

Abstract
To investigate whether an open-to-closed transition before the chemical step and induced-fit mechanism exist in DNA polymerase μ (pol μ), we analyze a series of molecular-dynamics simulations with and without the incoming nucleotide in various forms, including mutant systems, based on pol μ's crystal ternary structure. Our simulations capture no significant large-scale motion in either the DNA or the protein domains of pol μ. However, subtle residue motions can be distinguished, specifically of His329 and Asp330 to assemble in pol μ's active site, and of Gln440 and Glu443 to help accommodate the incoming nucleotide. Mutant simulations capture a DNA frameshift pairing and indicate the importance of Arg444 and Arg447 in stacking with the DNA template, and of Arg448 and Gln440 in helping to stabilize the position of both the DNA template and the incoming nucleotide. Although limited sampling in the molecular-dynamics simulations cannot be ruled out, our studies suggest an absence of a large-scale motion in pol μ. Together with the known crystallization difficulties of capturing the open form of pol μ, our studies also raise the possibility that a well-defined open form may not exist. Moreover, we suggest that residues Arg448 and Gln440 may be crucial for preventing insertion frameshift errors in pol μ.
Introduction
To maintain the integrity of genetic information, DNA polymerases play an important role in DNA replication, damage repair, recombination, and somatic hypermutation (1,2). Although DNA polymerases share a similar structure with palm, thumb, and finger subdomains (3), their functions vary considerably. X-family DNA polymerases, including polymerase μ (pol μ), polymerase β (pol β), polymerase λ (pol λ), polymerase X (pol X), and terminal deoxynucleotidyl transferase (Tdt) (4), participate mainly in DNA repair rather than replication (5). The repair function of DNA polymerases is crucial. Numerous diseases, including various cancers and neurological conditions, are associated with DNA polymerase errors that are not accurately repaired (6,7).
Pol μ and two other X-family members—Tdt and pol λ—are associated with nonhomologous DNA end joining (NHEJ) of double-strand breaks in DNA (8–11). Thus, repair can proceed even without the guidance of a homologous sequence. In addition to DNA repair, NHEJ is also responsible for the V(D)J recombination (9,11), which functions to assemble the antigen receptor gene segments required for adaptive immune responses (12).
Experimental data show that pol μ has unusual primer-template specificity. Like pol β and pol λ, pol μ can conduct DNA synthesis in a template-dependent manner (Table 1, a and b) (13), with comparable insertion accuracy (fidelity) (14,15). Pol μ also shares with Tdt the ability to catalyze template-independent synthesis (Table 1 d) (16). However, pol μ is only efficient when adding specific types of nucleotides (e.g., 2′-deoxythymidine 5′-triphosphate (dTTP)), and performs nucleotide insertion more efficiently in the presence of a template strand (14,17). In addition, a unique property of pol μ is that it can direct template-based DNA synthesis if there is at least one homologous basepair upstream from the template base, without requiring that all upstream primer bases be paired (Table 1 c) (9,13). This substrate flexibility may signal a special role in the NHEJ process, namely, to promote accurate immunoglobulin κ light-chain recombination (11).
Table 1.
Template strand requirements of X-family polymerases
| Proposed activities | Template strand requirement | pol β | pol λ | pol μ | Tdt |
|---|---|---|---|---|---|
a
|
Continuous template | Yes | Yes | Yes | No |
b
|
Gapped template, primer terminus paired with template | No | Yes | Yes | No |
c
|
Primer terminus not paired with template, incoming nucleotide instructed by template | No | No | Yes | No |
d
|
Incoming nucleotide not instructed by template | No | No | Limited | Yes |
Kinetic, structural, and computational studies of several DNA polymerases (18–31), including pol X (18), pol β (19,27–29), and pol λ (21,30) in the X family, have shown a common nucleotide insertion pathway characterized by transitions between open and closed forms. After DNA binding occurs, the DNA polymerase forms an open binary complex. Upon incorporation of the incoming nucleotide, the complex undergoes conformational changes to rearrange the catalytic groups and forms a closed ternary complex. The nucleotidyl transfer chemical reaction then occurs to extend the primer strand as a closed ternary product, followed by a reopening of product to the binary form, allowing another cycle to begin.
However, variations in the pathway to regulate the transition exist. For example, pol β and pol X show a large-scale protein motion in the thumb subdomain before chemistry (see Fig. S1 a in the Supporting Material) (18,19); this motion is absent in pol λ. Instead, a shift of the DNA template (Fig. S1 b) is indicated by both crystal complexes (30) and simulations (21). Differences in key residues involved in the conformational change are also observed across different polymerases.
In 2007, the Kunkel group crystallized pol μ in a ternary closed complex (PDB entry 2IHM) (32) comprising the polymerization domain (Pro132–Ala496) of murine pol μ with a gapped DNA duplex and bound incoming nucleotide (Fig. 1 a). The overall structure of pol μ is characteristic of other X-family polymerases, consisting of fingers (Val228–Thr288), palm (Pro289–Thr424), thumb (Pro425–Ala496), and N-terminal 8-kDa domain (Leu149–Gln227). Compared to pol λ and pol β, pol μ has a shorter loop in the thumb domain (thumb loop, Asp465–Val471) and two longer loops in its palm (Loop 1: His366–Arg389; Loop 2: Pro397–Cys411). Experiments as well as structural analyses indicate that Loop 1 in pol μ interacts with the template DNA (32,33). The specific amino-acid sequence of Loop 1 in pol μ has been suggested to be crucial for the enzyme's ability to conduct both template-dependent and template-independent synthesis (33,34). In addition, a fidelity controlling function of Loop 1 may exist in pol μ, as suggested by a similar function of Loop 1 in pol λ (35). However, Loop 1 is invisible in the electron density map of the pol μ complex, suggesting that it is disordered. Furthermore, because a binary open complex structure of pol μ is not yet available, the specific transition pathway between the binary and ternary state for pol μ is unknown.
Figure 1.
