

Abstract
Background and aims
Helicobacter pylori infection of gastric mucosa causes gastritis and transient hypochlorhydria, which may provoke emergence of a mucosal precancer phenotype; H pylori strains containing a cag pathogenicity island (PAI) augment cancer risk. Acid secretion is mediated by the catalytic α subunit of parietal cell H,K-ATPase (HKα). In AGS gastric epithelial cells, H pylori induces nuclear factor-κB (NF-κB) binding to and repression of transfected HKα promoter activity. This study sought to identify bacterial genes involved in HKα repression and to assess their impact on acid secretion.
Methods and results
AGS cells transfected with an HKα promoter construct or human gastric body biopsies were infected with wild-type (wt) or isogenic mutant (IM) H pylori strains. AGS cell HKα promoter activity, and biopsy HKα mRNA, protein and H+ secretory activity were measured by luminometry, reverse transcription—PCR, immunoblotting and extracellular acidification, respectively. Wt H pylori and ΔvacA, ΔureA, Δslt and ΔflaA IM strains repressed HKα promoter activity by ~50%, a ΔcagA IM strain repressed HKα by ~33%, and ΔcagE, ΔcagM and ΔcagL IM strains elicited no HKα repression. Wt H pylori-infected biopsies had markedly reduced HKα mRNA and protein compared with IM strain infections or mock-infected controls. Histamine-stimulated, SCH28080-sensitive biopsy acid secretion was significantly inhibited by wt but not by ΔcagL IM H pylori infection compared with vehicle-only controls.
Conclusions
It is concluded that H pylori cag PAI gene products CagE, CagM, CagL and, possibly, CagA are mechanistically involved in repression of HKα transcription. Further, acute H pylori infection of human gastric mucosa downregulates parietal cell H,K-ATPase expression, significantly inhibiting acid secretion.
INTRODUCTION
Helicobacter pylori infection of human gastric mucosa is associated with corpus-predominant gastritis, hypochlorhydria and multifocal gastric atrophy, a pathological progression that can lead to intestinal metaplasia, dysplasia and gastric adenocarcinoma.1 2 H pylori has been reported to induce profound hypochlorhydria within 3 days of infection,3-6 facilitating initial colonisation and promoting activation of proinflammatory pathways involved in development of disease.7 8 Mechanistic understanding of acute H pylori-induced hypochlorhydria is incomplete. Transient hypochlorhydria is not caused by parietal cell ablation, because histologically normal parietal cells are abundant in gastric biopsies of patients with hypochlorhydric epidemic gastritis, and gastric permeability is normal.9 Also, acute H pylori infection in Mongolian gerbils, which mimics the pathophysiological course of human H pylori infection, causes concurrent hypochlorhydria and gastritis with no glandular atrophy.10 The inflammatory cytokine interleukin 1β (IL-1β), secreted by monocytes and neutrophils recruited by H pylori-induced mucosal IL-8 production, inhibits gastric proton pump (H,K-ATPase) activity by impairing phosphatidylinositol 3-kinase (PI3K)-mediated increases in [Ca2+]i.11 12 Indeed, host IL-1 gene polymorphisms leading to increased IL-1β production in H pylori infection pose an increased risk of hypochlorhydria and gastric cancer.13 Vacuolating toxin (VacA) secreted by H pylori disrupts actin interaction with parietal cell apical membranes, preventing recruitment and fusion of H,K-ATPase-containing tubulovesicles and causing hypochlorhydria.14
These inhibitory mechanisms target H,K-ATPase post-translationally, but several lines of evidence point to H pylori-induced transcriptional repression of proton pumps. H pylori eradication in humans lowers gastric juice pH, and increases H,K-ATPase α subunit (HKα) mRNA content, consistent with H pylori-mediated inhibition of H,K-ATPase synthesis.15 We have reported that H pylori infection of AGS gastric epithelial cells represses the activity of transfected HKα promoter—reporter constructs,16-19 and that binding of nuclear factor-κB (NF-κB) p50 homodimeric transcription factor to a cognate HKα promoter cis-response element underlies this repression.17 The specific H pylori gene products mediating NF-κB-dependent HKα transcriptional repression are unknown.
H pylori strains containing the cytotoxin-associated gene (cag) pathogenicity island (PAI) are associated with severe gastric inflammation, ulceration and increased risk of gastric cancer.20-22 Several cag PAI genes encode proteins that assemble into a type IV secretion system (T4SS) or pilus spanning the bacterial inner and outer membranes, mediating H pylori adherence to host cells and enabling transfer of virulence factors. T4SS proteins include cytoplasmically oriented CagE (HP0544), an ATPase which energises transfer of virulence factors into host cells,20 CagM (HP0537) which forms the periplasmic and extracellular portions of the pilus,23 and CagL (HP0539) at the pilus tip.23 A CagL RGD motif contributes to bacterial adherence by interacting with α5β1 integrins on the host cell surface.24 CagA, encoded by cagA, the terminal gene of the cag PAI, is translocated into host cells through the T4SS where it undergoes tyrosine phosphorylation by Src kinase and stimulates SHP-2 tyrosine phosphatase activity,25 26 inducing abnormal epithelial cell proliferation and a transformed phenotype.26 Phosphorylated CagA promotes NF-κB p50/p65-mediated IL-8 induction and secretion, provoking inflammation and markedly augmenting the risk of adenocarcinoma27; however, CagA has not previously been tied to modulation of gastric acid secretion.
This study sought to identify H pylori virulence factors involved in HKα repression, and to assess its physiological significance. The results indicate that a small subset of H pylori cag PAI genes induce repression of HKα transcription and translation in human gastric mucosa leading to inhibition of acid secretion.
