti-donor CTL response, both within the graft, and systemically. By day 14, these responses were profoundly suppressed. Cells were pooled from groups of eight normal or three transplanted animals. Results, calculated as described in Materials and Methods, are means:± 1 SD, and are representative of two separate experiments.
Compared with GIC at the same time point, anti-donor CTL activity in spleens of graft recipients was very low on day 8. However, a much higher level, similar to that seen in the graft, was detected in spleens of untreated animals on day 14. These responses were suppressed to normal control values (low negligible CTL activity) in mice preconditioned with immature donor DC+anti-CD40L mAb.
Inhibition of antidonor CTL responses is correlated with increased numbers of apoptotic cells within GIC and spleen T cell populations
We next compared the incidence of apoptotic cells detected by TUNEL staining, both in heart GIC populations (located in the myocardium and surrounding vessels) and in the spleen of mice given immature donor DC plus anti-CD40L mAb, compared with untreated controls, or with mice given either immature DC, or mature DC alone. As shown in Figure 3, the incidence of apoptotic cells in the graft was increased in the combined treatment group, compared with that in animals given immature DC alone. The incidence of apoptotic cells in graft infiltrates was strikingly reduced in mice given mature donor DC that rejected their grafts in an accelerated fashion (Fig. 3). Treatment with immature donor DC+anti-CD40L mAb also resulted in comparatively high incidence of apoptotic (TUNEL+) cells in T cell areas and surrounding red pulp of recipient spleen (Fig. 4; Table 1). Spectrofluorometric assay of DNA fragmentation in spleen cell populations confirmed the high levels of apoptotic activity in this group (Table 1).
Figure 3.
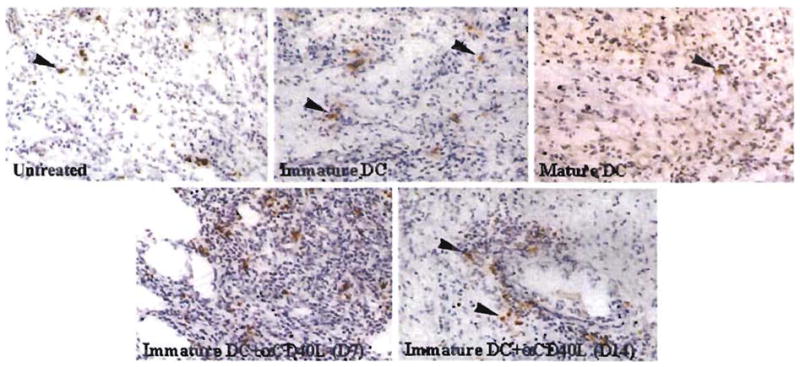
Detection of TUNEL+ apoptotic cells within B10 cardiac allografts of untreated C3H mice or mice pretreated (day –7) with either immature donor-derived DC±anti-CD40L mAb, or mature donor DC. Results obtained on day 7 posttransplant, with the exception of bottom right (day 14). The comparatively high incidence of apoptotic cells seen in the graft-infiltrating cell (GIC) population of mice pretreated with immature donor DC+anti-CD40L mAb correlated with significant elevations (2-fold) in DNA fragmentation detected by spectrofluorometric assay in freshly isolated GIC compared with untreated controls 7 days post transplant (see Table 1) (magnification × 100). Counterstained with hematoxylin.
Figure 4.

A–D, Detection of TUNEL+ (apoptotic cells) in spleen T cell and red pulp areas of (A) normal C3H mice or (B–D) C3H recipients of B10 cardiac allografts 7 days posttransplant. Heart-transplanted animals were either untreated (B), pretreated (day –7) with mature donor-derived DC (C), or immature donor DC + anti-CD40L mAb (D). The comparatively high numbers of apoptotic cells seen in mice given immature donor DC+mAb (D) correlates with the 2- to 3-fold increase in apoptotic activity (DNA fragmentation) detected by spectrofluorometric assay in spleen cells from this group, compared with untreated heart-grafted controls (Table 1). Pretreatment with donor mature DC, that accelerated graft rejection, was associated with an apparent reduction in the incidence of apoptotic cells in the spleen. Magnification: main, ×100, inset, ×400. Counterstained with hematoxylin.
Table 1.
Recipient pretreatment with immature donor DC+anti-CD40L mAb markedly increases the incidence of apoptotic cells in graft-infiltrating and spleen cell populations
| Cells | Treatment | % apoptotic cells |
|---|---|---|
| GICa | Normal | 5.5±2.3b |
| Untreated | 14.8±2.6 | |
| Mature DC | 7.9±1.5 | |
| Immature DC | 18.6±3.8 | |
| Immature DC+anti-CD40L mAb | 30.6±2.7 | |
| Spleen cells | Normal | 4.5±2.7c |
| Untreated | 14.8±5.6 | |
| Mature DC | 7.2±2.4 | |
| Immature DC | 18.2±6.2 | |
| Immature DC+anti-CD40L mAb | 32.4±8.6 |
GIC, Graft-infiltrating cells.