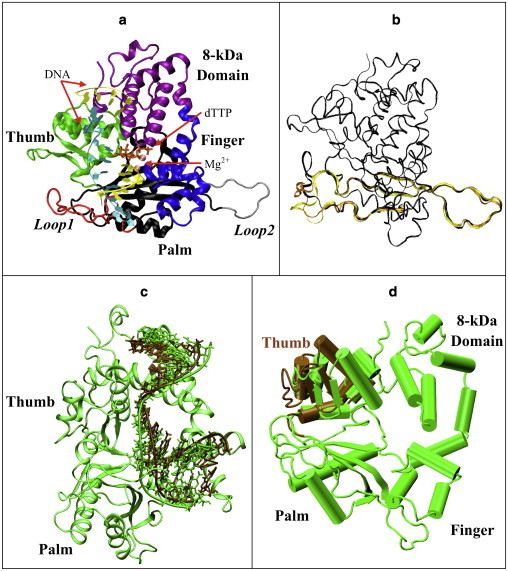
Our model of the murine pol μ ternary complex. (a) Protein domains, DNA strands, and Mg2+. (b) Starting loop conformations. Structure I (dark), from regular MD, used in simulations I, II, and VII–XVII; structure II (light), from 299 K REMD replica, used in simulations III and IV; structure III (medium), from 301 K REMD, used in simulations V and VI. Residues 360–420 of each structure are shown; other protein residues are drawn with a smaller bond radius. (c) Shifted-DNA model. DNA in the shifted-DNA model (dark) was taken from pol λ's binary structure, superimposed with DNA in pol μ's ternary structure (light). The protein is also shown as green. (d) Open-thumb model. The thumb region of pol μ (dark) was shifted to an open form comparable to pol β's binary structure and compared with pol μ's original ternary structure (light).
All-atom dynamics simulations can help unravel polymerase mechanisms by linking structures to generate pathways, which in turn can be related to function. Insights into geometry and structural specificity could also help elucidate the specific functional features of pol μ, such as catalyzing both template-dependent and template-independent DNA synthesis. However, simulations are challenged by the large computational requirements, approximations in force fields, and limited sampling (36). Yet, modeling need not be perfect to be meaningful, as demonstrated by many successes to date (37).
In this work, we seek to answer the following two questions: what is the nature of the conformational changes involved in pol μ before chemistry, and how are they related to those that occur in other polymerases? To treat the possible flexibility of Loop 1, we used replica exchange molecular dynamics (REMD) (38) simulations to enhance sampling. We performed molecular and Langevin dynamics from different starting structures, including forced open-like forms (Table 2), and analyzed simulations to delineate structural and dynamical changes before chemistry. In contrast to results in pol λ or pol β, we observed no large-scale thumb subdomain or DNA motion in pol μ. However, we captured subtle residue rearrangements at the active site, including His329 and Asp330 associated with active-site assembly, as well as Gln440 and Glu443 involved in nucleotide accommodation. Interestingly, motions in residues analogous to Asp330, Gln440, and Glu443 in pol β/λ/X (Table 3) were not observed for the other polymerases (18,19,21).
Table 2.
Summary of all simulations performed
| Simulation | dTTP/Mg2+ | Starting loops conformation | Residues mutated/other changes | Running time |
|---|---|---|---|---|
| I | Present | From regular MD | No | 120 ns |
| II | Absent | From regular MD | No | 120 ns |
| III | Present | From 299 K REMD replica | No | 40 ns |
| IV | Absent | From 299 K REMD replica | No | 40 ns |
| V | Present | From 301 K REMD replica | No | 40 ns |
| VI | Absent | From 301 K REMD replica | No | 40 ns |
| VII | Present | From regular MD | Langevin damping coefficient: 10 ps−1 → 15 ps−1 | 40 ns |
| VIII | Absent | From regular MD | Langevin damping coefficient: 10 ps−1 → 15 ps−1 | 40 ns |
| IX | Present | From regular MD | CHARMM force field without CMAP | 40 ns |
| X | Absent | From regular MD | CHARMM force field without CMAP | 40 ns |
| XI | Present | From regular MD | Arg444 → Ala444 | 40 ns |
| XII | Present | From regular MD | Arg447 → Ala447 | 40 ns |
| XIII | Present | From regular MD | Arg448 → Ala448 | 40 ns |
| XIV | Absent | From regular MD | DNA taken from crystal binary structure of pol λ | 40 ns |
| XV | Absent | From regular MD | Thumb shifted to open form | 40 ns |
| XVI | Present | From regular MD | Protonation state of His329: Nδ → Nɛ | 40 ns |
| XVII | Absent | From regular MD | Protonation state of His329: Nδ → Nɛ | 40 ns |
Table 3.
Key residues around the active site, their analog residues across the X family, and suggested functions
| pol μ | pol β | pol λ | pol X | Function in pol μ | Function in other polymerases |
|---|---|---|---|---|---|
| Asp330 | Asp190 | Asp427 | Asp49 | Coordinates with Mg2+ for chemical reaction | Coordinates with Mg2+ for chemical reaction |
| Asp332 | Asp192 | Asp429 | Asp51 | Coordinates with Mg2+ for chemical reaction | Coordinates with Mg2+ for chemical reaction |
| Asp420 | Asp256 | Asp490 | Asp100 | Coordinates with Mg2+ for chemical reaction | Coordinates with Mg2+ for chemical reaction |
| His329 | Gly189 | Gly426 | Asn48 | Flips to trigger the flip of Asp330 | |
| Gly435 | Tyr271 | Tyr505 | His115 | Contacts DNA bases, thereby correctly positioning the primer terminus for catalysis | |
| Trp436 | Phe272 | Phe506 | Phe116 | Flips to initiate DNA or subdomain motion | |
| Val422 | Arg258 | Ile492 | Phe102 | Interacts with other key residues to control their conformational change | |
| Gln440 | Asp276 | Ala510 | Val120 | Participates in accommodating the incoming nucleotide | Weakens the binding affinity with the nucleotide (pol β) |
| Glu443 | Asn279 | Asn513 | Leu123 | Participate in accommodating the incoming nucleotide through water | Hydrogen-bonds to the nucleotide and is directly related to catalytic ability (pol β) |
| Arg444 | Lys280 | Arg514 | Ile124 | Stabilizes DNA in closed form by stacking with the template | First residue to respond to thumb motion (pol λ) stacks with template base in closed state |
| Arg447 | Arg283 | Arg517 | Arg127 | Stabilizes the DNA template and Loop 1 in closed form | Stabilizes DNA in closed form and helps check for correct basepairing |
| Arg448 | Ala284 | Ala518 | Ala128 | Stabilizes DNA to prevent insertion errors; stabilizes Loop 1 in closed form |
∗Residues in bold indicate motion detected in wild-type MD simulations.
