

Abstract
A new (to our knowledge) de novo design framework with a ranking metric based on approximate binding affinity calculations is introduced and applied to the discovery of what we believe are novel HIV-1 entry inhibitors. The framework consists of two stages: a sequence selection stage and a validation stage. The sequence selection stage produces a rank-ordered list of amino-acid sequences by solving an integer programming sequence selection model. The validation stage consists of fold specificity and approximate binding affinity calculations. The designed peptidic inhibitors are 12-amino-acids-long and target the hydrophobic core of gp41. A number of the best-predicted sequences were synthesized and their inhibition of HIV-1 was tested in cell culture. All peptides examined showed inhibitory activity when compared with no drug present, and the novel peptide sequences outperformed the native template sequence used for the design. The best sequence showed micromolar inhibition, which is a 3–15-fold improvement over the native sequence, depending on the donor. In addition, the best sequence equally inhibited wild-type and Enfuvirtide-resistant virus strains.
Introduction
Worldwide, as of 2007, an estimated 33,000,000 people are living with HIV, and the annual number of new HIV infections is estimated at ∼2,700,000 (1). Recent work (2,3) has outlined the progress and setbacks that have occurred in the pursuit of an HIV vaccine and the advances and options for controlling HIV. One of the reasons stated for the limited progress and the setbacks is that many of the vaccine candidates are based upon empirical trials, as opposed to predictive, scientifically based trials. In contrast, excitement has been generated for the development of entry and fusion inhibitors that prevent infection of susceptible cells.
The infection of host cells by HIV-1 is a multistep process which begins with the proteolytic cleavage of gp160 into two subunits by a cellular protease. The resulting subunits consist of a surface subunit (gp120) and a transmembrane subunit (gp41) (4). The surface subunit gp120 binds to the host cell receptor CD4, which leads to a conformational change in gp120, causing the extension of the V3 loop of gp120 which then binds to chemokine receptor CCR5 or CXCR4 or both. This latter binding leads to the exposure of the fusion domain of the transmembrane subunit, gp41. The transmembrane subunit gp41 is considered a class I membrane fusion protein and can be divided into the following domains: extracellular, transmembrane, and cytoplasmic (5). The extracellular domain can be further divided into four functional domains: fusion peptide, N-terminal heptad repeat (NHR or HR1), C-terminal heptad repeat (CHR or HR2), and a tryptophan-rich region (5). Both NHR and CHR contain a number of 4-3 heptad repeats, and these repeats generally form coiled-coil structures (6). After gp41 has been exposed, the NHR and CHR come together to form a six-helix bundle. Once the six-helix bundle is formed, the viral and cell membranes fuse.
The first U.S. Food and Drug Administration (FDA)-approved HIV fusion inhibitor, Enfuvirtide (IC50 = 36 nM (7)), was developed by Roche (Hoffmann-La Roche, Nutley, NJ) and Trimeris (Morrisville, NC), and has been in use since 2003. Enfuvirtide's sequence is based on the sequence of gp41 CHR and targets gp41 NHR. Despite its success as an inhibitor, it is a 36-amino-acid helical peptide and its length makes the drug difficult and costly to manufacture. As a result, it has three weaknesses:
-
1.
It can be easily degraded by proteolytic enzymes in the blood, leading to short lifetimes of 2 h in vivo.
-
2.
It has a high cost of production due to its size.
-
3.
It lacks oral bioavailability, resulting in inconvenient dosage form and schedule (90 mg twice daily).
The other fusion inhibitor on the market, Maraviroc (IC50 of 0.40 nM (8)), was developed by Pfizer (New York, NY) and was approved in 2007. As with Enfuvirtide, it is used in patients that have developed resistance to standard HIV treatments. Maraviroc differs from Enfuvirtide in that it is a small molecule and not a peptide and it targets gp120 as opposed to gp41. Maraviroc binds to CCR5, one of the chemokine receptors needed in the infection process. It is available in tablet form, making it a more convenient drug to take (150 mg and 300 mg doses twice daily). It has one weakness, however. Maraviroc is only effective in patients whose virus populations utilize the CCR5 receptor. The drug has limited activity in patients whose virus population has evolved the ability to utilize the CXCR4 receptor or both receptors.
In addition to the two FDA-approved fusion inhibitors, there are many more that have been published in the open literature. Welch et al. (9) presented a very potent peptide (IC50 = 0.25 nM) of length 45, whereas in contrast Sia et al. (10) tested a number of shorter peptides (14-amino-acids long), with the best obtaining an IC50 of 35 μM. Other work has also focused on finding peptidic inhibitors of Enfuvirtide-resistant HIV strains (11–13), which have developed due to the increased use of Enfuvirtide. Third-generation fusion inhibitors such as T-2635 (38-amino-acids long) (14) and Sifuvirtide (36-amino-acids long) (15) are peptides based upon the original Enfuvirtide sequence that have been improved using rational design. Sifuvirtide showed a sixfold improvement over Enfuvirtide whereas T-2635 showed up to 3600-fold improvement against Enfuvirtide-resistant viruses. In addition, various small molecules are being investigated to find new HIV-1 inhibitors that may have increased bioavailability and reduced cost of production over peptidic inhibitors (5,16–20). There have also been studies that add sugar or cholesterol groups to peptides to increase inhibitory potencies and make the peptidic inhibitors more druglike (5,21,22).
De novo protein design has been applied to a number of systems, including HIV-1 (23,24). The goal is to determine a specific amino-acid sequence that will fold into a given rigid or flexible three-dimensional backbone template. Reviews by Fung et al. (25), Floudas et al. (26), Kortemme and Baker (27), and Kuhlman and Baker (28) highlight the methodologies and successes in the field. Some examples of protein design methods include dead-end elimination (29–31), self-consistent mean field (32,33), Monte Carlo (34–37), and methods that use flexible backbone templates (38–40).
Methods and Materials
De novo design framework
What we believe are novel inhibitors of fusion were designed using a de novo protein design framework. An overview of the de novo design framework is presented in Fig. 1.