Determined by TUNEL staining.
Determined by spectrofluorometric DNA fragmentation assay.
C3H mice received either no treatment, or 2 × 106 mature B10 DC, or 2 × 106 immature B10 DC ±200 μg anti-CD40L mAb, 7 days before heterotopic vascularized B10 heart transplant. Seven days post-transplant, heart graft-infiltrating cells were isolated, as described in Materials and Methods, and stained for apoptosis by TUNEL. The incidence of TUNEL+ cells in counts of 500 cells was determined by a “blinded” observer. Spleen cell suspensions were prepared at the same time, cultured overnight in complete medium, and DNA fragmentation determined by spectrofluorometric assay, as described in Materials and Methods. Results as means ±1 SD obtained from groups of three animals.
Enhancement of heart allograft survival by donor immature DC+anti-CD40L mAb treatment is associated with increased microchimerism
To determine the relation between graft survival and microchimerism, donor I-Ab+ cells were identified and enumerated in host spleens at various times post transplant (days 0, 7, 14, and 100) (Fig. 5). I-Ab+ cells with DC morphology were readily detected on day 0 in the spleens of heart recipients pretreated with immature DC ± anti-CD40L mAb. Compared with untreated, heart-grafted controls, similar numbers of donor cells were detected in these groups at day 7. By day 14, the level of microchimerism had declined, except in the group given donor cells + mAb, in which it was sustained (Fig. 6A). Moreover, in the latter group, animals with beating grafts >100 days posttransplant exhibited the highest levels of microchimerism detected throughout the study (P<0.05, compared with values at all other time points) (Figs. 5 and 6B).
Figure 5.
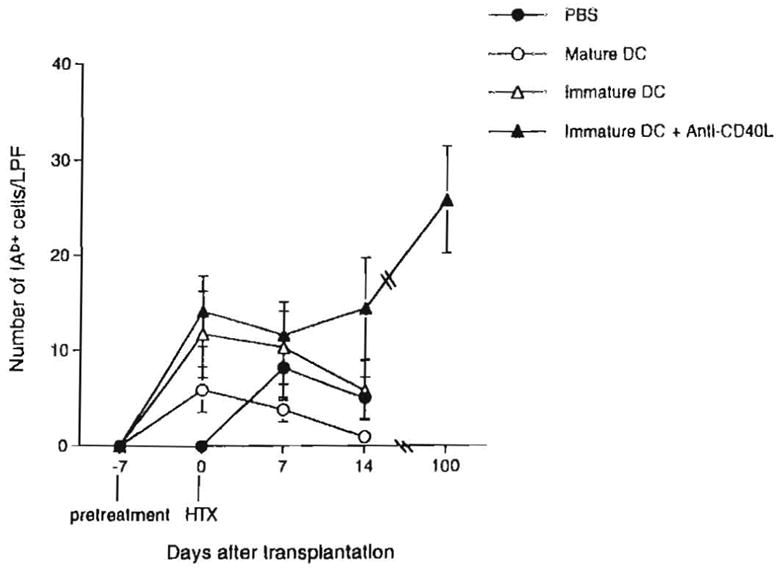
Quantitation of microchimerism (donor MHC class II+ cells) in spleens of C3H recipients of B10 cardiac allografts pretreated (day –7) with either immature donor-derived DC±tanti-CD40L mAb, or mature donor DC. The total number of donor MHC class II+ (IAb+) cells in cryostat sections of spleens at three separate levels, was determined under high power examination (×40 objective) for each of three mice in each group, at each time point. Results are expressed as mean number of IAb+ cells per low power field± 1 SD.
Figure 6.

A, Microchimerism (donor MHC class II+ cells) detected by immunohistochemistry 14 days posttransplant, in spleens of C3H recipients of B10 cardiac allograft pretreated (day –7) with either immature donor-derived DC±anti-CD40L mAb, or mature donor DC. Whereas infusion of donor mature DC 7 days before heart transplant resulted in reduced levels of microchimerism, pretreatment with immature donor DC, especially in combination with anti-CD40L mAb, resulted in the highest observed levels of donor class II+ cells. B, This effect became more pronounced with time posttransplant, and the highest incidences of microchimerism were detected in animals with long-surviving grafts, 100 days posttransplant. Anti-CD40 mAb administration alone did not affect the incidence of donor MHC class II+ cells compared with PBS controls. Magnification, A×200, B×100 (left); ×1000 (right).