In addition, we also investigated the roles of residues Arg444, Arg447, and Arg448 of pol μ through mutant simulations. Our simulations suggest that Arg444 and Arg447 help to maintain the position of the DNA template in pol μ and stabilize the ternary state. We also observed that Arg448 cooperates with Gln440 to stabilize the position of both the DNA template and the incoming nucleotide, and therefore may help to prevent frameshift insertion errors. Overall, our modeling studies suggest a lack of significant DNA or protein motion in pol μ, but point to subtle side-chain rearrangements unique to pol μ.
Materials and Methods
Initial model
The pol μ initial model was prepared based on the murine crystal pol μ ternary (PDB entry 2IHM) complex. Missing protein residues His366–Val386 (in Loop 1) and Ala403–Ala405 (in Loop 2) were inserted with the InsightII package (Accelrys, San Diego, CA). A hydroxyl group was added to the 3′ carbon of the 2′,3′-dideoxythymidine 5′-triphosphate (ddTTP) sugar moiety to form dTTP. All other missing atoms from the crystal structure were similarly added. The Na+ occupying the catalytic ion site in the crystal structure was modified to Mg2+.
The system was solvated with explicit TIP3 water model in a water box with the use of the VMD program (39). The smallest image distance between the solute and the faces of the periodic cubic cell was 7 Å. In addition to the water molecules in the crystal structure, 13,625 water molecules were added via the VMD program. The total number of water molecules was 13,716. To obtain a neutral system at an ionic strength of 150 mM, 46 Na+ and 28 Cl− ions were added to the system. All of the Na+ and Cl− ions were placed at least 8 Å away from both the protein and DNA atoms and from each other.
The initial model contained 47,612 atoms, 91 crystallographically resolved water molecules from the ternary complex, 13,625 bulk water molecules, two Mg2+ ions, incoming nucleotide dTTP, and 46 Na+ and 28 Cl− counterions.
Based on this initial model, we also prepared three mutant models, a shifted-DNA model, and an open-thumb model. These models are discussed in more detail below.
Minimization and equilibration
Initial energy minimizations and equilibration simulations were performed using the CHARMM program (version c35b2) (40) with the CHARMM all-atom force field including the cross-term energy correction map (CMAP) specification for proteins (41–43). The system was minimized with fixed positions for all heavy atoms of protein or nucleotides, using steepest descent (SD) for 10,000 steps followed by the adopted basis Newton-Raphson (ABNR) method for 20,000 steps. Then the atoms of added residues (His366–Val386 and Ala403–Ala405) were released. Another cycle of minimization was performed for 10,000 steps using SD followed by 20,000 steps of ABNR. The equilibration process was started with a 100 ps simulation at 300 K using single-time step Langevin dynamics, while keeping all the heavy atoms of protein or nucleotides fixed. The SHAKE algorithm (44) was employed to constrain the bonds involving hydrogen atoms. This was followed by unconstrained minimization consisting of 10,000 steps of SD and 20,000 steps of ABNR.
Modeling the loops
Missing-loop construction and subsequent production dynamics were performed using the program NAMD (37) with the CHARMM force field. All protein or DNA atoms, except those from the added residues (His366–Val386 and Ala403–Ala405) to relax the added loops and the water around our complex, were fixed. The system was equilibrated for 1 ns at constant pressure and temperature. Pressure was maintained at 1 atm using the Langevin piston method (45) with a piston period of 100 fs, a damping time constant of 50 fs, and a piston temperature of 300 K. The temperature was maintained at 300 K using weakly coupled Langevin dynamics of nonhydrogen atoms with a damping coefficient of 10 ps−1. Bonds to all hydrogen atoms were kept rigid using SHAKE, permitting a time step of 2 fs. The system was simulated in periodic boundary conditions with full electrostatics computed using the particle mesh Ewald method (46) with grid spacing on the order of ≤1 Å. Short-range nonbonded terms were evaluated at every step using a 12 Å cutoff for van der Waals interactions and a smooth switching function. The final dimensions of the system were 78.95 Å × 74.61 Å × 79.91 Å.
Two different procedures were then followed for further equilibration of the loops. Regular MD (simulation 0) was performed at a constant temperature and volume for 4 ns, using the same constraints as above. Independently, REMD (38) simulations (see Supporting Material) were performed on a duplicate sample frame. REMD was performed under a temperature range of 270–380 K with 64 replicas. The time interval between two exchanges was set to 2 ps. All other conditions were the same as in the regular MD (simulation 0). The REMD simulation totaled 4.2 ns per replica (cumulative time of 268.8 ns).
The REMD simulations were run on the Dell computer cluster at New York University.
Production dynamics
Table 2 summarizes all of the production simulations performed. The last frame from regular MD utilized in loop modeling (simulation 0) was used as one starting structure (starting structure I; Fig. 1 b). From the loop modeling REMD simulations, the two last frames corresponding to replicas with the final temperature of 299 K and 301 K were utilized as two other starting structures (starting structures II and III; Fig. 1 b). For each of the three starting geometries, two simulations were performed. In one set, the original complex was left unchanged (simulations I, III, and V). In the other set, the incoming dTTP as well as two active-site Mg2+ ions were removed (simulations II, IV, and VI), in an attempt to trigger an open form of pol μ. Simulations VII and VIII correspond to simulations I and II, except that a damping coefficient of 15 ps−1 in Langevin dynamics, instead of 10 ps−1, was utilized to enhance sampling; similarly, simulations IX and X explored a variant CHARMM force-field version.
To examine the roles of several arginines of pol μ, we prepared three mutant models based on starting structure I. Arg444, Arg447, and Arg448 were mutated to alanine in simulations XI, XII, and XIII, respectively. In all mutant simulations, the dTTP and Mg2+ moieties were retained as described above.
Because a binary crystal structure of pol μ is not available, we prepared two models to mimic the open form of pol λ or pol β. In simulation XIV, the primer-template DNA in the pol μ complex was replaced with the DNA in the crystal binary structure of pol λ to mimic the possible binary structure of pol μ characterized by DNA sliding (shifted-DNA model; Fig. 1 c). The protein conformation was as in simulation I. In simulation XV, we shifted the thumb region (Pro425–Ala496) of pol μ to an open form (open-thumb model; Fig. 1 d). The distance for each residue to shift was determined by the positional difference of its corresponding residue's Cα atom in pol β's crystal ternary and binary structure. All other regions of the protein and DNA atoms were held as in starting structure I. In both simulations XIV and XV, the incoming dTTP and two Mg2+ were removed. Table 2 summarizes all of the production simulations performed. Finally, simulations XVI and XVII explored variations in protonation states of His329.