MATERIALS AND METHODS
Cells, bacteria and transient transfection
Human AGS gastric adenocarcinoma cells (ATCC, Manassas, Virginia, USA) were maintained in culture as described.17 H pylori wild-type (wt) cag+ strains (60190, 7.13, 8823 and P12) were grown on Brucella broth (Difco Laboratories, Detroit, Michigan, USA) plates containing 10% fetal bovine serum (FBS) and 2.4% agar (Brucella—agar plates) at 37°C using a microaerophilic gas pack system (BD Biosciences, Sparks, Maryland, USA). H pylori isogenic mutant strains 60190 ΔcagA, 60190 ΔvacA, 60190 ΔflaA, 60190 ΔureA, 7.13 ΔcagPAI, 7.13 ΔcagE, 7.13 ΔcagA, 7.13 ΔcagM and 7.13 Δslt were grown in kanamycin-containing (25 μg/ml) Brucella—agar plates. The P12 ΔcagL mutant strain was grown on chloramphenicol-containing (5 μg/ml) Brucella—agar plates. Bacterial multiplicities of infection (MOI) were calculated as described.17 A 2179 bp segment of human gastric HKα 5′-flanking sequence (HKα2179) was integrated into the luciferase reporter plasmid pGL2-Basic Vector as described.18 AGS cells were co-transfected with HKα2179 and pMaxGFP, and promoter—reporter activities were measured and normalised as described.18
Human gastric biopsies
Gastric endoscopic biopsies were acquired from consenting patients (21—60 years old) undergoing oesophagogastroduodenoscopy or endoscopic ultrasound at the MUSC Digestive Disease Center (IRB protocol HR16941). Exclusion criteria included patients with positive urea breath and CLO tests. Four full-thickness biopsies (6—42 mg each) per patient were obtained from normal-appearing corpus mucosa on the greater curvature of the stomach. Single biopsies were incubated in individual wells of 96-well culture plates with F12 culture medium (100 μl, 1h, 37°C), infected for varying periods of time with 24 h cultures of H pylori (1—2×105 bacteria/mg wet weight biopsy), and then rinsed 3× with F12 medium. Same-patient biopsies incubated with F12 medium alone served as mock infection controls.
Real-time reverse transcription—PCR (RT—PCR)
Total RNA was isolated from gastric biopsies using STAT-60 reagent (Tel Test, Friendswood, Texas, USA) and reverse-transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, California, USA). Biopsy HKα mRNA was measured by RT—PCR (iCycler iQ, iQ SYBR Green Super mix; Bio-Rad). Forward and reverse HKα and β-microglobin primer (used for data normalisation) sequences were: HKα-F, 5′-GGAGGACCACCACCTACAAGAT-3′; HKα-R, 5′-ATGCTGATGAAGAACACGGTGT-3′; β-microglobin-F, 5′-AGATGAGTATGCCTGCCGTGTG-3′; and β-microglobin-R, 5′-TCAAACCTCCATGATGCTGCTTAC-3′.
Immunoblotting analysis
Gastric biopsies were infected in vitro with H pylori (1—2×105 bacteria/mg wet weight biopsy, 24 h), washed in ice-cold buffer (50 mM Tris, pH 7.2, 5 mM EGTA) and disaggregated using a Potter—Elvjheim glass—Teflon homogeniser. The homogenate was centrifuged at 7000 g for 10 min, and aliquots of supernatant (5 μg of protein) were heated at 55°C for 5 min with equal volumes of sodium dodecyl sulfate—polyacrylamide gel electrophoresis (SDS—PAGE) sample buffer. Biopsy HKα content was assessed by immunoblotting using HK 12.18 antibody as described28 and β-actin antibody (Sigma-Aldrich, St Louis, Missouri, USA) as a gel loading control.
Biopsy acid secretion
Gastric biopsies were infected in vitro with H pylori (3×106 bacteria/biopsy), divided into four parts along the mucosal—serosal axis, immobilised in wells of a 24-well XF24 culture plate pretreated overnight with Cell-Tak (1 μl; BD Biosciences, Bedford, Massachusetts, USA), and overlaid with pyruvate- and bicarbonate-free Dulbecco’s modified Eagle’s medium (DMEM; 600 μl; Mediatech, Manassas, Virginia, USA). Before physiological measurements, biopsies were treated for 30 min with 5-(N-ethyl-n-isopropyl) amiloride (EIPA; 150 μM) to block Na+/H+ exchanger activity. Gastric biopsy medium acidification was measured in 37°C-thermostatted 24-well plates using a XF24 Extracellular Flux Analyser (Seahorse Biosciences, Billerica, Massachusetts, USA). Automated placement of fibre-optic pH-sensitive fluorescent hydrogel probes within ~300 μm of the biopsies created a ‘virtual chamber’ enclosing ~28 μl of medium immediately overlying a biopsy. [H+] in this limited diffusion region was measured once every 10—20 s over a period of 90—120 min, and the rate of medium acidification was calculated from the slope of change in [H+]. The probes were then retracted and vibrated to mix and re-equilibrate formerly enclosed medium with bulk medium. pH measurements were made without significant depression of oxygen tension or medium acidification, achieving microphysiometer-like sensitivity. Constitutive H+ secretion was measured for ~30 min before programmed remote injection of histamine (1 mM final).
Data analysis
All values are reported as the mean ±SD. Data comparisons between control and treatment groups were assessed by two-way analysis of variance using the Bonferroni post-test method as implemented in the statistical software package GraphPad PRISM version 4. p<0.05 was considered statistically significant.