Up-regulation of costimulatory molecule (CM) expression on donor-derived immature DC is not prevented by anti-CD40L mAb
Immature DC that are deficient in cell surface expression of CD80 and CD86 can prolong allograft survival, but fail to induce tolerance (16–18). The eventual rejection of the grafts may be due to “late” up-regulation of CM on the donor DC after presumed interaction with recipient T cells. Under these conditions, ligation of CD40 on DC and CD40L on activated T cells may result in up-regulated CM expression, with consequent T cell proliferation, and CTL generation. We therefore explored possible mechanisms involved in the enhancement of allograft survival by combined immature DC and anti-CD40L mAb therapy. We investigated CM expression on donor-derived DC in vitro and in situ, 7 days after their systemic infusion. In vitro studies showed that expression of CD40, CD80, and CD86 was increased significantly by exposure of immature DC to allogeneic T cells for 18–36 hr (Fig. 7). This up-regulation of CM expression was not prevented by adding anti-CD40L mAb (10 μg/ml) at the start of culture (Fig. 7). The concentration of mAb used achieved maximal inhibition of T cell proliferation (30–40%) in 72-hr primary MLR. This finding was confirmed in vivo by injecting immature donor DC (I-Ab+, CD40lo, CD80lo, CD86lo)±anti-CD40L mAb, and performing double immunohistochemical staining for donor MHC class II and CD86. As shown in Figure 8, more than 80% of I-Ab+ cells in spleens of mice given immature DC+anti-CD40L mAb were also CD86+ 7 days after their injection (compared with only approximately 10% before injection). This indicated that expression of CD86 was up-regulated in host lymphoid tissue, despite the presence of anti-CD40L mAb.
Figure 7.

Up-regulation of costimulatory molecule expression on immature DC after exposure to allogeneic T cells is not inhibited in the presence of anti-CD40L mAb. Immature B10 (H-2b) DC were cocultured with purified allogeneic naive C3H (H-2k) T cells (DC: T cells = 1:2) for 18 hr (CD40, CD86) or 36 hr (CD80) in the absence or presence of anti-CD40L mAb (10 μg/ml). Expression of CD40, 80, and 86 was then determined on gated cells by flow cytometric analysis (T cells were gated out by staining with anti-CD3 mAb). Open profiles denote Ig isotype controls.
Figure 8.
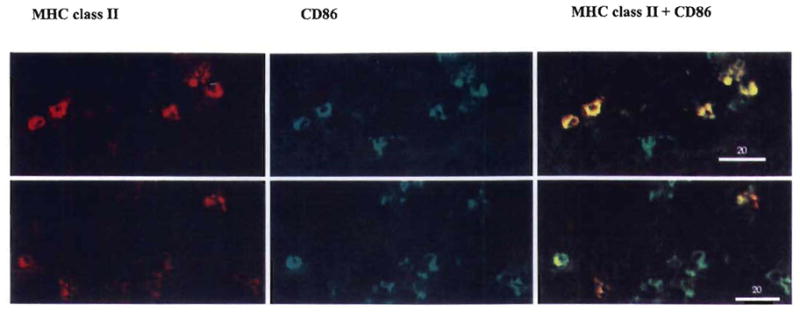
Infusion of immature donor DC+anti-CD40L mAb 7 days before transplant does not result in inhibition of B7 (CD86) molecule expression on donor cells in spleens of allogeneic recipients 7 days postinfusion. Sections were stained by two-color immunohistochemistry, for either donor MHC class II (red), or B7-2 (CD86) (green), or both molecules (yellow denotes positive cells) as described in Materials and Methods. Animals received either immature DC+anti-CD40L mAb (upper row), or immature DC alone. Results are representative of groups of three mice in each treatment group. Magnification ×400.
Immature DC+anti-CD40L mAb pretreatment reduces IL-2 production in the spleens of heart allograft recipients
Next, we investigated the possible role of IL-2 in relation to the apoptosis observed in lymphoid tissue of the graft recipients. Because apoptosis of activated T cells may be attributable to insufficiency of IL-2 production, with consequent reduction in cell viability/growth, we investigated IL-2 production in situ in the spleens of heart-allografted mice at various times (days 0, 7, and 14) posttransplant. Administration of immature DC reduced the number of IL-2+ cells and the intensity of IL-2 staining in T cell and red pulp areas compared with untreated graft recipients (Fig. 9), whereas in contrast, administration of mature donor DC 7 days before organ grafting increased the incidence of IL-2+ cells and the intensity of IL-2 staining. The inhibitory effect of the immature cells was rendered more pronounced by coadministration of anti-CD40L mAb. Thus, the comparatively high incidence of apoptotic cells of lymphoid tissue in the combination therapy group was correlated both with a reduction in local IL-2 production, and with comparatively high levels of donor DC microchimerism (or donor MHC class II+ cells).