In all of these trajectories, all heavy atoms were free to move. Each simulation was run for at least 40 ns. Regular MD simulations using the NAMD package were run on the IBM Blue Gene/L at Rensselaer Polytechnic Institute.
Results and Discussion
Wild-type pol μ systems
Global motion
No substantial protein subdomain motions
During all of our wild-type simulations, no large-scale protein motions were captured as in pol β or pol X (Fig. 2 a), where a large open-to-closed transition was observed (18,19). Note that in simulation XV, where we shifted the thumb to an open-like configuration, the thumb reverted to the closed form (Fig. 2 c). This suggests that an open form of protein is not favored in pol μ.
Figure 2.
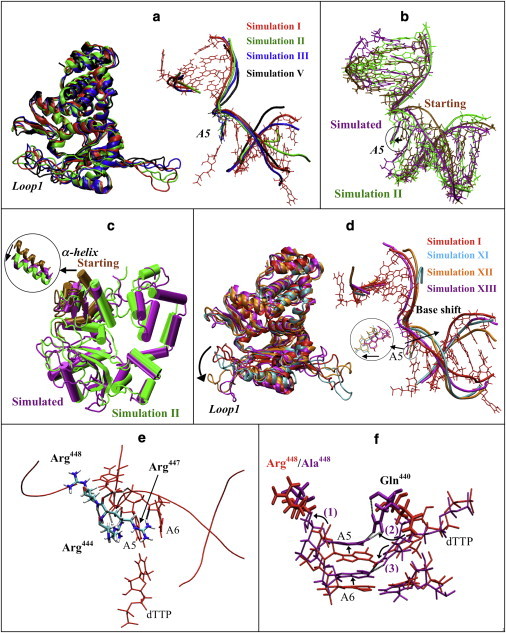
Protein and DNA motions in pol μ simulations. (a) Protein motions (left) and DNA motions (right) of pol μ in selected simulations (red, simulation I; green, simulation II; blue, simulation III; black, simulation V). For clarity, DNA bases other than A5 in simulations II, III, and V are not shown. (b) DNA position in simulation XIV (shifted-DNA model), before (brown) and after simulation (purple), compared with simulation II (green). (c) Protein conformation in simulation XV (open-thumb model), before (brown) and after (purple) simulation, compared with simulation II (green). The motion of α-helix N in the thumb is shown. (d) Protein motions (left) and DNA motions (right) of pol μ in mutant simulations compared with wild-type simulation I (red, simulation I; cyan, simulation XI; orange, simulation XII; purple, simulation XIII). DNA bases other than A5 in simulations XI, XII, and XIII are not shown. The shift of A5 base is indicated. (e) Interactions between DNA and residues Arg444, Arg447, and Arg448. Only residues A5–A6 in the DNA template are shown. Dashed lines indicate hydrogen bonds. (f) The insertion frameshift error observed in simulation XIII (R448A, purple) compared to wild-type simulation I (red). The frameshift behavior can be described in three steps: 1), A5 and A6 rotate/shift toward the downstream side; 2), Gln440 flips to A5; and 3), dTTP shifts toward the upstream side and flips to A6. Nucleotides are drawn with a smaller bond radius. Dashed lines indicate hydrogen bonds.
Loop 1 (His366–Arg389) appeared to be quite flexible in all of the simulations. However, we observed that the conformation of Loop 1 did not significantly depend on the position of dTTP/Mg2+ (Fig. S2). The flexibility we observed is consistent with the absence of Loop 1 in the electron density map.
No substantial DNA motion
Without dTTP/Mg2+, the DNA template did not shift as much as observed in pol λ (21), and the template base A5 at the gap pairing with dTTP did not move significantly (Fig. 2 a and Fig. S2). In simulation XIII, where we shifted the DNA to a binary-like position, both the DNA template strand and the A5 base shifted back toward the ternary position (Fig. 2 b), even though dTTP and Mg2+ were absent. This suggests that the DNA in the pol μ complex tends to stay in the ternary position, and that the DNA sliding motion noted for pol λ may not exist in pol μ.
Binding free energies further support the lack of DNA sliding motion in pol μ. Using the molecular-mechanics Poisson-Boltzmann surface area approach (47–49) (see Supporting Material), we computed the binding free energies between pol μ and the DNA, and compared them with corresponding values in the pol λ system (50) (Table S1). The lower binding free energy between pol μ and the DNA by ∼55 kcal/mol compared to that between pol λ and the DNA suggests that pol μ binds the DNA more tightly than pol λ. Hence, the DNA is less likely to shift.
We also performed a principle components analysis to better analyze pol μ's lack of global motion (Fig. S3). Our simulations, in a time frame more than six times the length of the previous work on related polymerases (18,19,21,31), suggest that pol μ is more difficult to open compared to other polymerases in the same family. Although limited sampling in MD cannot be ruled out, these observations, together with the previously noted crystallization difficulties of capturing the open form of pol μ (T. A. Kunkel, National Institutes of Health, personal communication, 2010), raise the possibility that a well-defined open form of pol μ, as characterized by DNA sliding or protein domain rearrangements from the closed form, may not exist.
To better compare our results with previous findings on other polymerases obtained with the non-CMAP version of the CHARMM force field, we performed simulations of pol μ with the CHARMM force field without the CMAP specification (simulations IX and X). No substantial difference was observed (Fig. S2 b). Our previous studies on pol λ also revealed no significant difference between these different versions of CHARMM force fields (M. Foley and T. Schlick, unpublished). Our results suggest that force-field variations are not a factor in explaining the different biological observations.