RESULTS
The H pylori cag PAI represses HKα promoter activity
To test the hypothesis that H pylori cag PAI genes are required for HKα repression, AGS cells were transfected with a HKα2179 promoter—Luc reporter construct, independently infected (MOI 25, 6 h) with wt H pylori strains or their corresponding isogenic mutant (IM) strains 7.13 ΔcagPAI and 8823 ΔcagPAI, and promoter activity was measured as normalised relative light units (RLU) of luciferase activity. Wt H. pylori strains 7.13 and 8823 suppressed HKα2179 promoter activity by 30% and 45%, respectively (figure 1). Infection with 7.13 ΔcagPAI and 8823 ΔcagPAI showed no statistically significant repression of HKα2179 activity compared with mock-infected cells, suggesting that genes in the H pylori cag PAI are involved in repression of HKα promoter activity.
Figure 1.
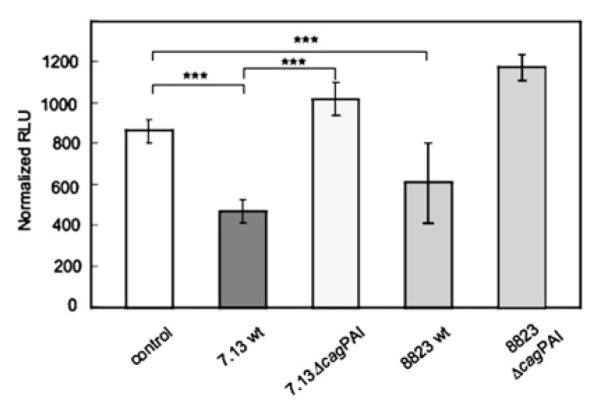
The H. pylori cag pathogenicity island (PAI) represses H,K-ATPase α subunit (HKα) promoter activity in AGS cells. Cells were transiently transfected with the HKα2179 promoter—Luc reporter construct and infected (multiplicity of infection 25, 6 h) with wild-type (wt) H pylori strains 7.13, 8823, 7.13 ΔcagPAI or 8823 ΔcagPAI. HKα2179 promoter activity was measured as normalised relative light units (RLU) of luciferase activity. Data represent the mean ±SD from three independent experiments (***p<0.001).
Specific cag PAI genes are implicated in AGS cell HKα promoter repression
To identify H pylori genes responsible for HKα repression, AGS cells transfected with the HKα2179 promoter—Luc reporter construct were independently infected with wt H pylori or IM strains lacking cag PAI genes (cagM, cagE, cagL and cagA) or non-cag PAI genes (slt, vacA, flaA and ureA) encoding known H pylori virulence factors. The slt gene encodes a lytic transglycosylase that sheds a peptidoglycan alanine—glumate—diaminopimelic acid tripeptide agonist of the intracellular receptor Nod1, thereby inducing NF-κB activation in AGS cells.29 VacA permeabilisation of parietal cells induces Ca2+-activated calpain proteolysis of ezrin, which disrupts actin organisation and prevents fusion of H,K-ATPase-containing tubulovesicles, causing hypochlorhydria.14 FlaA, a flagellin subunit in the filamentous core of flagella, enables H pylori motility towards the neutral pH microenvironment of surface epithelial cells.30 UreA, a urea transporter subunit, together with UreI (a periplasmic membrane H+-gated urea channel) and carbonic anhydrase, serves to neutralise the acidic microenvironment of H pylori.31 32
AGS cells infected with wt H pylori 60190 showed ~50% repression of transfected HKα2179 promoter activity (figure 2), comparable with the repression exerted by wt H pylori 7.13 and 8823 (figure 1) and by wt H pylori P12 (data not shown). In contrast, the ΔcagM, ΔcagE and ΔcagL mutants showed no significant HKα promoter repression compared with mock-infected AGS cells. The non-cag PAI mutants (Δslt, ΔvacA, ΔflaA and ΔureA) repressed the HKα promoter as much as wt H pylori strains. The ΔcagA mutant repressed HKα promoter activity significantly less than the wt strain and the Δslt, ΔflaA and ΔureA IMs (p<0.05). These data indicate that cagM, cagE and cagL encode proteins involved in HKα repression, and that cagA may require participation of other factor(s) to effect HKα repression. The data are not manifestations of non-specific H pylori—AGS cell interactions because the non-cag PAI genes tested did not repress HKα activity.
Figure 2.
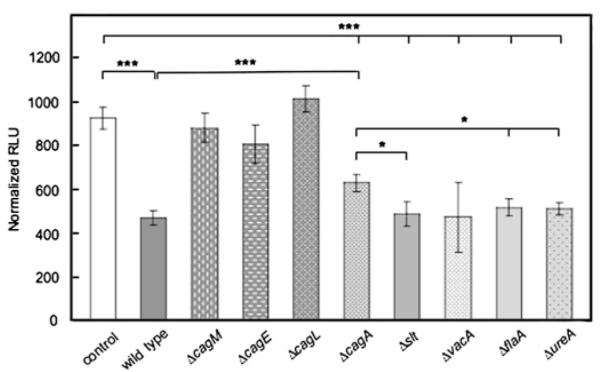
The cag pathogenicity island (PAI)-specific genes cagM, cagE, cagL and cagA are responsible for H,K-ATPase α subunit (HKα) promoter repression in AGS cells. AGS cells were transfected with the HKα2179 promoter—Luc reporter construct and infected (multiplicity of infection 25, 6 h) with wild-type H pylori or one of eight H pylori isogenic mutant strains (ΔcagM, ΔcagE, ΔcagA, ΔcagL, Δslt, ΔvacA, ΔflaA and ΔureA). HKα2179 promoter activity was expressed in normalised relative light units (RLU) of luciferase activity. Data represent the mean ±SD from three independent experiments (***p<0.001; *p<0.05).