Figure 9.
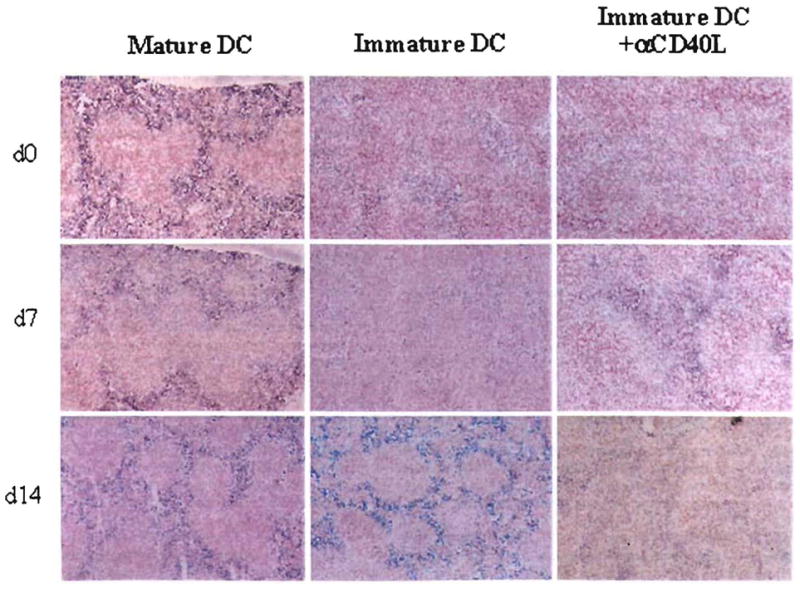
IL-2 synthesis detected by immunohistochemical staining in the spleen of heart allograft recipients, at various times post heart transplant (HTX). Whereas infusion of donor mature DC 7 days before HTX was associated with high levels of IL-2 expression (blue staining) on day 0, 7, and 14 posttransplant, animals given immature donor DC did not exhibit strong staining for IL-2 until 7 days post-HTX. Infusion of immature donor DC+anti-CD40L mAb suppressed IL-2 production at each time point examined. Magnification ×100.
DISCUSSION
These observations lend credibility to the hypothesis that clonal exhaustion-deletion of lymphocytes, linked to the presence of migratory donor leukocytes (including DC) is the seminal basis of organ allograft acceptance and of transplantation tolerance generally (1–4). Different from the spontaneous tolerance induced by the liver in the same strain combination (7), the prolonged survival of cardiac allografts required assistance by concomitant pretreatment with a highly targeted immunosuppressant (CD40L mAb) and an additional source of modified donor cells.
The supplementary cells were MHC class II+ donor DC that had propagated under conditions that inhibit their expression of the CM molecules CD40, 80, and 86, and reduce their immunogenicity (16, 23). The second component of pretreatment was with the CD40L mAb, which is directed against the CD40L up-regulated by activated T cells (24). This mAb is thought to act by blocking CD40 receptor engagement of CD40L by DC, B cells, and other antigen-presenting cells (APC), thus preventing up-regulation of B7 and other CM (24–27). A possible explanation for the previously reported efficacy of the two modalities in combination (16) was that the CD40L mAb perpetuated counter-regulatory (i.e., proapoptotic) signals from DC (21). Instead, the infused DC differentiated after their infusion. This was evident from their up-regulation of B7.2 (CD86), consistent with recently reported observations in a cardiac transplant model in which resting B cells were the APC (26), and also with the earlier demonstration that blockade of the CD40 pathway did not inhibit B7 transcripts in murine heart allografts (27).
The prolonged acceptance of cardiac allografts in the pretreated animals (> 100 days in 40% of recipients) appeared to occur in essentially the same sequential steps as the tolerance induced spontaneously by liver allografts. The first phase was acute donor-specific clonal activation of CTL after migration of graft-derived donor leukocytes and the infused donor-derived DC to host lymphoid organs. This was succeeded by apoptosis associated with IL-2 deficiency (that may also reflect T cell anergy) in the microenvironment of the activated cells. Ongoing studies in our laboratory using limiting dilution analysis will provide an accurate estimate of the frequency of donor-reactive CTL and of IL-2-producing cells. The apoptosis at the graft site was a direct reflection of the apoptosis in the host spleen. Finally, a more or less stable state was established with low level (micro)chimerism and residual low grade antigraft activity in the lymphoid organs.