Subtle residue motions
Coordination adjustment around Mg2+ (Asp330 and His329)
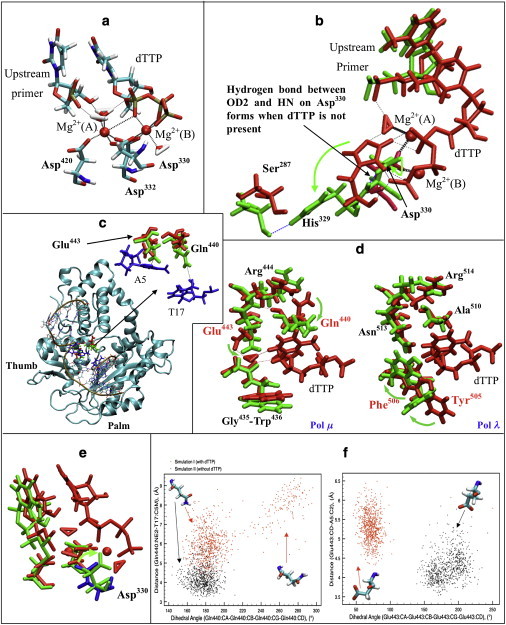
The structure of the active site is highly conserved among DNA polymerases (3). Two metal ions (usually Mg2+) are coordinated with a few acidic residues (51,52). In family X, three aspartates bind the ions at the active site. The geometry of the active site shown in Fig. 3 a is similar to geometries in other X-family polymerases (Table S2) (18,19,21). By the end of the simulation, the geometry around Mg2+ had not significantly deviated from the crystal structure, except that two water molecules moved in to coordinate with the catalytic Mg2+ [Mg2+ (A)].
Figure 3.
Active-site and dTTP-binding pocket coordination and rearrangement in pol μ. (a) Representative active-site conformation of pol μ from simulation I. Bold dashed lines indicate coordination around Mg2+; thin dashed lines indicate hydrogen bonds between water molecules and the DNA/dTTP. Mg2+(A), catalytic ion; Mg2+(B), nucleotide-binding ion. (b) Comparison of His329-Asp330 in pol μ in simulations I (red, with dTTP) and II (green, without dTTP). Bold dashed lines indicate coordination around Mg2+, black thin dashed lines indicate hydrogen bonds formed in simulation I, and blue thin dashed lines indicate hydrogen bonds formed in simulation II. (c) The location of the dTTP-binding pocket in pol μ (red, Gln440 and Glu443 in simulation I; green, Gln440 and Glu443 in simulation II; blue, A5 and T17 in DNA). Dashed lines indicate the distances used in cluster analysis (Fig. 3f). (d) Comparison of the dTTP-binding pocket of pol μ (left) and pol λ (right). Structures with dTTP are marked red (simulation I of pol μ, and ternary crystal structure of pol λ); structures without dTTP are marked green (simulation II of pol μ, and binary crystal structure of pol λ). Dashed lines indicate hydrogen bonds. The names of residues controlling the accommodating process are shown in red. (e) Comparison of active-site conformation of pol μ in simulations I (red and blue, with dTTP) and II (green, without dTTP). Asp330 in simulation I is marked blue. (f) Cluster analysis of Gln440 (left) and Glu443 (right) in simulations I (red) and II (black) based on dihedral angles (CA-CB-CG-CD) and distances to the DNA (Gln440:NE2-T17:C5M or Glu443:CD-A5:C2).
Previous experiments have shown that alanine mutations for Asp330 and Asp332 disrupt pol μ's activity (53). To investigate the roles of the three catalytic aspartates (Asp330, Asp332, and Asp420) in active-site assembly, we compared the active-site coordination with and without dTTP/Mg2+. Our simulations suggest that Asp330 in pol μ may control the active-site assembly: when dTTP and Mg2+ are removed, the Asp330 coordinating with both Mg2+ in ternary state rotates away (Fig. 3 e), thereby distorting the active site. The rearrangement of the aspartates between the ternary and binary states is commonly observed within the X-family, but different aspartates are involved. Asp490 in pol λ or Asp100 in pol X, analogous to Asp420 in pol μ, flip to coordinate with Mg2+ and/or other key residues (18,21), whereas Asp192 in pol β, analogous to Asp332 in pol μ, contributes most to the rearrangement (19). However, in other X-family polymerases, significant motion of residues analogous to Asp330 in pol μ (Asp190 in pol β, Asp427 in pol λ, and Asp49 in pol X) was not observed. Our study shows that the flip of Asp330 is related to the conformational change of His329, an active-site residue that is not conserved in pol β, pol λ, or pol X.
His329 was previously reported to be crucial for pol μ's catalytic activity when the substrate is single-stranded DNA (32). Our simulations suggest that His329 may also contribute to the function of pol μ when the substrate is double-stranded DNA. Fig. 3 b compares the geometry of His329–Asp330, with and without dTTP/Mg2+. When dTTP/Mg2+ is absent, His329 prefers to hydrogen-bond to the nearby Ser287, and Asp330 bends slightly to form a hydrogen bond between its OD2 and HN. With the incoming dTTP, His329 flips to dTTP and binds to it. The steric hindrance helps Asp330 to flip to coordinate with Mg2+. Because our simulations reveal the motion of His329–Asp330 to be independent of the loop conformation, we suggest that the motion of His329–Asp330 is a characteristic feature of pol μ's active-site assembly.
To illustrate the possible impact of the protonation states of His329, we changed the initial protonation state of His329 from Nδ to Nɛ (Fig. S4 a) and performed simulations (simulations XVI and XVII). The new active-site conformation is shown in Fig. S4 b. Our results illustrate that the changed state of His329 still triggers hydrogen-bond formation with the incoming dTTP. The motions of Asp330 also remain the same.
The time evolution of key dihedral angles of His329 and Asp330 in simulations I–VIII is presented in Fig. S5. Interestingly, in simulation VII (with a larger damping coefficient for sampling), although the dTTP/Mg2+ is present at the active site, His329 flips through an intermediate conformation, whereas Asp330 remains in its original conformation to coordinate with Mg2+ (Fig. S6). The intermediate conformation of His329 is also observed in simulation III, and in simulation XVI (Fig. S4 b) after the change in the protonation state of His329. This rearrangement suggests an inherent flexibility in the active site of pol μ.
Incoming nucleotide-binding pocket (Gln440 and Glu443)
The conserved Tyr-Phe motif in pol β and pol λ, and His-Phe in pol X participate in the binding of the incoming nucleotide to correctly position the primer terminus (30). In simulations of other X-family polymerases, a flip of the phenylalanine is associated with the protein or DNA motion (18,19,21), whereas the neighboring tyrosine or histidine directly contacts the bases of the DNA. However, this Tyr/His-Phe motif is lacking in pol μ, where Gly435–Trp436 occupies the corresponding location. No significant motion of Gly435–Trp436 was captured in any of our simulations. However, motions of Gln440 and Glu443, which correspond to residues Asp276 and Asn279 in pol β, occurred (Fig. 3 c). Experiments in pol β indicate that Asp276 weakens the binding affinity with the incoming nucleotide (26,54), whereas Asn279 hydrogen-bonds with dTTP to affect catalytic ability (55). In our simulations, we captured a similar interaction between Glu443 and dTTP through a water molecule (Fig. 3 d). This suggests that Glu443 in pol μ may also be related to pol μ's catalytic ability.