Figure 1.
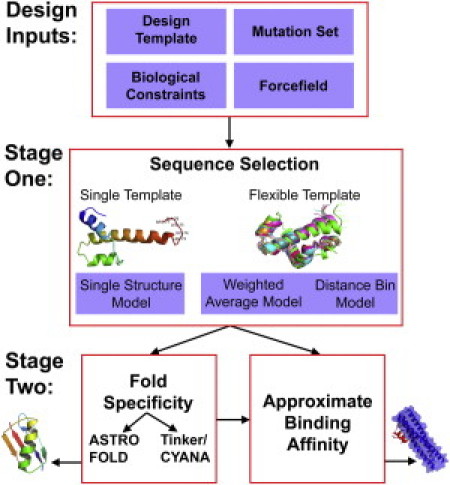
Overview of the de novo design framework.
Design template
The design template for entry inhibitors is the crystal structure of C14linkmid, a 14-residue crosslinked peptide, in complex with the hydrophobic core of gp41 (PDB code: 1GZL). The hydrophobic core of gp41 consists of residues Leu29, Leu30, Leu32, Thr33, Val34, Trp35, Gly36, Ile37, Lys38, Leu40, and Gln41 (numbered using positions in 1GZL) and has been shown to be a viable drug target (41–44). C14linkmid was confirmed by Sia et al. (10) to be a highly potent inhibitor to gp41 with an IC50 value of 35 μM for cell-cell fusion. For the design, the crosslinker was not incorporated. Sia et al. (10) reported an IC50 for the unlinked peptide (C14unlinkmid) of >500 μM. Because the PDB file contained two identical monomer units (A-C and B-D), only chains A and C were used for the design template. The crystal structure of the complex is shown in Fig. 2.
Figure 2.
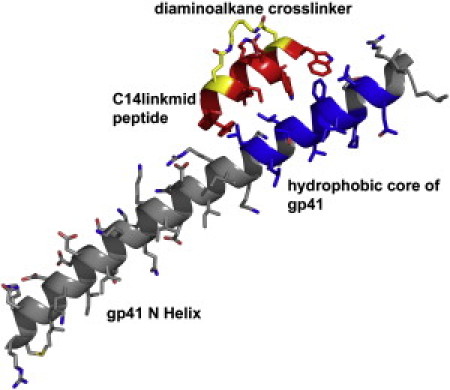
Crystal structure of C14linkmid in complex with the hydrophobic core of gp41, PDB code: 1GZL. The diaminoalkane crosslinker and the hydrophobic core of gp41, consisting of residues Leu29, Leu30, Leu32, Thr33, Val34, Trp35, Gly36, Ile37, Lys38, Leu40, and Gln41are colored blue (the residues are numbered according to their position in 1GZL). Figure created using PyMOL v0.99 (73).
Mutation set and biological constraints
Although the original sequences of C14linkmid and C14unlinkmid are 14-residues long, only 12 of the residues were present in the crystal structure
All positions were varied according to solvent-accessible surface area (SASA) patterning, which was also employed in the full-sequence design of human β-defensin-2 (45). If the residue in the crystal structure has a SASA >50%, a set of hydrophilic amino acids are allowed. If the residue has a SASA <20%, a set of hydrophobic residues are allowed. If the residue has an SASA in between, then all residues are allowed. Proline was not allowed, because it creates kinks or it breaks an α-helix, which is the structure of C14linkmid. Positions 628, 631, 635, and 638 were allowed to mutate to the hydrophobic set
Position 629 was allowed to mutate to the hydrophobic set
Positions 630, 632, 634, 637, and 639 were allowed to mutate to all amino acids except Cys and Pro. Positions 633 and 636 were allowed to mutate to all amino acids except Pro. No residues of the hydrophobic core of gp41 were allowed to mutate, as this is the target protein we wish to inhibit. This mutation set leads to a problem complexity of 2.51 × 1013.
In addition, the charge of the section of the peptidic inhibitor from position 630 to position 636 was fixed to be within plus- or minus-one from the native charge of −3.
Force field
The force field used was derived by solving a linear programming parameter estimation problem which requires the low energy high-resolution decoys for a large training set of proteins to be energetically less favorable than their native conformations (46–48). In the de novo design of entry inhibitors for HIV-1, we used the high-resolution centroid-centroid force field (47) because, based on our previous studies on human β-defensin-2 (45), it was more accurate than the Cα-Cα force field (46).
Sequence selection
Because the design template is a single crystal structure, the basic model for sequence selection was used in the sequence selection stage; see Eq. 1 below (for the formulations of the flexible template models, the reader is referred to (40,45)):
subject to
| (1) |
The set i = 1,…,n defines the number of amino-acid positions along the backbone of the design template. Set j{i} = 1,…,mi defines the mutations that are allowed at each position i, with, for the general case, mi = 20 ∀ i. The sets k and l are equivalent to the sets i and j, respectively. In the objective function,
is the pairwise energy parameter (force field). It is dependent upon the distance between residues i and k, (xi, xk); the type of amino acid j at position i; and the type of amino acid l at position k. Two binary variables are introduced, yji and ylk, which take the value of one if amino acid j (or l) is selected at position i (or k) and zero otherwise. The final binary variable, wjlik, is an interaction variable and is defined as the product of yji and ylk. It is 1 if amino acid j is selected at position i and amino acid l is selected at position k.
Using Eq. 1, we generated 500 low-energy sequences for which we calculated the fold specificities.
Fold specificity calculations
Fold specificity calculations are used to rank the sequences from the sequence selection stage based upon a measure of how well the sequence folds into the design template. As seen in Fig. 1, the fold specificity calculations can take on one of two approaches: the ASTRO-FOLD approach and the Tinker/CYANA approach. For this study, the Tinker/CYANA approach was used. For details about the ASTRO-FOLD approach, the reader is referred to Klepeis et al. (49–53) and the review by C. A. Floudas (54).