The persistence of chimerism after transplantation has been explained by the presence of precursor and stem cells in the passenger leukocytes of organs (8, 28, 29). The continuous presence of donor-derived leukocytes in lymphoid areas, Or their leakage there from nonlymphoid repositories has been suggested as an explanation for the maintenance of clonal exhaustion (1, 30, 31), comparable to the stable equilibrium between destructive and nondestructive immunity described in an analogous model of autoimmune diabetes mellitus (32, 33). This view is consistent with the historical antigen-dependent state (34, 35). The experiments of Ehl et al. (5) have demonstrated that the migratory donor a leukocytes (i.e., microchimerism) rather than the peripheral graft are the critical source of this antigen.
Before the presence of persistent chimerism in organ recipients was recognized, allograft acceptance was explained by T cell receptor ligation of host lymphocytes by organ parenchymal cells that are incapable of delivering an effective costimulatory signal. The postulated result was antigen-specific clonal “silencing” (anergy) (36–41), defined by the restoration of T cell reactivity when IL-2 is added to host lymphocytes in vitro or in vivo. We have argued instead that both the culmination of the afferent phase and the effector response leading to antigen-specific nonreactivity occur in the host lymphoid organs where antigen-reactive T cells are activated within the first few days and differentiate to effector cells (1). It is in these organized lymphoid collections where DC and other APCs that have captured and processed antigen present the peptide fragment of the antigen to antigen-specific T cell receptors in the context of their up-regulated host MHC complex peptide. The lymphoid organs provide the unique architectural structure and cellular milieu in which factors that constitute the necessary immunogenic-tolerogenic environment are present in abundance; e.g., cytokines, other molecules, cell to cell proximity, and homing mechanisms that subserve transport of the antigen to the lymphoid organs (31, 42).
Reverse delivery from the lymphoid organs to the peripheral organ of antigen-activated CTL that already have received a death signal is assured by chemokines and other mechanisms that ensure homing of these effector cells to the allograft site (43). The rapidity with which this occurs is evident in our experiments from the essentially equal rate of apoptosis in the host cells infiltrating the graft and in the spleen cells (Table 1). More than 30% of lymphocytes in both locations were undergoing apoptosis at the peak period of 8 posttransplant days (Table 1). Because the spleen constitutes only a fraction of the organized host lymphoid tissue (all of which share the same basic architecture and function) to which donor leukocytes migrate (6,8,10,11,38,39,44) the magnitude of the cumulative apoptosis in the global lymphoid system can be reasonably inferred. Indeed, the comparatively high incidence of apoptotic lymphoid cells in the spleens of animals given donor cells + anti-CD40L mAb was also observed in the mesenteric lymph nodes (Lu L, Thomson AW, unpublished observations). The presence of an organ allograft is not essential for the deletion by apoptosis described herein, as has been shown by Sykes et al. (45) in mouse recipients of bone marrow allografts using treatment with sublethal total body irradiation and CD40L mAb and CTLA4 Ig.
The molecular and cellular pathways responsible for the apoptosis are incompletely understood, but may include those of replicative exhaustion (11, 12) [e.g., telemere shortening when the Hayflick proliferative limit is exceeded (46)], and Fas/Fas ligand or other mechanisms of programed cell death (13, 21). Indeed whatever the precise explanation, it has been shown that endogenous cytokines classically associated with immune activation and rejection (e.g., IL-2, interferon-γ, and IL-12) can not be removed from the immunological response system by gene deletion or other means without loss of tolerogenesis (47–52). Recently, IL-2 has been shown to enhance activation-induced cell death mediated by the Fas pathway in CD4+ cells (53). It also has been established in a variety of models that the clonal exhaustion-deletion is more rapidly accomplished with larger amounts of antigen (reviewed in Ref. 1). This accounts for the unusual tolerogenicity of the leukocyte-rich liver and explains why adjunct donor leukocytes or purified donor antigen facilitate the engraftment in most experimental transplantation models (5) including several used to test the efficacy of CD40L mAb (25, 26, 54–57).
The collective evidence from in vivo research in transplantation immunology has shown that if donor cells from the allograft (or cells infused separately) do not reach host lymphoid organs, a specific cytotoxic T cell response is either not induced or cannot be maintained, thereby precluding the succeeding step of donor specific tolerance (1, 30, 31, 58). With activation, the transition from destructive immunity to nonreactivity appears to occur by the final common pathway described, the diverse cellular and molecular targets of the immunosuppressants notwithstanding (1). Consequently, the clinical applicability of drugs under preclinical development ultimately hinges on their relative efficacy, toxicity, and compatibility with other agents.
It is noteworthy that immunosuppression is a two-edged sword, controlling destructive consequences of immunity, but potentially eroding the basis of allograft acceptance, i.e., immune activation. For example, the administration of adrenal cortical steroids prevents spontaneous tolerance in a liver transplant model (11), and of current interest, cyclosporine appears to abrogate the tolerogenic effects of costimulatory blockade (57).