H pylori attenuates HKα mRNA and protein expression in human gastric mucosa
To assess the significance of these cag PAI genes for parietal cell H,K-ATPase expression, human gastric corpus biopsies were infected with wt H pylori 60190 (2×105 bacteria/mg wet weight biopsy, 24 h). Biopsy HKα mRNA and HKα protein content was measured by RT—PCR and immunoblotting, respectively. In eight out of 10 biopsies examined, H pylori infection repressed HKα mRNA expression by two- to sevenfold compared with mock-infected biopsies (figure 3A). Compared with freshly acquired biopsies, those incubated for 24 h with wt H pylori showed histological evidence of ischaemic damage, although parietal cells appeared morphologically normal (figure 3B,C). Three additional patient biopsies infected with wt H pylori 60190 (2×105 bacteria/mg wet weight biopsy, 24 h) were lysed and their HKα protein content was assessed by immunoblotting. Antibody specificity was confirmed by single-band (94 kDa) positive reactivity with H,K-ATPase purified from pig gastric microsomes (figure 3B, lane 1). Mock-infected biopsies showed the same band at 94 kDa (figure 3B, lane 2); quantitative densitometry (data not shown) indicated a biopsy HKα protein content of ~0.1 mg/mg wet weight. HKα protein expression was undetectable in biopsies infected for 24 h with wt H pylori 60190 (figure 3B, lane 3). These data clearly demonstrate that ex vivo H pylori infection of human gastric mucosa causes strain-and host-dependent reduction of parietal cell HKα protein expression.
Figure 3.

H pylori represses H,K-ATPase α subunit (HKα) levels in human gastric biopsies. Biopsies were infected (1—2×105 bacteria/mg wet weight biopsy, 24 h) with wild-type H pylori strain 60190. (A) RNA was extracted from biopsies and the HKα mRNA content was measured by reverse transcription—PCR. Open bars represent mock-infected biopsies, and shaded bars represent patient-matched infected biopsies (means, SD, n=3). (B, C) Gastric biopsies before (B) and after (C) 24 h infection with wild-type H pylori (H&E stain, ×40 magnification; arrows indicate parietal cells). (D) Biopsies were lysed and HKα protein content was assessed by immunoblotting using antibody HK 12.18 against HKα and β-actin antibody as gel loading control (representative gel of three individual patient replicates).
The cag PAI is required for H pylori-induced HKα transcriptional repression in human gastric mucosa
To investigate further the role of the cag PAI in HKα mRNA repression, gastric corpus biopsies from three patients were independently infected (1—2×105 bacteria/mg wet weight of biopsy, 24 h) with wt H pylori 7.13 or a ΔcagPAI mutant. Wt H pylori infection repressed HKα mRNA expression by two- to sevenfold compared with mock infection (figure 4A). Infection with the ΔcagPAI mutant had no effect on HKα mRNA expression in two patients, and caused ~50% reduction of HKα mRNA expression in a third. To investigate the role of specific cag PAI genes in HKα transcriptional repression, biopsies were infected with ΔcagL, ΔcagE, ΔcagA or ΔcagM mutants or their background wt strains. Mock-infected biopsies from seven different patients showed widely different HKα mRNA content (figure 4B—D). Infection of matching biopsies with wt P12 (figure 4B) or wt 7.13 (figure 4C) markedly reduced HKα mRNA content compared with the mock-infected same-patient control. Infection of matching patient biopsies with P12 ΔcagL IM (figure 4B) partially reduced HKα mRNA content in one patient, and significantly increased HKα mRNA in two others. Infection with 7.13 ΔcagE non-significantly reduced HKα mRNA in two patients, and significantly reduced HKα mRNA in a third (figure 4C). Infection with 7.13 ΔcagA non-significantly reduced HKα mRNA, while infection with 7.13 ΔcagM and 7.13 Δslt increased HKα mRNA threefold and 3.5-fold, respectively, compared with mock-infected same-patient controls (figure 4D). The partial abrogation of H pylori-induced HKα mRNA repression by 7.13 ΔcagA suggests that CagA may play a role in repression together with other factors, and is consistent with the 7.13 ΔcagA-induced partial abrogation of transfected HKα promoter repression in AGS cells (figure 2). Taken together, these data indicate that H pylori cag PAI gene products participate in bacterial and host cell pathways that regulate HKα gene expression.
Figure 4.

cag pathogenicity island (PAI)-specific genes are necessary for H,K-ATPase α subunit (HKα) repression in human gastric mucosa. Gastric biopsies were infected ex vivo (1—2×105 bacteria/mg wet weight biopsy, 24 h) with (A) wild-type (wt) H pylori strain 7.13 or 7.13 ΔcagPAI isogenic mutant (IM) strain; (B) wt H pylori strain P12 or P12 ΔcagL IM strain; (C) wt H pylori strain 7.13 or 7.13 ΔcagE IM strain; and (D) wt H pylori strain 7.13 (open bar), 7.13 ΔcagA, 7.13 ΔcagM or 7.13 Δslt (shaded bars; all biopsies from the same patient). RNA was extracted from biopsies and the HKα mRNA content was measured by reverse transcription—PCR (means, SD, n=3; ***p<0.001, **p<0.01, *p<0.1).
cag PAI genes downregulate HKα protein expression in human gastric mucosa
Gastric biopsy HKα protein content in response to H pylori infection was assessed by HKα-specific immunoblotting. Infection of biopsies with wt H pylori 7.13 or 60190 virtually eliminated the immunoreactive 94 kDa HKα signal (figure 5A—D,F), while wt H pylori P12 markedly reduced HKα protein expression (figure 5E, lane 2). Biopsy infections with P12 ΔcagPAI, 60190 ΔcagA, 7.13 ΔcagM, 7.13 ΔcagE and P12 ΔcagL (figure 5A—D) caused no reduction in biopsy HKα content; in contrast, infection with the 60190 ΔvacA mutant completely eliminated the 94 kDa HKα band (figure 5F). Thus, VacA is not involved in H pylori-induced repression of HKα transcription. The complete abrogation of H pylori-induced HKα protein repression by 7.13 ΔcagA (figure 5B) is inconsistent with the ΔcagA-induced partial abrogation of transfected HKα promoter activity in AGS cells (figure 2) and of HKα mRNA expression in gastric biopsies (figure 4D). This inconsistency may reflect mechanistic differences in AGS cell and parietal cell regulation of HKα promoter activity, and/or H pylori CagA-induced post-translational regulation of HKα protein expression. Taken together, the HKα protein immunoblot data indicate that the cag PAI genes cagA, cagM, cagE and cagL participate in downregulation of HKα protein expression in gastric parietal cells.