Fung et al. (45) proposed an approximate method for fold specificity calculation which is computationally efficient. First, a flexible template is defined based on the upper and lower bounds on both the distances between α-carbons and the ϕ- and ψ-angles between residues. An ensemble of hundreds of random conformers is then generated within the confines of the flexible template using the CYANA 2.1 software package for NMR structure refinement (55,56). For each structure in the ensemble, local minimizations are then performed by the TINKER package (57) as directed by gradients in the fully atomistic force field AMBER (58). AMBER is used to evaluate the potential energy of the structure. These ensembles are generated for the native sequence of the fold and for each candidate mutant sequence. The specificity of each mutant sequence (set novel) to the target fold is then calculated relative to the native sequence (set native) using the Boltzmann distribution from statistical mechanics (Eq. 2), where
Both the predicted energy of each conformer and its root mean-square deviation from the template structure are used in this calculation:
| (2) |
Fold specificity calculations using the Tinker/CYANA approach were performed on all 500 sequences from the sequence selection stage, and the 30 top-ranked sequences were used in the approximate binding affinity stage.
Approximate binding affinity calculations
Bellows et al. (59) recently presented what they consider a novel protein design framework based upon approximate binding affinity calculations. The approximate binding affinity calculations are used to rank the designed sequences that are in complex with a target protein. These calculations can be done on the sequences directly from the sequence selection stage, or can be performed on the high fold specificity sequences obtained from the fold specificity stage. Because this stage is computationally demanding, approximate binding affinity calculations are not done on all 500 sequences, but rather a smaller subset. In this case, the 30 top-ranked sequences based upon fold specificity were sent to this stage.
Lilien et al. (60) proposed an approach for the calculation of approximate binding affinities of protein-ligand complexes. It is based on generating rotamerically based ensembles of the protein, the ligand, and the protein-ligand complex and using those ensembles to calculate partition functions. This approximate binding affinity is denoted as K∗ and is defined by
| (3) |
In Eq. 3, qPL is the partition function of the protein-ligand complex, qP is the partition function of the free protein, and qL is the partition function of the free ligand. The partition functions are defined in Eq. 4, where the sets B, F, and L contain the rotamerically based conformations of the bound protein-ligand complex, the free protein, and the free ligand, respectively. En is the energy of conformation n, R is the gas constant, and T is the temperature:
| (4) |
K∗ is an approximation of the true binding affinity (KA) because of the approximation of the partition functions. Because K∗ uses the Boltzmann probability distribution, it satisfies the Ergodic hypothesis and can be proved to approximate KA (see Lilien et al. (60) and Bellows et al. (59) for derivation). If the partition functions are exactly calculated, K∗ = KA. Because enumerating all possible conformers of complex molecular species is computationally demanding, K∗ is approximate.
The rotamerically based conformation ensembles (sets B, F, and L) are generated using the Rosetta++ software package and OREO, a clustering algorithm. An ensemble of 1000 structures is predicted for each sequence using RosettaAbinitio (61–63). The 1000 structures are clustered based upon their ϕ- and ψ-angles using OREO (64,65), giving 11 representative backbone structures for each peptide sequence. These 11 structures are docked to the target protein using RosettaDock (66–68), generating 1000 complexes for each peptide structure. The final rotamerically based conformation ensembles are generated using RosettaDesign (69). For the peptide and complex ensembles, 110 starting structures from the structure prediction ensemble and the docking ensemble are selected and expanded for final ensembles of 22,000 conformers each. For the protein ensemble, one starting structure is used for a final ensemble of 2000 conformers. Details on ensemble generation are provided in the Supporting Material.
Single round infectivity assay
The single-round infectivity assay is carried out at The Johns Hopkins University School of Medicine (laboratory of Professor R. F. Silicianos). It is based on an assay developed by Zhang et al. (70) and further modified by Shen et al. (7). Briefly, pseudotyped virus capable of single-round infection is prepared by transfecting HEK293T cells with a GFP-tagged, envelope-defective HIV-1 vector (pNL43-DE-EGFP) along with an envelope expression vector carrying either HIV-1 CXCR4 or envelope protein G of vesicular stomatitis virus (VSVG) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturers protocol. Resistance mutation gp41 N43D was made by site-directed mutagenesis (Stratagene, La Jolla, CA). At 48 h after transfection, cell debris is cleared and virus particles are isolated by ultracentrifugation as described previously. Virus amounts are standardized by p24 using an ELISA (PerkinElmer, Norwalk, CT). Peripheral blood mononuclear cells are obtained from healthy blood donors by Hypaque-Ficoll gradient centrifugation and then activated with phytohemagglutinin (0.5 mg/mL) and interleukin-2 (100 U/mL) for three days. CD4+ T cells are selected by magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) and seeded in a 96-well plate at 105 cells per well in RPMI1640 supplemented with 10% fetal bovine serum, interleukin-2 (100 U/mL), and cytokine-rich supernatant. Drugs are added at this step, and then infection with recombinant virus is carried out by spinoculation at 1200g, 30 1C for 2 h.
Infected cells are incubated at 37°C for three days, washed, and then fixed in 2% formaldehyde. Infectivity is quantified as the percentage of GFP+ cells by FACS analysis (BD Biosciences, San Jose, CA). Viability is approximated by cell morphology as measured by forward and side scatter. Initial screening experiments were done in duplicate with cells from two different donors. Envelope specificity experiments were done in duplicate with cells from one donor. Followup screening of SQ435 against wild-type and N43D resistant virus was done in triplicate with cells from three different donors, and measurements with Enfuvirtide were done in triplicate with cells from two donors. All healthy blood donors gave their informed consent, and the study was approved by the Institutional Review Board of The Johns Hopkins University.