Acknowledgments
The authors thank Professor Rolf Zinkemagel (Zurich) for critical review of the manuscript, Ms. Alison Logan for skillful flow cytometric analyses, Ms. Jennifer Little for assistance with preparation of figures, the Immunex Corporation for M158 mAb, and Schering-Plough Research Institute for recombinant cytokines.
Footnotes
Presented at the 17th Annual Meeting of the American Society of Transplant Physicians, May 10–13, 1998, Chicago, IL.
Supported by National Institutes of Health grants DK49745, AI41011, and DK 29961.
Abbreviations: CML, cell-mediated lymphocytotoxicity; Ab, antibody; APC, antigen-presenting cell; B10, C57BL/10J; BM, bone marrow; C3H, C3HJHeJ; CDC, complement-dependent cytotoxicity; CM, costimulatory molecule(s); CTL, cytotoxic T lymphocyte; CTLA4, cytotoxic T lymphocyte antigen 4; DC, dendritic cell; FITC, fluorescein isothiocyanate; GIC, graft-infiltrating cell; GM-CSF, granulocyte-macrophage colony-stimulating factor; HBSS, Hanks’ balanced salt solution; IL, interleukin; mAb, monoclonal antibody; MHC, major histocompatibility complex; MLR, mixed leukocyte reaction; MST, median survival time; NPC, nonparenchymal cell; r, recombinant; RT, room temperature; TGFβ, transforming growth factor-β; Th, T helper; TUNEL, terminal deoxynucleotidyl transferase-catalyzed dUTP-digoxigenin nick-end labeling.
References
- 1.Starzl TE, Zinkernagel R. Antigen localization and migration in immunity and tolerance. N Engl J Med. 1998;339:1905. doi: 10.1056/NEJM199812243392607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Starzl TE, Demetris AJ, Murase N, Ildstad S, Ricordi C, Trucco M. Cell migration, chimerism, and graft acceptance. Lancet. 1992;1:1579. doi: 10.1016/0140-6736(92)91840-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Starzl TE, Demetris AJ, Trucco M, et al. Cell migration and chimerism after whole-organ transplantation: the basis of graft acceptance. Hepatology. 1993;17:1127. [PMC free article] [PubMed] [Google Scholar]
- 4.Starzl TE, Demetris AJ, Murase N, Trucco M, Thomson AW, Rao AS. The lost chord: microchimerism. Immunol Today. 1996;17:577. doi: 10.1016/s0167-5699(96)10070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehl S, Aichele P, Ramseier H, et al. Antigen persistence and time point of T cell tolerization determine the efficacy of tolerization protocols for prevention of skin graft rejection. Nature Med. 1998;4:1015. doi: 10.1038/2001. [DOI] [PubMed] [Google Scholar]
- 6.Demetris AJ, Murase N, Fujisaki S, Fung JJ, Rao AS, Starzl TE. Hematolymphoid cell trafficking. microchimerism, and GVHD reactions after liver, bone marrow, and heart transplantation. Transplant Proc. 1993;25:3337. [PMC free article] [PubMed] [Google Scholar]
- 7.Qian S, Demetris AJ, Murase N, Rao AS, Fung JJ, Starzl TE. Murine liver allograft transplantation: tolerance and donor cell chimerism. Hepatology. 1994;19:916. doi: 10.1002/hep.1840190418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu L, Rudert WA, Qian S, et al. Growth of donor-derived den. dritic cells from the bone marrow of murine liver allograft recipients in response to granulocyte/macrophage colony-stimulating factor. J Exp Med. 1995;182:379. doi: 10.1084/jem.182.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun J, McCaughan GW, Gallagher ND, Sheil AGR, Bishop GA. Deletion of spontaneous rat liver allograft acceptance by donor irradiation. Transplantation. 1995;60:233. doi: 10.1097/00007890-199508000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Murase N, Starzl TE, Tanabe M, et al. Variable chimerism, graft versus host disease, and tolerance after different kinds of cell and whole organ transplantation from Lewis to Brown-Norway rats. Transplantation. 1995;60:158. doi: 10.1097/00007890-199507000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bishop GA, Sun J, DeCruz DJ, et al. Tolerance to rat liver allografts. III. Donor cell migration and tolerance-associated cytokine production in peripheral lymphoid tissues. J Immunol. 1996;156:4925. [PubMed] [Google Scholar]
- 12.Bishop GA, Sun JN, Sheil AGR, McCaughan GW. High-dose/activation-associated tolerance. A mechanism for allograft tolerance. Transplantation. 1997;64:1377. doi: 10.1097/00007890-199711270-00001. [DOI] [PubMed] [Google Scholar]
- 13.Qian S, Lu L, Fu F, et al. Apoptosis within spontaneously accepted mouse liver allografts. J Immunol. 1997;158:4654. [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer D, Baumgardt S, Loeffeler S, et al. Apoptosis of T lymphocytes in liver and/or small bowel allografts during tolerance induction. Transplantation. 1998;66:1530. doi: 10.1097/00007890-199812150-00018. [DOI] [PubMed] [Google Scholar]