Figure 5.

cag pathogenicity island (PAI) genes markedly downregulate H,K-ATPase α subunit (HKα) expression in human gastric mucosa. Gastric biopsies were infected (1—2×105 bacteria/mg wet weight biopsy, 24 h) with wild-type (wt) H pylori strains 7.13, 60190 or P12 or the corresponding isogenic mutant strains 7.13 ΔcagPAI, 60190 ΔcagA, 7.13 ΔcagM, 7.13 ΔcagE, P12 ΔcagL or 60190 ΔvacA. Biopsies were lysed and HKα protein content was assessed by immunoblotting using antibody HK 12.18 against HKα and β-actin antibody as gel loading control (representative gels of three individual patient replicates).
H pylori inhibits acid secretion in human gastric mucosa
To investigate the physiological consequence of H pylori-induced repression of HKα transcription and translation, human gastric corpus biopsies were infected (H pylori wt 60190, MOI ~50, 15 h) or mock infected in XF24 culture wells. The biopsies were treated with EIPA (150 μM, 30 min), and biopsy medium pH was recorded continuously for 3 min at intervals of 7 min. After 21 min, histamine (1 μM final) or vehicle alone was added to some biopsies. Figure 6A shows changes in extracellular pH of three representative biopsies as a function of time. Although addition of vehicle caused an abrupt transient medium alkalinisation (figure 6A, left traces), the rate of biopsy-mediated medium acidification was minimally affected. Histamine addition (figure 6A, middle traces) also caused a transient medium alkalinisation, but was followed by significant medium acidification. In contrast, biopsies infected with wt H pylori displayed markedly reduced medium acidification on histamine addition (figure 6A, right traces). The slopes of the initial rates of change of extracellular pH were transformed for buffer capacity to yield the proton production rate (PPR; pmol H+/min). As shown in figure 6B, mock-infected biopsies maintained a stable constitutive PPR of ~400 pmol H+/min for 50 min. Histamine addition to mock-infected biopsies increased the PPR to ~1650 pmol H+/min within 10 min, declining to ~1150 pmol H+/min over the next 20 min. In contrast, histamine addition to H pylori-infected biopsies transiently increased the PPR to ~600 pmol H+/min within 10 min, declining to <500 pmol H+/min over the next 20 min. Significantly, biopsy H pylori infection also reduced constitutive PPR compared with mock-infected controls. Pretreatment of biopsies with 50 μM SCH28080 for 30 min abrogated histamine-stimulated medium acidification (figure 6C), confirming mechanistic involvement of biopsy H,K-ATPase activity in this acidification.
Figure 6.

Gastric acid secretion by human gastric mucosal biopsies is inhibited by wild-type H pylori strains. Gastric biopsies were infected with wild-type H pylori strain 60190, P12 or P12 ΔcagL (1—2×105 bacteria/mg wet weight biopsy, 15 h) or Brucella broth alone, and then incubated with 150 μM 5-(N-ethyl-n-isopropyl) amiloride (EIPA) with or without 50 μM SCH28080 for 30 min. The pH of the medium bathing the biopsies was measured continuously for 3 min at 7 min intervals. After 21 min, histamine (or vehicle) was added to some biopsies to a 1 mM final concentration (arrows). (A) Changes in extracellular pH of three representative biopsies are shown as a function of time. Open squares, biopsy treated with vehicle alone; open triangles, biopsy treated with histamine; and open circles, biopsy infected with wt H pylori strain 60190 and treated with histamine. (B) The slopes of the initial rates of change of extracellular pH of medium bathing the biopsies as shown in A were transformed for buffer capacity to yield the biopsy proton production rate (PPR; pmol H+/min). Open squares, biopsies treated with vehicle alone; open triangles, biopsies treated with histamine; and open circles, biopsies infected with wild-type H pylori strain 60190 and treated with histamine. (C) Open triangles, biopsy treated with histamine in the absence of SCH28080; open diamonds, biopsy treated with 50 μM SCH28080 for 30 min and then treated with histamine. (D) Open triangles, biopsy treated with histamine; open diamonds, biopsy infected with wild-type H pylori strain P12; asterisks, biopsy infected with the H pylori P12 ΔcagL isogenic mutant. Data points in (B) and (C) are the mean PPR ±SD, n=3 biopsies; data points in (D) are measurements of single biopsies.
Finally, to confirm the specificity of H pylori-mediated inhibition of biopsy acid secretion, and to probe the dependency of such secretion on the cag PAI gene cagL, biopsies were infected (2 h) with wt H pylori P12 or P12 ΔcagL. Figure 6D shows that within 15 min of histamine administration, a mock-infected biopsy responded with a 4.5-fold increase in PPR. Infection of a second biopsy from the same patient with wt H pylori strain P12 (figure 6D) markedly reduced constitutive PPR and completely abrogated histamine-stimulated PPR. In contrast, infection of a third biopsy from the same patient with the P12 ΔcagL mutant (figure 6D) had no effect on constitutive PPR, and was permissive of 3.3-fold histamine stimulation of PPR. Taken together, these data demonstrate inhibition of parietal cell H,K-ATPase-mediated acid secretion as a physiological consequence of H pylori infection, and implicate the cag PAI gene cagL as a mechanistic intermediary in the inhibitory pathway.