Results
Computational results
Table 1 shows the top 10 sequences ranked based upon approximate binding affinity. Included in this set is the native sequence of C14unlinkmid. The sequences show some remarkable conservation in certain positions. The overall mutation pattern obtained from the 10 sequences is
Positions 637 and 639 consistently mutated to Arg and positions 628, 630, 631, 634, 636, and 638 always mutated to one of two amino acids, with one of the choices often being the native amino acid. Only position 629 showed much variability. In addition, there was a strong prevalence for Trp or Tyr selection, seen in positions 628, 631, 635, and 638. The native sequence has only two Trp and one Tyr, yet a number of the mutant sequences have a combination of Trp and Tyr up to five. Six of the 30 sequences are predicted to be better binders than the native.
Table 1.
Computational results for the design of novel HIV-1 fusion inhibitors
| Name | Sequence selectionrank | Fold specificityrank | Approximate bindingaffinity rank | Sequence |
||
|---|---|---|---|---|---|---|
| 628 | 639 | |||||
| SQ044 | 44 | 29 | 1 | Y | CDYEDRYERW | R |
| SQ435 | 435 | 15 | 2 | W | CDWRDEWERY | R |
| SQ175 | 175 | 11 | 3 | W | ADWRDEWERY | R |
| SQ486 | 486 | 20 | 4 | Y | MEYDERYDRW | R |
| SQ323 | 323 | 17 | 5 | Y | WEYDDRYDRW | R |
| SQ318 | 318 | 3 | 6 | Y | VEYDDRYDRW | R |
| Native | n/a | n/a | 7 | W | EEWDREIENY | T |
| SQ321 | 321 | 2 | 8 | Y | LEYDDRYDRW | R |
| SQ322 | 322 | 6 | 9 | Y | IEYDDRYDRW | R |
| SQ457 | 457 | 1 | 10 | W | ADWRDEWDRY | R |
Experimental results
The five top-ranked sequences based on approximate binding affinities plus the native sequence itself were selected for synthesis and tested using a single round infectivity assay. Peptide synthesis was performed by GenScript (Piscataway, NJ). The peptides were ordered in quantities of 1–4 mg at >95% purity. The N-termini were acetylated and the C-termini were amidated. We then verified peptide molecular weights and checked for side products by matrix-assisted laser desorption/ionization mass spectrometry. All peptides were found to be as expected.
Fig. 3 shows the experimental results for the initial peptide screen. Every mutant sequence except SQ044 showed experimental inhibitory activity better than the native sequence, as was predicted by the de novo protein design approach. SQ435 was shown to be the most potent inhibitor, with a viral replication (% of control) of 0.05–1.12% at 500 μM and 20–35% at 50 μM. The IC50 of SQ435 was then determined against cells from two additional donors in duplicate and found to be 29.7 μM (Fig. 4 A, experiment 140). In addition, almost all the peptides showed minimal toxicity, with >90% viability at a concentration of 500 μM. Fig. S1 in the Supporting Material shows the viability of the cells treated with each peptide at two concentrations, 50 μM and 500 μM.
Figure 3.
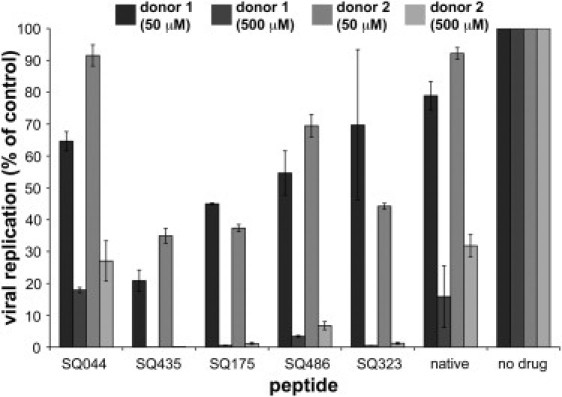
Inhibition of entry of wild-type HIV into primary CD4+ T cells by the top five mutant sequences and the native sequence. Experiments were done in duplicate with cells from two donors and at two peptide concentrations, 50 μM and 500 μM. Replication (% of control) was determined by single-round infectivity assay with GFP-encoding recombinant HIV. (Error bars) Span of values obtained.
Figure 4.
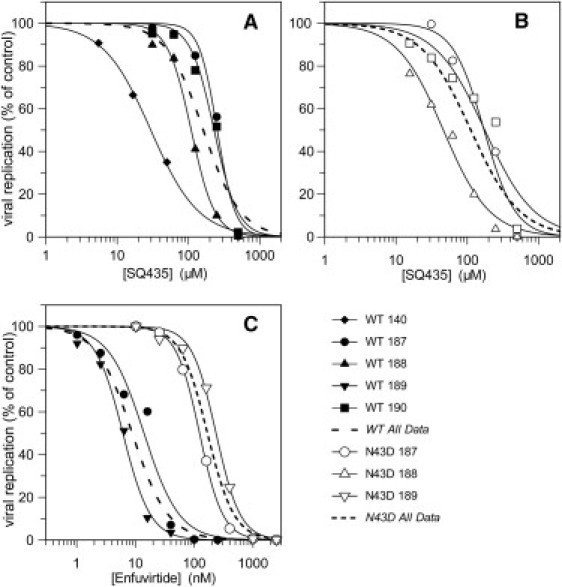
Inhibition of entry of wild-type and gp41 N43D mutant HIV into primary CD4+ T cells by SQ435 (A and B) and Enfuvirtide (C). Replication (% of control) was determined by single-round infectivity assay with GFP-encoding recombinant HIV. Individual donors illustrated with unique symbols and separate curves to illustrate the donor-to-donor variability in potency. (Solid symbol) Wild-type virus. (Open symbol) N43D mutant. (Dashed lines) Aggregate IC50 curve across all donors, and two different dash styles distinguish the two viruses used.
To determine whether the inhibition of HIV-1 replication is, in fact, at the entry stage, sequences SQ435 and SQ175 were selected for testing against HIV pseudoviruses carrying either HIV-1 envelope or envelope protein G of VSVG (Fig. S2). Although SQ435 and SQ175 do not show as great an inhibitory effect in this experiment as compared to the two donors in Fig. 3, there is still an effect at a concentration of 500 μM with a viral replication (% of control) for SQ435 of 0.25% and for SQ175 of 6%. In comparison, there is no effect when the VSVG envelope is used, with viral replication (% of control) close to 100% for both peptides. This shows that the peptides are HIV-1-envelope-specific.