Fig. 3 d also shows that Gln440 and Glu443 together with Gly435–Trp436 form a dTTP-binding pocket. When dTTP is present, Gln440 flips toward Arg444, whereas Glu443 interacts with dTTP through a water-mediated hydrogen bond, allowing the pocket to accommodate the incoming dTTP. We observe that Gln440 and Glu443 in simulations without dTTP/Mg2+ (simulations II, IV, VI, and VIII) tend to tighten the dTTP-binding pocket compared to the dTTP/Mg2+-free simulations (simulations I, III, V, and VII) shown in Fig. S7. The cluster analysis for Gln440 and Glu443 from simulations I and II (Fig. 3 f) shows that with dTTP/Mg2+, Glu443 shifts away from the incoming nucleotide, whereas Gln440 also shifts and/or rotates to favor the accommodation of the incoming nucleotide. Without dTTP/Mg2+, both Gln440 and Glu443 remain closer to the DNA, thereby tightening the binding pocket.
In pol β, pol λ, and pol X, similar incoming nucleotide-binding pockets exist, but the side of Gly435–Trp436 (closer to the upstream primer strand) shifts and flips to accommodate dTTP (Fig. 3 d). In pol μ, motions are observed along the side of Gln440 and Glu443 instead (closer to the downstream primer strand). Furthermore, Glu443 and Gln440 in pol μ also interact with other key residues, such as Arg444 and Arg447, so further interactions may be formed.
Mutant systems (Arg444, Arg447, and Arg448)
We designed mutant simulations XI (R444A), XII (R447A), and XIII (R448A) to investigate the importance of these residues in pol μ. Arg444 and Arg447 are analogous to Arg514 and Arg517 in pol λ, which trigger both the DNA motion and the thumb loop motion (21), and Arg517 also controls fidelity (56). In pol β, pol μ's Arg447's analog Arg283 is also important for fidelity (57–59). Arg448 is not conserved in pol β or pol λ, where an alanine occupies the corresponding location. Our goal here was to determine how these arginines (Arg444, Arg447, and Arg448) contribute to the function of pol μ.
We first investigated the interactions between the three arginines and the DNA template, as shown in Fig. 2 e. Arg444 and Arg447 interact with the DNA template through a series of hydrogen bonds and stacking interactions, in similarity to a mechanism in pol λ, and Arg448 interacts with the DNA template through electrostatic interactions. In simulations XI and XII, we observe that the DNA template shifts away from the ternary position for ∼6 Å, and the A5 base also shifts away for ∼3 Å (Fig. 2 d), in a manner comparable to the previously observed shift in pol λ (21) between binary and ternary crystal structures. This may suggest the importance of Arg444 and Arg447 in maintaining the active (ternary) form of pol μ, via stabilization of the DNA template. Our suggestion is also supported by the stronger interaction energy between the thumb and DNA template in the wild-type simulation (I) than in the mutant simulations (XI and XII), as shown in Fig. S8.
In mutant simulations XI and XII, the motions of other key residues in the active site, such as His329, Asp330, Gln440, and Glu443, are not affected. Thus, the ability of Arg444/Arg447 in pol μ to trigger the motion of other key residues may be limited. However, Arg447 may participate in the active-site assembly when an incorrect nucleotide exists at the active site, as suggested by the similar fidelity checking function of Arg517 in pol λ or Arg283 in pol β (24,56).
In simulation XIII (R448A), the dTTP moves closer to base A6. We compare the distances of dTTP-A5 and dTTP-A6 in simulations I and XIII in Fig. S9. In Fig. 2 f, we show that dTTP tends to pair with A6 instead of the correct base, A5. We observe that this frameshift occurs in three steps. First, the decreased steric hindrance between Ala448 and the DNA template triggers several bases in the DNA template, including A5 and A6, to shift toward the downstream side. Second, because the shifted A5 is closer to Gln440, Gln440 is attracted by A5 and flips to hydrogen-bond to it, thereby deforming the nucleotide-binding pocket. Third, dTTP shifts and rotates toward the upstream side to pair with A6. Overall, the DNA template shifts toward the downstream side, whereas the dTTP shifts toward the upstream side, leading to an incorrect frameshift basepairing. Thus, we suggest that Arg448 with the help of Gln440 in pol μ is important for maintaining the positions of both the DNA template and the incoming nucleotide, particularly to prevent insertion frameshift errors in DNA synthesis. Insertion errors in pol μ were also observed during the template-dependent synthesis with a nonpaired primer in vitro (9). Thus, we suggest the possibility that Gln440 and Arg448 also participate in nonpaired primer DNA synthesis, a unique function of pol μ.
In simulations XII and XIII (R447A and R448A), the final thumb loop-Loop 1 distance is unusually large (i.e., 34.7 Å and 29.4 Å; Fig. 2 d and Fig. S10). Such an open Loop 1 conformation may hamper the various interactions between Loop 1 and the DNA (9,33), thereby interfering with the chemical reaction. Because Arg444, Arg447, and Arg448 are also conserved in Tdt, which only catalyzes with single-stranded DNA with no template strands, these residues may also be important for interactions with other portions of the DNA or protein.
Conclusions
Our MD simulations of pol μ with and without the incoming nucleotide and catalytic ions (Mg2+) in various forms, including mutant systems, probed the transition of pol μ upon binding of the correct nucleotide. The results suggest that, in contrast to several other X-family polymerases, the catalytic cycle of pol μ may not include large-scale protein subdomain or DNA motion. Based on a comparison with other dynamics studies of DNA polymerases, including pol X (18), pol β (19), and pol λ (21), we conclude that the length of our pol μ simulations is not likely a major factor that can explain the different observations. This raises the possibility that an induced-fit mechanism characterized by an open-to-closed transition before chemistry may not exist in pol μ. Of course, we cannot exclude the possibility that other transitions could occur on longer timescales.
Subtle motions of key residues were captured, suggesting several active-site rearrangements upon dTTP binding. Rearrangements of His329 and Asp330 are important for the active-site assembly of pol μ. The dTTP accommodation characterized by the flip of Glu443 and Gln440 was also captured.