DISCUSSION
This study identified the cag PAI as instrumental in H pylori-induced HKα repression. Infection of AGS cells with cag PAI-deficient or with cagE-, cagM- and cagL-deficient H pylori strains had no effect on transfected HKα promoter activity. Inactivation of cagE, which encodes a cytoplasmically oriented ATPase, prevents CagA translocation into the host cell and consequent induction of IL-8.23 33 CagM is a key structural protein of the periplasmic and extracellular portions of the T4SS pilus. CagL contributes to bacterial adherence and T4SS function, specifically translocation of CagA into host cells.24 Thus the insensitivity of HKα promoter constructs to AGS cell infection with ΔcagPAI, ΔcagE, ΔcagM or ΔcagL strains suggests that H pylori-induced HKα repression requires a structurally intact, functional T4SS pilus and specifically implicates a mechanistic role for CagE, CagM and CagL in this repression.
The partial sensitivity of HKα promoter constructs to infection with the ΔcagA strain also implicates CagA translocation in HKα repression. Unphosphorylated CagA binds to growth factor receptor-bound 2 (Grb2) which activates the Ras/MEK/ERK mitogen-activated protein kinase (MAPK) pathway, also promoting cell proliferation.34 A role for unphosphorylated CagA in HKα repression is therefore consistent with our previous finding that H pylori inhibits HKα gene expression by ERK1/2-mediated NF-κB p50 homodimer binding to the HKα promoter.17 Our finding here that ΔcagA infection partially abrogates wt H pylori-induced HKα repression, compared with the complete abrogation conferred by ΔcagE, ΔcagM or ΔcagL infections, suggests that bacterial virulence factor(s) other than those identified here may, independently or together with CagA, activate NF-κB signalling. The data indicate that, like CagA, delivery of such factor(s) to AGS cells requires a structurally intact functional T4SS. H pylori ΔvacA, ΔureA, Δfla and Δslt strains, deficient in non-cag PAI virulence factors, repressed HKα promoter activity as much as corresponding wt strains. These data exonerate VacA, UreA-dependent bacterial urea flux and flagellin A as causative factors in HKα transcriptional repression. Furthermore, our data showing HKα repression by the Δslt mutant suggest minimal participation of Nod1 signalling in H pylori-mediated NF-κB p50 homodimer binding to the HKα promoter.
Previous studies of H pylori-mediated HKα promoter regulation exclusively utilised transfected AGS cells.16-18 Although widely used in H pylori pathophysiology studies, AGS cells do not express H,K-ATPase and so are imperfect parietal cell surrogates. To assess the physiological consequences of H pylori-mediated HKα gene repression, human gastric biopsy HKα mRNA, HKα protein and acid secretion were measured in response to H pylori infection. The relative HKα mRNA content of biopsies varied widely, and could arise from differing penetration or grasp of the biopsy forceps recovering variable numbers of parietal cells. Also, proton pump inhibitor (PPI)-induced changes in HKα gene expression could potentially affect HKα mRNA levels. However, regardless of the relative baseline HKα mRNA content of mock-infected gastric biopsies, wt H pylori infection of biopsies markedly and significantly repressed HKα mRNA, whereas infection of same-patient biopsies with ΔcagPAI, ΔcagL, ΔcagM, ΔcagE and ΔcagA mutant strains failed to do so. The suppressive effect of H pylori on HKα expression is thus independent of the initial transcriptional status of the HKα gene.
The ΔcagL and ΔcagM mRNA data (figure 4B,D) are provocative, showing that infection of gastric biopsies with CagL- or CagM-deficient H pylori may activate HKα expression, a response we observed when AGS cells transfected with HKα2179 promoter were infected with Escherichia coli DH5α (unpublished). CagL and CagM expression may represent a first line of H pylori defence against gastric acid, which may otherwise be upregulated by the presence of CagL/CagM-deficient Gram-negative bacteria. Host-specific factors also appear to play a role in calibrating the acid secretory response to bacterial infection.
Also provocative was the finding that wt H pylori infection of human gastric biopsies significantly attenuates HKα protein expression. The subunit is readily detected by immunoblotting in mock-infected biopsy homogenates, and in biopsies infected with certain H pylori mutants, as shown in figure 5. H pylori-induced HKα disappearance 24 h postinfection cannot be attributed to cessation of HKα gene transcription and translation because the half-life of HKα is 48 h.35 We hypothesise that H pylori infection induces unscheduled proteasomal degradation of HKα, which would rapidly deplete parietal cells of acid secretory capacity, ameliorating the hostile acidic environment and facilitating H pylori gastric colonisation. Consistent with this hypothesis is our observation that histamine-stimulated, SCH28080-sensitive gastric biopsy H+ secretion was markedly inhibited by 2 h co-incubation with wt H pylori P12 (figure 6D).