- 15.Thomson AW, Lu L, Murase N, Demetris AJ, Rao AS, Starzl TE. Microchimerism, dendritic cell progenitors and transplantation tolerance. Stem Cells. 1995;13:622. doi: 10.1002/stem.5530130607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu L, Li W, Fu F, et al. Blockade of the CD40-CD40 ligand pathway potentiates the capacity of donor-derived dendritic cell progenitors to induce long-term cardiac allograft survival. Transplantation. 1997;64:1808. doi: 10.1097/00007890-199712270-00031. [DOI] [PubMed] [Google Scholar]
- 17.Rastellini C, Lu L, Ricordi C, Starzl TE, Rao AS, Thomson AW. GM-CSF stimulated hepatic dendritic cell progenitors prolong pancreatic islet allograft survival. Transplantation. 1995;60:1366. [PMC free article] [PubMed] [Google Scholar]
- 18.Fu F, Li Y, Qian S, et al. Costimulatory molecule-deficient dendritic cell progenitors (MHC class II+, B7-1dimB7-2−) prolong cardiac allograft survival in non-immunosuppressed recipients. Transplantation. 1996;62:659. doi: 10.1097/00007890-199609150-00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu L, Woo J, Rao AS, et al. Propagation of dendritic cell progenitors from normal mouse liver using GM-CSF and their maturational development in the presence of type-1 collagen. J Exp Med. 1994;179:1823. doi: 10.1084/jem.179.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu L, Qian S, Hershberger P, Rudert WA, Lynch DH, Thomson AW. Fas ligand (CD95L) and B7 expression on dendritic cells provide counter-regulatory signals for T cell survival and proliferation. J Immunol. 1997;158:5676. [PubMed] [Google Scholar]
- 22.Thomson AW, Lu L, Subbotin VM, et al. In vitro propagation and homing of liver-derived dendritic cell progenitors to lymphoid tissues of allogeneic recipients. Transplantation. 1995;59:544. doi: 10.1097/00007890-199502270-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu L, McCaslin D, Statzl TE, Thomson AW. Mouse bone marrow-derived dendritic cell progenitors (NLDC 145+, MHC class II+, B7–1dim, B7–2−) induce alloantigen-specific hyporespon-siveness in murine T lymphocytes. Transplantation. 1995;60:1539. doi: 10.1097/00007890-199560120-00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durie FH, Foy TM, Masters SR, Laman JE, Noelle RJ. The role of CD40 in the regulation of humoral and cell-mediated immunity. Immunol Today. 1994;15:406. doi: 10.1016/0167-5699(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 25.Hancock WW, Sayegh MH, Zheng X-G, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA. 1996;93:13967. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niimi M, Pearson TC, Larsen CP, et al. The role of the CD40 pathway in alloantigen-induced hyporesponsiveness in vivo. J Immunol. 1998;161:5331. [PubMed] [Google Scholar]
- 27.Larsen CP, Alexander DZ, Hollenbaugh D, et al. CD40-gp39 interactions play a critical role during allograft rejection: suppression of allograft rejection by blockade of the CD40-gp39 pathway. Transplantation. 1996;61:4. doi: 10.1097/00007890-199601150-00002. [DOI] [PubMed] [Google Scholar]
- 28.Murase N, Starzl TE, Ye Q, et al. Multilineage hematopoietic reconstitution of supralethally irradiated rats by syngeneic whole organ transplantation: with particular reference to the liver. Transplantation. 1996;61:1. doi: 10.1097/00007890-199601150-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taniguchi H, Toyoshima T, Fukao K, Nakauchi H. Presence of hematopoietic stem cells in the adult liver. Nature Med. 1996;2:198. doi: 10.1038/nm0296-198. [DOI] [PubMed] [Google Scholar]
- 30.Zinkemagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18:14. doi: 10.1016/s0167-5699(97)80008-4. [DOI] [PubMed] [Google Scholar]
- 31.Zinkemagel RM, Ehl S, Aichele P, Oehen S, Kundig T, Hengartner H. Antigen localization regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol Rev. 1997;156:199. doi: 10.1111/j.1600-065x.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- 32.Ohashi PS, Oehen S, Buerki K, et al. Ablation of “tolerance” and induction of diabetes by viruses infection in viral antigen transgenic mice. Cell. 1991;65:305. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 33.Ohashi PS, Oehen S, Aichele P, et al. Induction of diabetes is influenced by the infectious virus and local expression of MHC class I and tumor necrosis factor-α. J Immunol. 1993;150:5185. [PubMed] [Google Scholar]