Our mutant simulations for R444A, R447A, and R448A suggest that although Arg444 and Arg447 appear to stack with the DNA template in a mechanism similar to that of pol λ, they trigger much less motion in other key residues. Arg448 and Gln440, neither of which are conserved in pol β or pol λ, are important for maintaining the correct basepairing of the incoming nucleotide in pol μ, and therefore to prevent insertion frameshift errors. They may also participate in nonpairing primer DNA synthesis. Arg447 and Arg448 also affect the conformation of Loop 1 and thus may be important for maintaining the Loop 1-DNA interactions that are crucial for template-independent synthesis (33). A natural extension of this work would be to determine whether these residues play important roles in template-independent synthesis or controlling fidelity in pol μ.
Acknowledgments
The computations performed in this study were conducted using the resources of the Computational Center for Nanotechnology Innovations supported by the New York State Foundation for Science, Technology and Innovation, and the Dell computer cluster supported by New York University Information Technology Services. Molecular images were generated using the VMD program (39).
This work was supported in part by Philip Morris USA Inc. and Philip Morris International, the National Science Foundation (MCB-0316771), the National Institutes of Health (R01 ES012692), and the American Chemical Society's Petroleum Research Fund (PRF No. 39115-AC4 to T. S.).
Supporting Material
References
- 1.Faili A., Aoufouchi S., Weill J.C. DNA polymerase η is involved in hypermutation occurring during immunoglobulin class switch recombination. J. Exp. Med. 2004;199:265–270. doi: 10.1084/jem.20031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hubscher U., Maga G., Spadari S. Eukaryotic DNA polymerases. Annu. Rev. Biochem. 2002;71:133–163. doi: 10.1146/annurev.biochem.71.090501.150041. [DOI] [PubMed] [Google Scholar]
- 3.Steitz T.A. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 4.Yamtich J., Sweasy J.B. DNA polymerase family X: function, structure, and cellular roles. Biochim. Biophys. Acta. 2010;1804:1136–1150. doi: 10.1016/j.bbapap.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchiyama Y., Takeuchi R., Sakaguchi K. Distribution and roles of X-family DNA polymerases in eukaryotes. Biochimie. 2009;91:165–170. doi: 10.1016/j.biochi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Sweasy J.B., Lauper J.M., Eckert K.A. DNA polymerases and human diseases. Radiat. Res. 2006;166:693–714. doi: 10.1667/RR0706.1. [DOI] [PubMed] [Google Scholar]
- 7.Mirkin S.M. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 8.Capp J.P., Boudsocq F., Canitrot Y. The DNA polymerase λ is required for the repair of non-compatible DNA double strand breaks by NHEJ in mammalian cells. Nucleic Acids Res. 2006;34:2998–3007. doi: 10.1093/nar/gkl380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nick McElhinny S.A., Havener J.M., Ramsden D.A. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol. Cell. 2005;19:357–366. doi: 10.1016/j.molcel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Bertocci B., De Smet A., Reynaud C.A. Nonoverlapping functions of DNA polymerases μ, λ, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006;25:31–41. doi: 10.1016/j.immuni.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Bertocci B., De Smet A., Reynaud C.A. Immunoglobulin κ light chain gene rearrangement is impaired in mice deficient for DNA polymerase μ. Immunity. 2003;19:203–211. doi: 10.1016/s1074-7613(03)00203-6. [DOI] [PubMed] [Google Scholar]
- 12.Dudley D.D., Chaudhuri J., Alt F.W. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv. Immunol. 2005;86:43–112. doi: 10.1016/S0065-2776(04)86002-4. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Wu X., Wang Z. Highly frequent frameshift DNA synthesis by human DNA polymerase μ. Mol. Cell. Biol. 2001;21:7995–8006. doi: 10.1128/MCB.21.23.7995-8006.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Covo S., Blanco L., Livneh Z. Lesion bypass by human DNA polymerase μ reveals a template-dependent, sequence-independent nucleotidyl transferase activity. J. Biol. Chem. 2004;279:859–865. doi: 10.1074/jbc.M310447200. [DOI] [PubMed] [Google Scholar]
- 15.Roettger M.P., Fiala K.A., Suo Z. Pre-steady-state kinetic studies of the fidelity of human DNA polymerase μ. Biochemistry. 2004;43:13827–13838. doi: 10.1021/bi048782m. [DOI] [PubMed] [Google Scholar]
- 16.Domínguez O., Ruiz J.F., Blanco L. DNA polymerase μ (Pol μ), homologous to TdT, could act as a DNA mutator in eukaryotic cells. EMBO J. 2000;19:1731–1742. doi: 10.1093/emboj/19.7.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahajan K.N., Nick McElhinny S.A., Ramsden D.A. Association of DNA polymerase μ (pol μ) with Ku and ligase IV: role for pol μ in end-joining double-strand break repair. Mol. Cell. Biol. 2002;22:5194–5202. doi: 10.1128/MCB.22.14.5194-5202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sampoli Benítez B.A., Arora K., Schlick T. In silico studies of the African swine fever virus DNA polymerase X support an induced-fit mechanism. Biophys. J. 2006;90:42–56. doi: 10.1529/biophysj.105.071944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arora K., Schlick T. In silico evidence for DNA polymerase-β's substrate-induced conformational change. Biophys. J. 2004;87:3088–3099. doi: 10.1529/biophysj.104.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora K., Schlick T. Conformational transition pathway of polymerase β/DNA upon binding correct incoming substrate. J. Phys. Chem. B. 2005;109:5358–5367. doi: 10.1021/jp0446377. [DOI] [PubMed] [Google Scholar]
- 21.Foley M.C., Arora K., Schlick T. Sequential side-chain residue motions transform the binary into the ternary state of DNA polymerase λ. Biophys. J. 2006;91:3182–3195. doi: 10.1529/biophysj.106.092080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radhakrishnan R., Schlick T. Orchestration of cooperative events in DNA synthesis and repair mechanism unraveled by transition path sampling of DNA polymerase β's closing. Proc. Natl. Acad. Sci. USA. 2004;101:5970–5975. doi: 10.1073/pnas.0308585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beard W.A., Shock D.D., Wilson S.H. Loss of DNA polymerase β stacking interactions with templating purines, but not pyrimidines, alters catalytic efficiency and fidelity. J. Biol. Chem. 2002;277:8235–8242. doi: 10.1074/jbc.M107286200. [DOI] [PubMed] [Google Scholar]