H pylori-mediated modulation of HKα transcription and translation in AGS cells was complemented by measuring biopsy acid secretion. Our data provide direct evidence that acute H pylori infection of human gastric corpus biopsies inhibits both basal and histamine-stimulated acid secretion. Histamine-induced, SCH28080-sensitive stimulation of H+ secretion also showed that the biopsies are still functionally active and H pylori-induced inhibition of acid secretion is not simply a manifestation of biopsy deterioration. Clearly, biopsies have compromised epithelial integrity, and serosal access of bacteria to gastric glands is greatly facilitated. Moreover, H pylori induces matrix metalloproteinase-7 (MMP-7) expression in gastric epithelial cells,36 and disrupts cell adhesion junctions37; both events clearly disturb epithelial integrity and facilitate mucosal infiltration by H pylori. Although adhesion of H pylori to biopsy parietal cells was not investigated in this study, H pylori has been shown to invade gastric epithelial intercellular and intracellular sites,38 39 consistent with direct H pylori-mediated mobilisation of parietal cell NF-κB, but not excluding paracrine influences from other epithelial cells. Our observations of gastric biopsy H,K-ATPase activity demonstrate the feasibility of studying H pylori-mediated acid inhibition in human gastric mucosa under conditions allowing contolled time- and dose-dependent exposure to H pylori. Importantly, genomic DNA microarray studies have shown that H pylori gene expression patterns are different in human gastric mucosa and in vitro,40 emphasising that mechanistic findings in AGS cells require validation in a human gastric mucosal setting.
In summary, this study established that the H pylori cag PAI genes cagL, cagM and cagE are implicated in repression of HKα transcription following acute infection, and that this repression is reflected in markedly diminished HKα translation and ensuing H,K-ATPase activity in H pylori-infected gastric biopsies. The study introduces a novel and potentially informative model for studying the molecular pathophysiology of human H pylori infection, allowing for the first time controlled exposure of human gastric mucosa to different H pylori strains and refined pharmacological interventions to dissect the affected cellular signalling pathways.
Significance of this study.
What is already known about this subject?
Gastric mucosal H pylori infection causes transitory hypochlorhydria and gastritis that may progress to adenocarcinoma
H pylori strains with a cag PAI augment cancer risk
Parietal cell proton pump α subunit (HKα) mediates acid secretion
H pylori-induced NF-κB represses HKα promoter activity in gastric epithelial cells
What are the new findings?
H pylori mutants deficient in cag PAI genes cagE, cagM and cagL failed to repress HKα promoter activity
cagA deficiency partially repressed HKα promoter
Human gastric biopsy HKα mRNA and protein expression was markedly reduced by wild-type H pylori infection
Biopsy acid secretion was significantly inhibited by wild-type but not CagL-deficient H pylori infection
How might it impact on clinical practice in the foreseeable future?
Knowledge of H pylori genotype may inform therapeutic decision-making by identifying high-risk patients who warrant eradication therapy.
Acknowledgements
We thank Dr Gary Shull (University of Cincinnati) for genomic DNA incorporating a portion of the human gastric H,K-ATPase α subunit 5′-flanking sequence, Dr Steffen Backert (University College, Dublin) for provision of H pylori strain P12 and the corresponding cagL− isogenic mutant, and for valuable discussions, and Drs Brenda Hoffman and Marcello Vela (Division of Gastroenterology and Hepatology, MUSC) and April Wood (Clinical Coordinator, Digestive Disease Center, MUSC) for provision of human gastric biopsies. We thank also Gyda Beeson (Department of Pharmaceutical Sciences, MUSC) for her expertise and guidance in measuring biopsy extracellular acidification.
Funding NIH grant DK064371 to AJS.
Footnotes
Competing interests None.
Ethics approval This study was conducted with the approval of the MUSC Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.El-Omar EM, Oien K, El-Nujumi A, et al. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15–24. doi: 10.1016/s0016-5085(97)70075-1. [DOI] [PubMed] [Google Scholar]
- 2.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 3.Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol. 1987;82:192–9. [PubMed] [Google Scholar]
- 4.Graham DY, Alpert LC, Smith JL, et al. Iatrogenic Campylobacter pylori infection is a cause of epidemic achlorhydria. Am J Gastroenterol. 1988;83:974–80. [PubMed] [Google Scholar]
- 5.Sobala GM, Crabtree JE, Dixon MF, et al. Acute Helicobacter pylori infection: clinical features, local and systemic immune response, gastric mucosal histology, and gastric juice ascorbic acid concentrations. Gut. 1991;32:1415–18. doi: 10.1136/gut.32.11.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harford WV, Barnett C, Lee E, et al. Acute gastritis with hypochlorhydria: report of 35 cases with long term follow up. Gut. 2000;47:467–72. doi: 10.1136/gut.47.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spicer Z, Miller ML, Andringa A, et al. Stomachs of mice lacking the gastric H,K-ATPase alpha-subunit have achlorhydria, abnormal parietal cells, and ciliated metaplasia. J Biol Chem. 2000;275:21555–65. doi: 10.1074/jbc.M001558200. [DOI] [PubMed] [Google Scholar]
- 8.Zavros Y, Eaton KA, Kang W, et al. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene. 2005;24:2354–66. doi: 10.1038/sj.onc.1208407. [DOI] [PubMed] [Google Scholar]
- 9.Ramsey EJ, Carey KV, Peterson WL, et al. Epidemic gastritis with hypochlorhydria. Gastroenterology. 1979;76:1449–57. [PubMed] [Google Scholar]
- 10.Takashima M, Furuta T, Hanai H, et al. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut. 2001;48:765–73. doi: 10.1136/gut.48.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace JL, Cucala M, Mugridge K, et al. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol. 1991;261:G559–64. doi: 10.1152/ajpgi.1991.261.4.G559. [DOI] [PubMed] [Google Scholar]