We then selected peptide SQ435 for more rigorous IC50 measurement and also tested it against virus bearing the well-known Enfuvirtide resistance mutation gp41 N43D (71). Enfuvirtide (Hoffmann-La Roche) was used as a control. A new batch of peptide (100 mg) was purchased from GenScript, dissolved to 5 mM, and confirmed by matrix-assisted laser desorption/ionization mass spectrometry. The results show high donor-to-donor variability in potency against both wild-type virus (Fig. 4 A and Table 2, 29–253 μM) and N43D virus (Fig. 4 B and Table 2, 46–176 μM). Interestingly, the N43D mutation causes no change in susceptibility to entry inhibition by SQ435, with IC50 calculated across all experiments of 162 μM and 112 μM for wild-type and N43D virus, respectively. In fact, the mutation causes a very slight increased susceptibility to inhibition by SQ435. In contrast, this mutation causes a 19-fold increase in the IC50 of Enfuvirtide, with IC50 8.8 nM versus wild-type virus and 167 nM versus N43D virus (Fig. 4 C).
Table 2.
Experimental IC50 values for SQ435 and Enfuvirtide against both wild-type HIV-1 and the N43D mutant
| Donor | IC50 SQ435 (mM) |
IC50 Enfuvirtide (nM) |
||
|---|---|---|---|---|
| Wild-type | N43D | Wild-type | N43D | |
| 140 | 29 | |||
| 187 | 253 | 172 | 14.0 | 122.9 |
| 188 | 109 | 46 | ||
| 189 | 6.1 | 234.6 | ||
| 190 | 229 | 176 | ||
| All data | 162 ± 28 | 112 ± 20 | 8.8 ± 1.3 | 167.8 ± 15.8 |
To further test the efficacy of what we consider a novel de novo protein design framework, we calculated the approximate binding affinity between gp41 and SQ435 both with and without the N43D resistance mutation. We made the assumption that the mutation causes no change in the overall fold of gp41. The model predicts very little change in the approximate binding affinity of SQ435, with values of 2.74 × 10−01 for gp41 N43D and 2.52 × 10−01 for wild-type, correlating well with the experimental results.
To ensure that the effects of SQ435 were not specific to our standard laboratory strain NL4-3 carrying CXCR4-tropic envelope HXB2, entry inhibition by SQ435 and Enfuvirtide was analyzed against virus bearing four different envelopes. Two CCR5-tropic reference sequences YU-2 and SF162 and two clinically isolated CCR5-tropic sequences, C98v1e4 and C109v1e4, were selected (72), and HXB2 was included as a control. The data are shown in Fig. 5 and IC50 values are reported in Table 3. The results show that SQ435 is active against all envelopes tested with similar potency, and the IC50 of 172 mM across all envelopes is comparable to 162 μM measured previously against HXB2. Interestingly, virus with envelope from CCR5-tropic strain YU-2 was found to be approximately twofold more sensitive to SQ435 than virus with HXB2, whereas Enfuvirtide was found to be ∼3–10-fold less potent against all CCR5-tropic envelopes.
Figure 5.
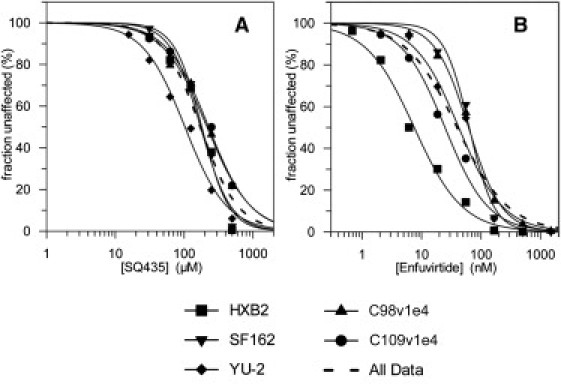
Inhibition of entry of HIV bearing five different envelopes into primary CD4+ T cells by SQ435 (A) and Enfuvirtide (B). Envelopes tested include CXCR4-tropic HXB2, two CCR5-tropic reference envelopes YU-2 and SF162, and two clinically isolated CCR5-tropic envelopes C98v1e4 and C109v1e4. Replication (% of control) was determined by a single-round infectivity assay with GFP-encoding recombinant HIV. Each envelope isolate is illustrated with a unique symbol and separate curve. (Dashed lines) Aggregate IC50 curve across all clinical isolates.
Table 3.
Experimental IC50 values for SQ435 and Enfuvirtide against five clinical isolates of HIV-1 envelope
| Envelope | IC50 SQ435 (mM) |
IC50 Enfuvirtide (nM) |
||||
|---|---|---|---|---|---|---|
| Donor 195 | Donor 196 | All data | Donor 195 | Donor 196 | All data | |
| HXB2 | 193 | 156 | 174 | 4.3 | 14.4 | 7.5 |
| SF162 | 198 | 167 | 180 | 48 | 75 | 63 |
| YU-2 | 148 | 73 | 103 | 37 | 49 | 42 |
| C98v1e4 | 237 | 199 | 215 | 64 | 59 | 62 |
| C109v1e4 | 195 | 257 | 227 | 17 | 39 | 35 |
| All env | 188 ± 11 | 158 ± 15 | 172 ± 10 | 23 ± 5 | 66 ± 11 | 40 ± 6 |
Finally, due to the success of SQ435, five new sequences were designed based on the sequence of SQ435. Table 4 provides the new sequences along with their corresponding approximate binding affinity rank. NS-1 simply replaces the Tyr in position 638 of SQ435 with Trp. NS-2 swaps the amino acids at positions 637 and 638 of NS-1. NS-3, NS-4, and NS-5 are truncated versions of NS-1 and NS-2. Only the sequences with length 12 are predicted to bind better than the native. All five new sequences were synthesized and tested. Fig. S3 shows the inhibition results of the new sequences. NS-1 and NS-2 showed some activity at 500 μM; however NS-3, NS-4, and NS-5 showed almost no activity even at the higher concentration, which validates the approximate binding affinity prediction. This indicates that either the peptide length is too short to be able to effectively bind to gp41 or that the amino acids at positions 638 and 639 in particular are important for binding.