- 34.Aust JB, Rogers W, Guttmann R. The antigen persistence concept of homograft tolerance: immunologic response to tumor and spleen cells following transfer, reimplantation and parabiosis. Ann Surg. 1965;162:738. doi: 10.1097/00000658-196510000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morecki S, Leshem B, Eid A, Slavin S. Alloantigen persistence in induction and maintenance of transplantation tolerance. J Exp Med. 1987;165:1468. doi: 10.1084/jem.165.6.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenkins MR, Pardoll DM, Mizuguchi J, Chused TM, Schwartz RH. Molecular events in the induction of a nonresponsive state of interleukin-2 producing helper T lymphocyte clones. Proc Natl Acad Sci USA. 1987;84:5409. doi: 10.1073/pnas.84.15.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohn M. The wisdom of hindsight. Annu Rev Immunol. 1994;12:1. doi: 10.1146/annurev.iy.12.040194.000245. [DOI] [PubMed] [Google Scholar]
- 38.Janeway CA, Jr, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 39.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J Exp Med. 1996;184:1. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janeway CA, Travers P, Walport M, Capra JD. Immunobiology, the immune system in health and disease. 4. New York: Garland Publishing; 1999. p. 272. [Google Scholar]
- 42.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 System of T cell costimulation. Annu Rev Immunol. 1996;14:233. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 43.Picker LJ, Butcher EC. Physiological and molecular mechanisms of lymphocyte homing. Annu Rev Immunol. 1993;10:561. doi: 10.1146/annurev.iy.10.040192.003021. [DOI] [PubMed] [Google Scholar]
- 44.Murase N, Demetris AJ, Woo J, et al. Graft versus host disease (GVHD) after BN to LEW compared to LEW to BN rat intestinal transplantation under FK 506. Transplantation. 1993;55:1. doi: 10.1097/00007890-199301000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wekerle T, Sayegh MH, Hill J, et al. Extrathymic T cell deletion and allogeneic stem cell engraftment induced with costimulatory blockade is followed by central T cell tolerance. J Exp Med. 1998;187:2037. doi: 10.1084/jem.187.12.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Effros RB, Pawelec G. Replicative senescence of T cells: does the Hayflick limit lead to immune exhaustion? Immunol Today. 1997;18:450. doi: 10.1016/s0167-5699(97)01079-7. [DOI] [PubMed] [Google Scholar]
- 47.Dai Z, Konieczny BT, Baddoum FK, Lakkis FG. Impaired alloantigen-mediated T cell apoptosis and failure to induce long-term allograft survival in IL-2 deficient mice. J Immunol. 1998;161:1659. [PubMed] [Google Scholar]
- 48.Konieczny BT, Dai Z, Elwood ET, et al. IFN-gamma is critical for long-term allograft survival induced by blocking the CD28 and CD40 ligand T cell costimulation pathways. J Immunol. 1998;160:2059. [PubMed] [Google Scholar]
- 49.Sayegh MH, Akalin E, Hancock WW, et al. CD28-B7 blockade after alloantigenic challenge in vivo inhibits Th1 cytokines but spares Th2. J Exp Med. 1995;181:1869. doi: 10.1084/jem.181.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α-chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 51.Kneitz B, Herman T, Yonehara S, Schimpl A. Normal clonal expansion but impaired Fas-mediated cell death and anergy in IL-2-deficient mice. Eur J Immunol. 1995;25:2572. doi: 10.1002/eji.1830250925. [DOI] [PubMed] [Google Scholar]
- 52.Van Parijs L, Biuckians A, Ibragimove A, Alt FW, Willerford D, Abbas AK. Functional responses and apoptosis of CD25 nL-2Rα)-deficient T cells expressing a transgenic antigen receptor. J Immunol. 1997;158:3738. [PubMed] [Google Scholar]
- 53.Rafaeli Y, Parijs LV, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 54.Markees T, Phillips NE, Gordon EJ, et al. Long-term survival of skin allografts induced by donor splenocytes and anti-CD154 antibody in thymectomized mice requires CD4+T cells, interferon-gamma and CTLA4. J Clin Invest. 1998;101:2446. doi: 10.1172/JCI2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parker DC, Greiner DL, Phillips NE, et al. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA. 1995;92:9560. doi: 10.1073/pnas.92.21.9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buhlmann JE, Foy TM, Aruffo A, et al. In the absence of a CD40 signal, B cells are tolerogenic. Immunity. 1995;2:645. doi: 10.1016/1074-7613(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 57.Larsen CP, Elwood ET, Alexander DZ, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 58.Zinkernagel RM. Immunology taught by viruses. Science. 1996;271:173. doi: 10.1126/science.271.5246.173. [DOI] [PubMed] [Google Scholar]