- 12.Schepp W, Dehne K, Herrmuth H, et al. Identification and functional importance of IL-1 receptors on rat parietal cells. Am J Physiol. 1998;275:G1094–105. doi: 10.1152/ajpgi.1998.275.5.G1094. [DOI] [PubMed] [Google Scholar]
- 13.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 14.Wang F, Xia P, Wu F, et al. Helicobacter pylori VacA disrupts apical membrane-cytoskeletal interactions in gastric parietal cells. J Biol Chem. 2008;283:26714–25. doi: 10.1074/jbc.M800527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuta T, Baba S, Takashima M, et al. H+/K+-adenosine triphosphatase mRNA in gastric fundic gland mucosa in patients infected with Helicobacter pylori. Scand J Gastroenterol. 1999;34:384–90. doi: 10.1080/003655299750026399. [DOI] [PubMed] [Google Scholar]
- 16.Saha A, Hammond CE, Gooz M, et al. The role of Sp1 in IL-1beta and H. pylori-mediated regulation of H,K-ATPase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2008;295:G977–86. doi: 10.1152/ajpgi.90338.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saha A, Hammond CE, Trojanowska M, et al. Helicobacter pylori -induced H,K-ATPase {alpha}-subunit gene repression is mediated by NF-{kappa}B p50 homodimer promoter binding. Am J Physiol Gastrointest Liver Physiol. 2008;294:G795–807. doi: 10.1152/ajpgi.00431.2007. [DOI] [PubMed] [Google Scholar]
- 18.Saha A, Hammond CE, Gooz M, et al. IL-1beta modulation of H, K-ATPase {alpha}-subunit gene transcription in Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1055–61. doi: 10.1152/ajpgi.00338.2006. [DOI] [PubMed] [Google Scholar]
- 19.Gooz M, Hammond CE, Larsen K, et al. Inhibition of human gastric H(+)-K (+)-ATPase alpha-subunit gene expression by Helicobacter pylori. Am J Physiol Gastrointest Liver Physiol. 2000;278:G981–91. doi: 10.1152/ajpgi.2000.278.6.G981. [DOI] [PubMed] [Google Scholar]
- 20.Censini S, Lange C, Xiang Z, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996;93:14648–53. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blaser MJ, Perez-Perez GI, Kleanthous H, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–15. [PubMed] [Google Scholar]
- 22.Peek RM, Jr, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest. 1995;73:760–70. [PubMed] [Google Scholar]
- 23.Fischer W, Puls J, Buhrdorf R, et al. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42:1337–48. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 24.Kwok T, Zabler D, Urman S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–6. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 25.Bourzac KM, Guillemin K. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol. 2005;7:911–19. doi: 10.1111/j.1462-5822.2005.00541.x. [DOI] [PubMed] [Google Scholar]
- 26.Higashi H, Tsutsumi R, Muto S, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–6. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 27.Franco AT, Johnston E, Krishna U, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–87. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smolka AJ, Larsen KA, Hammond CE. Location of a cytoplasmic epitope for monoclonal antibody HK 12.18 on H, K-ATPase alpha subunit. Biochem Biophys Res Commun. 2000;273:942–7. doi: 10.1006/bbrc.2000.3031. [DOI] [PubMed] [Google Scholar]
- 29.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–74. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 30.Josenhans C, Labigne A, Suerbaum S. Comparative ultrastructural and functional studies of Helicobacter pylori and Helicobacter mustelae flagellin mutants: both flagellin subunits, FlaA and FlaB, are necessary for full motility in Helicobacter species. J Bacteriol. 1995;177:3010–20. doi: 10.1128/jb.177.11.3010-3020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cussac V, Ferrero RL, Labigne A. Expression of Helicobacter pylori urease genes in Escherichia coli grown under nitrogen-limiting conditions. J Bacteriol. 1992;174:2466–73. doi: 10.1128/jb.174.8.2466-2473.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weeks DL, Eskandari S, Scott DR, et al. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science. 2000;287:482–5. doi: 10.1126/science.287.5452.482. [DOI] [PubMed] [Google Scholar]
- 33.Tummuru MK, Sharma SA, Blaser MJ. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol Microbiol. 1995;18:867–76. doi: 10.1111/j.1365-2958.1995.18050867.x. [DOI] [PubMed] [Google Scholar]
- 34.Mimuro H, Suzuki T, Tanaka J, et al. Grb2 is a key mediator of Helicobacter pylori cagA protein activities. Mol Cell. 2002;10:745–55. doi: 10.1016/s1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 35.Gedda K, Scott D, Besancon M, et al. Turnover of the gastric H+, K(+)-adenosine triphosphatase alpha subunit and its effect on inhibition of rat gastric acid secretion. Gastroenterology. 1995;109:1134–41. doi: 10.1016/0016-5085(95)90571-5. [DOI] [PubMed] [Google Scholar]
- 36.Wroblewski LE, Noble PJ, Pagliocca A, et al. Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in human gastric epithelial cells: role in epithelial cell migration. J Cell Sci. 2003;116:3017–26. doi: 10.1242/jcs.00518. [DOI] [PubMed] [Google Scholar]
- 37.Weydig C, Starzinski-Powitz A, Carra G, et al. CagA-independent disruption of adherence junction complexes involves E-cadherin shedding and implies multiple steps in Helicobacter pylori pathogenicity. Exp Cell Res. 2007;313:3459–71. doi: 10.1016/j.yexcr.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 38.Bjorkholm B, Zhukhovitsky V, Lofman C, et al. Helicobacter pylori entry into human gastric epithelial cells: a potential determinant of virulence, persistence, and treatment failures. Helicobacter. 2000;5:148–54. doi: 10.1046/j.1523-5378.2000.00023.x. [DOI] [PubMed] [Google Scholar]
- 39.Necchi V, Candusso ME, Tava F, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009–23. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 40.Graham JE, Peek RM, Jr, Krishna U, et al. Global analysis of Helicobacter pylori gene expression in human gastric mucosa. Gastroenterology. 2002;123:1637–48. doi: 10.1053/gast.2002.36589. [DOI] [PMC free article] [PubMed] [Google Scholar]