Table 4.
New peptide sequences based upon the sequence of SQ435
| Name | Length | Approximate bindingaffinity rank | Sequence |
||
|---|---|---|---|---|---|
| 628 | 639 | ||||
| NS-1 | 12 | 1 | W | CDWRDEWE | RWR |
| NS-2 | 12 | 2 | W | CDWRDEWE | WRR |
| Native | 12 | 3 | W | EEWDREIE | NYT |
| NS-4 | 9 | 4 | W | CDWRDEWE | |
| NS-3 | 10 | 5 | W | CDWRDEWE | R |
| NS-5 | 9 | 6 | W | CDWRDEWR | |
Discussion
To our knowledge, in this article a novel method for de novo protein design is applied to the discovery of new HIV-1 entry inhibitors. The method consists of two main stages: a sequence selection stage and a validation stage. The validation stage can consist of either fold specificity calculations or approximate binding affinity calculations—or both. The sequence selection stage produced a lowest-energy, rank-ordered list of peptide sequences based upon the design template of a small peptide bound to gp41. The sequences were reranked based upon fold specificity calculations and then approximate binding affinity calculations. This provided a list of six mutant sequences that were predicted to be better binders than the native sequence.
Five of the mutant sequences plus the native sequence were selected for synthesis and testing in cell culture. Four out of the five mutant sequences (SQ435, SQ175, SQ486, and SQ323) showed inhibitory activity better than the native sequence, with SQ435 being the most potent. In addition, all of the peptide sequences were shown to be nontoxic to cells at concentrations up to 500 μM and were shown to be targeting the HIV-1 envelope. The effect of SQ435 was not limited to one reference isolate, and similar potency was shown against virus bearing five different envelopes. Finally, five new sequences were tested based upon the results of SQ435. The results indicated that, at least in this case, 12 residues were necessary to elicit an inhibitory response.
The IC50 of SQ435 spanned a range of 29–253 μM, showing donor variability. The IC50 calculated across all experiments for SQ435 was determined to be 162 μM, which is a 3–15-fold improvement over the native sequence depending on the donor (from IC50 > 500 mM to 29–253 μM). This is particularly exciting because SQ435 is two-residues shorter than C14unlinkmid. One donor also showed slight improvement in IC50 over the stapled version of the native peptide, C14linkmid (29 μM vs. 35 μM). Additionally, there is no loss of activity of SQ435 against the Enfuvirtide-resistant virus, whereas there is a 19-fold increase in the IC50 of Enfuvirtide against the Enfuvirtide-resistant virus. This may be because the mutation in the resistant virus, N43D, is located away from the hydrophobic core of gp41, which constitutes the binding pocket for this design. Although the IC50 of SQ435 is still several orders of magnitude greater than Enfuvirtide, the design is a success because SQ435 shows some potency despite being 24-residues shorter than Enfuvirtide and is just as potent against wild-type and Enfuvirtide-resistant strains. Incorporating nonnatural amino acids that would increase the α-helical propensity of the sequence or incorporating a carbon-chain staple that would constrain the peptide to be helical are possible ways to lower the IC50 of SQ435 by several orders of magnitude.
The de novo protein design framework successfully elucidated a number of peptide sequences that inhibit HIV-1 cell-cell fusion better than the native sequence, with the best sequence having a 3–15-fold improvement in IC50 over the native sequence, depending on the donor. This is a good first step in discovering short peptides that can effectively inhibit HIV-1 without the limitations of longer peptide such as poor bioavailability, high production cost, and short half-life.
Acknowledgments
C.A.F. gratefully acknowledges support from the National Science Foundation (grant No. CTS-0426691), R.F.S. gratefully acknowledges support from the National Institute of Health (grant No. RO1-081600) and the Howard Hughes Medical Institute, and P.A.C. gratefully acknowledges support from the National Cancer Institute (grant No. CA74305). A portion of this research was made possible with government support by the Department of Defense, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate Fellowship (No. 32 CFR 168a to M.L.B.).
Supporting Material
References
- 1.UNAIDS. 2008. 2008 Report on the Global AIDS Epidemic. XVII International AIDS Conference, International AIDS Society, Geneva, Switzerland.