- 24.Arora K., Beard W.A., Schlick T. Mismatch-induced conformational distortions in polymerase β support an induced-fit mechanism for fidelity. Biochemistry. 2005;44:13328–13341. doi: 10.1021/bi0507682. [DOI] [PubMed] [Google Scholar]
- 25.Beard W.A., Shock D.D., Wilson S.H. Influence of DNA structure on DNA polymerase β active site function: extension of mutagenic DNA intermediates. J. Biol. Chem. 2004;279:31921–31929. doi: 10.1074/jbc.M404016200. [DOI] [PubMed] [Google Scholar]
- 26.Vande Berg B.J., Beard W.A., Wilson S.H. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase β. Implication for the identity of the rate-limiting conformational change. J. Biol. Chem. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- 27.Kraynov V.S., Werneburg B.G., Tsai M.D. DNA polymerase β: analysis of the contributions of tyrosine-271 and asparagine-279 to substrate specificity and fidelity of DNA replication by pre-steady-state kinetics. Biochem. J. 1997;323:103–111. doi: 10.1042/bj3230103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krahn J.M., Beard W.A., Wilson S.H. Structural insights into DNA polymerase β deterrents for misincorporation support an induced-fit mechanism for fidelity. Structure. 2004;12:1823–1832. doi: 10.1016/j.str.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Sawaya M.R., Prasad R., Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Diaz M., Bebenek K., Pedersen L.C. A closed conformation for the Pol λ catalytic cycle. Nat. Struct. Mol. Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y., Arora K., Schlick T. Subtle but variable conformational rearrangements in the replication cycle of Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) may accommodate lesion bypass. Protein Sci. 2006;15:135–151. doi: 10.1110/ps.051726906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moon A.F., Garcia-Diaz M., Pedersen L.C. Structural insight into the substrate specificity of DNA polymerase μ. Nat. Struct. Mol. Biol. 2007;14:45–53. doi: 10.1038/nsmb1180. [DOI] [PubMed] [Google Scholar]
- 33.Juárez R., Ruiz J.F., Blanco L. A specific loop in human DNA polymerase μ allows switching between creative and DNA-instructed synthesis. Nucleic Acids Res. 2006;34:4572–4582. doi: 10.1093/nar/gkl457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delarue M., Boulé J.B., Papanicolaou C. Crystal structures of a template-independent DNA polymerase: murine terminal deoxynucleotidyltransferase. EMBO J. 2002;21:427–439. doi: 10.1093/emboj/21.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bebenek K., Garcia-Diaz M., Kunkel T.A. Loop 1 modulates the fidelity of DNA polymerase λ. Nucleic Acids Res. 2010;38:5419–5431. doi: 10.1093/nar/gkq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schlick T. Springer; New York: 2010. Molecular Modeling and Simulation: An Interdisciplinary Guide. [Google Scholar]
- 37.Schlick T., Collepardo-Guevara R., Xiao X. Biomolecular Modeling and Simulation: A Field Coming of Age. Q. Rev. Biophys. 2010 doi: 10.1017/S0033583510000284. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 39.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- 40.Brooks B.R., Bruccoleri R.E., Karplus M. Charmm—a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 41.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 42.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 43.MacKerell A.D., Banavali N.K. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000;21:105–120. [Google Scholar]
- 44.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of Cartesian equations of motion of a system with constraints—molecular-dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 45.Feller S.E., Zhang Y.H., Brooks B.R. Constant-pressure molecular-dynamics simulation—the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 46.Darden T., York D., Pedersen L. Particle mesh Ewald: An N · log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 47.Chong L.T., Duan Y., Kollman P.A. Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48G7. Proc. Natl. Acad. Sci. USA. 1999;96:14330–14335. doi: 10.1073/pnas.96.25.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorfe A.A., Jelesarov I. Energetics of sequence-specific protein-DNA association: computational analysis of integrase Tn916 binding to its target DNA. Biochemistry. 2003;42:11568–11576. doi: 10.1021/bi026937p. [DOI] [PubMed] [Google Scholar]
- 49.Adcock S.A., McCammon J.A. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foley M.C., Padow V.A., Schlick T. DNA pol λ's extraordinary ability to stabilize misaligned DNA. J. Am. Chem. Soc. 2010;132:13403–13416. doi: 10.1021/ja1049687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delarue M., Poch O., Argos P. An attempt to unify the structure of polymerases. Protein Eng. 1990;3:461–467. doi: 10.1093/protein/3.6.461. [DOI] [PubMed] [Google Scholar]
- 52.Wang J., Sattar A.K., Steitz T.A. Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 53.Capp J.P., Boudsocq F., Canitrot Y. Involvement of DNA polymerase μ in the repair of a specific subset of DNA double-strand breaks in mammalian cells. Nucleic Acids Res. 2007;35:3551–3560. doi: 10.1093/nar/gkm243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sweasy J.B. Fidelity mechanisms of DNA polymerase β. Prog. Nucleic Acid Res. Mol. Biol. 2003;73:137–169. doi: 10.1016/s0079-6603(03)01005-5. [DOI] [PubMed] [Google Scholar]
- 55.Martínek V., Bren U., Florián J. DNA polymerase β catalytic efficiency mirrors the Asn279-dCTP H-bonding strength. FEBS Lett. 2007;581:775–780. doi: 10.1016/j.febslet.2007.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foley M.C., Schlick T. Relationship between conformational changes in pol λ's active site upon binding incorrect nucleotides and mismatch incorporation rates. J. Phys. Chem. B. 2009;113:13035–13047. doi: 10.1021/jp903172x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Werneburg B.G., Ahn J., Tsai M.D. DNA polymerase β: pre-steady-state kinetic analysis and roles of arginine-283 in catalysis and fidelity. Biochemistry. 1996;35:7041–7050. doi: 10.1021/bi9527202. [DOI] [PubMed] [Google Scholar]
- 58.Beard W.A., Osheroff W.P., Wilson S.H. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β. J. Biol. Chem. 1996;271:12141–12144. doi: 10.1074/jbc.271.21.12141. [DOI] [PubMed] [Google Scholar]
- 59.Osheroff W.P., Beard W.A., Kunkel T.A. Base substitution specificity of DNA polymerase β depends on interactions in the DNA minor groove. J. Biol. Chem. 1999;274:20749–20752. doi: 10.1074/jbc.274.30.20749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.