- 2.Thayer A.M. Resetting priorities. Chem. Eng. News. 2008;86:17–28. [Google Scholar]
- 3.Thayer A.M. New antiretrovirals. Chem. Eng. News. 2008;86:29–36. [Google Scholar]
- 4.Chan D.C., Kim P.S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 5.Liu S.W., Wu S.G., Jiang S.B. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr. Pharm. Des. 2007;13:143–162. doi: 10.2174/138161207779313722. [DOI] [PubMed] [Google Scholar]
- 6.Qi Z., Shi W.G., Jiang S. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J. Biol. Chem. 2008;283:30376–30384. doi: 10.1074/jbc.M804672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen L., Peterson S., Siliciano R.F. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 2008;14:762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorr P., Westby M., Perros M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch B.D., VanDemark A.P., Kay M.S. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sia S.K., Carr P.A., Kim P.S. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. USA. 2002;99:14664–14669. doi: 10.1073/pnas.232566599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izumi K., Kodama E., Matsuoka M. Design of peptide-based inhibitors for human immunodeficiency virus type 1 strains resistant to T-20. J. Biol. Chem. 2009;284:4914–4920. doi: 10.1074/jbc.M807169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naito T., Izumi K., Matsuoka M. SC29EK, a peptide fusion inhibitor with enhanced α-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to Enfuvirtide. Antimicrob. Agents Chemother. 2009;53:1013–1018. doi: 10.1128/AAC.01211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishikawa H., Nakamura S., Matsuoka M. Electrostatically constrained α-helical peptide inhibits replication of HIV-1 resistant to Enfuvirtide. Int. J. Biochem. Cell Biol. 2009;41:891–899. doi: 10.1016/j.biocel.2008.08.039. [DOI] [PubMed] [Google Scholar]
- 14.Dwyer J.J., Wilson K.L., Delmedico M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against Enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA. 2007;104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y., Xiao Y., Zhang L. Design and evaluation of Sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008;283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 16.Balogh E., Wu D., Gochin M. NMR second site screening for structure determination of ligands bound in the hydrophobic pocket of HIV-1 gp41. J. Am. Chem. Soc. 2009;131:2821–2823. doi: 10.1021/ja8094558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu R.J., Tucker J.A., Sexton C. Design and synthesis of human immunodeficiency virus entry inhibitors: sulfonamide as an isostere for the α-ketoamide group. J. Med. Chem. 2007;50:6535–6544. doi: 10.1021/jm070650e. [DOI] [PubMed] [Google Scholar]
- 18.Li G., Haney K.M., Zhang Y. Comparative docking study of anibamine as the first natural product CCR5 antagonist in CCR5 homology models. J. Chem. Inf. Model. 2009;49:120–132. doi: 10.1021/ci800356a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madani N., Schön A., Sodroski J. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure. 2008;16:1689–1701. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu K., Lu H., Xie L. Design, synthesis, and biological evaluation of N-carboxyphenylpyrrole derivatives as potent HIV fusion inhibitors targeting gp41. J. Med. Chem. 2008;51:7843–7854. doi: 10.1021/jm800869t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maheshwari R., Levenson E.A., Kiick K.L. Manipulation of electrostatic and saccharide linker interactions in the design of efficient glycopolypeptide-based cholera toxin inhibitors. Macromol. Biosci. 2010;10:68–81. doi: 10.1002/mabi.200900182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ingallinella P., Bianchi E., Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA. 2009;106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berchanski A., Lapidot A. Computer-based design of novel HIV-1 entry inhibitors: neomycin conjugated to arginine peptides at two specific sites. J. Mol. Model. 2009;15:281–294. doi: 10.1007/s00894-008-0401-1. [DOI] [PubMed] [Google Scholar]
- 24.Imai M., Baranyi L., Okada H. Inhibition of HIV-1 infection by synthetic peptides derived CCR5 fragments. Biochem. Biophys. Res. Commun. 2007;353:851–856. doi: 10.1016/j.bbrc.2006.12.084. [DOI] [PubMed] [Google Scholar]
- 25.Fung H.K., Welsh W.J., Floudas C.A. Computational de novo peptide and protein design: rigid templates versus flexible templates. Ind. Eng. Chem. Res. 2008;47:993–1001. [Google Scholar]
- 26.Floudas C.A., Fung H.K., Rajgaria R. Advances in protein structure prediction and de novo protein design: a review. Chem. Eng. Sci. 2006;61:966–988. [Google Scholar]
- 27.Kortemme T., Baker D. Computational design of protein-protein interactions. Curr. Opin. Chem. Biol. 2004;8:91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Kuhlman B., Baker D. Exploring folding free energy landscapes using computational protein design. Curr. Opin. Struct. Biol. 2004;14:89–95. doi: 10.1016/j.sbi.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Desmet J., Maeyer M.D., Lasters I. The dead-end elimination theorem and its use in side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 30.Pierce N., Spriet J., Mayo S. Conformational splitting: a more powerful criterion for dead-end elimination. J. Comput. Chem. 2000;21:999–1009. [Google Scholar]
- 31.Georgiev I., Donald B.R. Dead-end elimination with backbone flexibility. Bioinformatics. 2007;23:i185–i194. doi: 10.1093/bioinformatics/btm197. [DOI] [PubMed] [Google Scholar]
- 32.Koehl P., Delarue M. Application of a self-consistent mean field theory to predict protein side-chains conformation and estimate their conformational entropy. J. Mol. Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 33.Saven J.G. Combinatorial protein design. Curr. Opin. Struct. Biol. 2002;12:453–458. doi: 10.1016/s0959-440x(02)00347-0. [DOI] [PubMed] [Google Scholar]
- 34.Zou J.M., Saven J.G. Using self-consistent fields to bias Monte Carlo methods with applications to designing and sampling protein sequences. J. Chem. Phys. 2003;118:3843–3854. [Google Scholar]
- 35.Koehl P., Levitt M. De novo protein design. I. In search of stability and specificity. J. Mol. Biol. 1999;293:1161–1181. doi: 10.1006/jmbi.1999.3211. [DOI] [PubMed] [Google Scholar]
- 36.Koehl P., Levitt M. De novo protein design. II. Plasticity in sequence space. J. Mol. Biol. 1999;293:1183–1193. doi: 10.1006/jmbi.1999.3212. [DOI] [PubMed] [Google Scholar]
- 37.Dantas G., Kuhlman B., Baker D. A large scale test of computational protein design: folding and stability of nine completely redesigned globular proteins. J. Mol. Biol. 2003;332:449–460. doi: 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- 38.Su A., Mayo S.L. Coupling backbone flexibility and amino acid sequence selection in protein design. Protein Sci. 1997;6:1701–1707. doi: 10.1002/pro.5560060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunders C.T., Baker D. Recapitulation of protein family divergence using flexible backbone protein design. J. Mol. Biol. 2005;346:631–644. doi: 10.1016/j.jmb.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 40.Fung H.K., Taylor M.S., Floudas C.A. Novel formulations for the sequence selection problem in de novo protein design with flexible templates. Optimiz. Meth. Softw. 2007;22:51–71. [Google Scholar]
- 41.Dong X.-N., Xiao Y., Chen Y.H. N- and C-domains of HIV-1 gp41: mutation, structure and functions. Immunol. Lett. 2001;75:215–220. doi: 10.1016/s0165-2478(00)00302-3. [DOI] [PubMed] [Google Scholar]
- 42.Eckert D.M., Malashkevich V.N., Kim P.S. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 43.Chan D.C., Chutkowski C.T., Kim P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan D.C., Fass D., Kim P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 45.Fung H.K., Floudas C.A., Morikis D. Toward full-sequence de novo protein design with flexible templates for human β-defensin-2. Biophys. J. 2008;94:584–599. doi: 10.1529/biophysj.107.110627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajgaria R., McAllister S.R., Floudas C.A. A novel high resolution Cα-Cα distance dependent force field based on a high quality decoy set. Proteins: Struct. Funct. Bioinf. 2006;65:726–741. doi: 10.1002/prot.21149. [DOI] [PubMed] [Google Scholar]
- 47.Rajgaria R., McAllister S.R., Floudas C.A. Distance dependent centroid to centroid force fields using high resolution decoys. Proteins. 2008;70:950–970. doi: 10.1002/prot.21561. [DOI] [PubMed] [Google Scholar]
- 48.Loose C., Klepeis J., Floudas C. A new pairwise folding potential based on improved decoy generation and side chain packing. Proteins: Struct. Funct. Bioinf. 2004;54:303–314. doi: 10.1002/prot.10521. [DOI] [PubMed] [Google Scholar]
- 49.Klepeis J.L., Floudas C.A., Lambris J.D. Integrated computational and experimental approach for lead optimization and design of compstatin variants with improved activity. J. Am. Chem. Soc. 2003;125:8422–8423. doi: 10.1021/ja034846p. [DOI] [PubMed] [Google Scholar]
- 50.Klepeis J.L., Floudas C.A., Lambris J.D. Design of peptide analogs with improved activity using a novel de novo protein design approach. Ind. Eng. Chem. Res. 2004;43:3817–3826. [Google Scholar]
- 51.Klepeis J.L., Floudas C.A. ASTRO-FOLD: A combinatorial and global optimization framework for ab initio prediction of three-dimensional structures of proteins from the amino acid sequence. Biophys. J. 2003;85:2119–2146. doi: 10.1016/s0006-3495(03)74640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klepeis J.L., Floudas C.A., Morikis D., Lambris J.D. Predicting peptide structures using NMR data and deterministic global optimization. J. Comput. Chem. 1999;20:1354–1370. [Google Scholar]
- 53.Klepeis J.L., Floudas C.A. Free energy calculations for peptides via deterministic global optimization. J. Chem. Phys. 1999;110:7491–7512. [Google Scholar]
- 54.Floudas C.A. Computational methods in protein structure prediction. Biotechnol. and Bioeng. 2007;97:207–213. doi: 10.1002/bit.21411. [DOI] [PubMed] [Google Scholar]
- 55.Güntert P., Mumenthaler C., Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 56.Guntert P. Automated NMR structure calculation with CYANA. Meth. Mol. Biol. J. Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 57.Ponder J. Washington University School of Medicine; St. Louis, MO: 1998. TINKER, software tools for molecular design. Department of Biochemistry and Molecular Biophysics. [Google Scholar]
- 58.Cornell W.D., Cieplak P., Kollman P.A. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 59.Bellows M.L., Fung H.K., Morikis D. New compstatin variants through two de novo protein design frameworks. Biophys. J. 2010;98:2337–2346. doi: 10.1016/j.bpj.2010.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lilien R.H., Stevens B.W., Donald B.R. A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. J. Comput. Biol. 2005;12:740–761. doi: 10.1089/cmb.2005.12.740. [DOI] [PubMed] [Google Scholar]
- 61.Lee M.R., Baker D., Kollman P.A. 2.1 and 1.8 Å average C(α) RMSD structure predictions on two small proteins, HP-36 and s15. J. Am. Chem. Soc. 2001;123:1040–1046. doi: 10.1021/ja003150i. [DOI] [PubMed] [Google Scholar]
- 62.Rohl C.A., Baker D. De novo determination of protein backbone structure from residual dipolar couplings using Rosetta. J. Am. Chem. Soc. 2002;124:2723–2729. doi: 10.1021/ja016880e. [DOI] [PubMed] [Google Scholar]
- 63.Rohl C.A., Strauss C.E.M., Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 64.DiMaggio P.A., Jr., McAllister S.R., Rabitz H.A. Biclustering via optimal re-ordering of data matrices in systems biology: rigorous methods and comparative studies. BMC Bioinformatics. 2008;9:458. doi: 10.1186/1471-2105-9-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DiMaggio P.A., McAllister S.R., Rabitz H.A. A network flow model for biclustering via optimal re-ordering of data matrices. J. Glob. Optim. 2010;47:343–354. [Google Scholar]
- 66.Daily M.D., Masica D., Gray J.J. CAPRI rounds 3–5 reveal promising successes and future challenges for RosettaDock. Proteins. 2005;60:181–186. doi: 10.1002/prot.20555. [DOI] [PubMed] [Google Scholar]
- 67.Gray J.J., Moughon S.E., Baker D. Protein-protein docking predictions for the CAPRI experiment. Proteins. 2003;52:118–122. doi: 10.1002/prot.10384. [DOI] [PubMed] [Google Scholar]
- 68.Gray J.J., Moughon S., Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 69.Kuhlman B., Baker D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. USA. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H., Zhou Y., Siliciano R.F. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J. Virol. 2004;78:1718–1729. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poveda E., Rodés B., Soriano V. Evolution of genotypic and phenotypic resistance to Enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J. Med. Virol. 2004;74:21–28. doi: 10.1002/jmv.20141. [DOI] [PubMed] [Google Scholar]
- 72.Bailey J.R., Lassen K.G., Siliciano R.F. Neutralizing antibodies do not mediate suppression of human immunodeficiency virus type 1 in elite suppressors or selection of plasma virus variants in patients on highly active antiretroviral therapy. J. Virol. 2006;80:4758–4770. doi: 10.1128/JVI.80.10.4758-4770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DeLano, W. L. 2002. The PyMOL molecular graphics system. http://www.pymol.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.