

Abstract
Systemic lupus erythematosis is an autoimmune disease of unknown etiology. Lupus pathology is thought to reflect autoantibody-mediated damage due to a failure of B lymphocyte tolerance. Since excessive B cell-activating factor belonging to the TNF family (BAFF) expression correlates with human and murine lupus, and BAFF signals B cell survival through BAFF-R, it is believed that excessive BAFF-R signaling can subvert B cell tolerance and facilitate lupus development. Here we report the unexpected finding that BAFF-R-mutant A/WySnJ mice develop a lupus-like syndrome. These mice carry the B cell maturation defect-1 (Bcmd-1) mutant allele of the Baffr gene. Bcmd-1 causes premature B cell death and profound B cell deficiency. Despite having 90% fewer splenic B cells than normal mice, A/WySnJ mice had an 18-fold increased frequency of splenocytes secreting IgM antibodies to dsDNA, and increased amounts of circulating IgM and IgG to dsDNA by 9 months of age. By age 11 months, most A/WySnJ mice displayed renal pathology characteristic of lupus, including proteinuria as well as periodic acid-Schiff-positive deposits and glomerular capillary bed destruction. Importantly, we genetically linked this autoimmunity to Bcmd-1, since congenic AW.Baffr+/+ mice carrying a wild-type allele developed none of these phenotypes. Our data provide the first evidence linking altered BAFF-R signaling to the development of B cell-mediated autoimmunity.
Keywords: Autoimmunity, BAFF-R, B lymphocytes, Lupus
Introduction
Systemic lupus erythematosis (SLE) is a genetically and immunologically complex autoimmune disorder whose etiology and pathogenesis are poorly understood [1]. Lupus predominantly strikes young women, especially non-Caucasian women, and shows alternating periods of illness (flares) and remission. A diagnosis of lupus is based on skin lesions (e.g. rashes, oral ulcerations), systemic autoimmune signs (non-deforming arthritis, pleuritis, proteinuria, neurological disorders), the appearance of circulating autoantibodies to chromatin components, tissue deposition of immune complexes (IC), and blood abnormalities (hemolytic anemia, leukopenia, lymphopenia) [2]. The loss of B and T lymphocyte self tolerance to nuclear components is commonly considered to have a causal role in SLE pathogenesis, but the mechanisms underlying the loss of self tolerance are debated and may be numerous [3, 4].
One suggested mechanism of subverting B lymphocyte self tolerance is excessive availability of B cell-activating factor belonging to the TNF family (BAFF) [4, 5]. BAFF is often elevated in the blood of SLE patients [6] and the serum of lupus-prone NZB/W F1 mice [7, 8]. Moreover, BAFF-Tg mice spontaneously develop a lupus-like syndrome, with elevated circulating Ig, rheumatoid factors, circulating IC, antibodies to dsDNA, and Ig deposition in the kidneys [5]. These data suggest that excessive BAFF can undermine B cell self tolerance mechanisms, resulting in systemic autoimmune disease.
How excess BAFF production may subvert B cell self tolerance mechanisms is not yet clear. BAFF is a B cell-specific survival factor that promotes survival through engagement of BAFF-R [5]. Since BAFF-R signaling induces pro-survival B cell lymphoma 2 (BCL-2) family proteins, and enforced Bcl-2 gene expression results in systemic autoimmunity [9], it is believed that excessive BAFF-R signaling can subvert B cell self tolerance [4]. Excessive BAFF-R signaling is thought to override the BCR-mediated activation of pro-apoptotic BIK and BIM in B cells undergoing self antigen stimulation [10]. Current models suggest that this excessive signaling is the result of an abnormally high BAFF:B cell ratio, established either by overexpression of BAFF (BAFF-Tg mice), or by a large decrease in B cell number (B lymphopenia) [11, 12].
Here we report that BAFF-R signaling-defective A/WySnJ mice develop systemic autoimmunity, in apparent contradiction with the model of excess BAFF-R signaling subverting B cell self tolerance in B lymphopenic strains [4, 13]. A spontaneous retrotransposon insertion event disrupted the chromosome 15 Baffr locus in A/WySnJ mice, generating the B cell maturation defect-1 (Bcmd-1) mutant allele [14–18]. A/WySnJ peripheral B-2 B cells express the mutant BAFF-R protein, but appear not to respond to BAFF in vivo [17, 19]. A/WySnJ B cells have excessive Bik gene expression and a short life span [16], so the A/WySnJ mice are severely B lymphopenic. Enforced Bcl-xL survival gene expression complemented the Bcmd-1 mutation and restored peripheral B cell development [20]. We find that A/WySnJ mice develop a spontaneous, late-onset, lupus-like systemic autoimmune syndrome. Moreover, the Bcmd-1 mutant allele of the Baffr locus appeared to control the lupus-like syndrome, since A/WySnJ-congenic mice with a wild-type Baffr locus did not develop the autoimmune syndrome. We discuss our findings in the context of current hypotheses for subversion of peripheral B cell self tolerance and the development of systemic auto-immunity.
Results
A/WySnJ mice develop autoantibodies to chromatin components
During genetic studies of A/WySnJ mice, we noted that many animals developed weight loss, patchy fur loss, skin lesions, a hunched posture, and occasionally splenomegaly as they aged. These signs are commonly associated with systemic autoimmune disease [1]. To test these mice for systemic autoimmunity, we evaluated them for an autoantibody response to dsDNA, which is a hallmark of systemic autoimmunity in mice and humans [1]. Serum samples were collected from 7–9-month-old male and female A/WySnJ mice, and age- and gender-matched A/J control mice, and an ELISA was performed to quantify IgM and IgG responses to dsDNA.
The IgM antibodies to dsDNA were readily apparent in A/WySnJ serum up to a dilution of 1:1600, and these mice produced significantly more IgM to dsDNA than A/J mice (p<0.01) at all dilutions tested (Fig. 1A). The A/WySnJ mice began to produce these IgM autoanti-bodies at about 7 months of age. At 9 months of age, 80% (18/20) of them had circulating dsDNA-specific IgM antibodies (ELISA absorbance 2 SD greater than the A/J mean value). Thereafter, the incidence did not increase. The elevation in circulating dsDNA-specific IgM antibodies does not represent a generalized B cell hyper-reactivity that is often seen in SLE, because the total IgM levels were equivalent in the A/WySnJ (359 ± 35 μg/mL) and A/J (340 ± 26 μg/mL) mice (seven to nine mice of each strain) [21]. We conclude that within the pool of circulating IgM antibodies, A/WySnJ mice have a significant amount of dsDNA-specific IgM, whereas A/J mice do not.
Figure 1.

A/WySnJ mice produce autoantibodies to dsDNA, whereas A/J and AW.Baffr+/+ mice do not. (A) IgM autoantibodies to dsDNA. (B) IgG autoantibodies to dsDNA. (C) IgM AFC specific for dsDNA. Serum autoantibodies to dsDNA were quantified by ELISA. AFC were quantified by ELISPOT assay. The ELISA data shown are the mean absorbance (± SD) for six to nine male and female mice at 7–9 months of age. The ELISPOT data shown are the mean (± SD) for six to 12 mice ≥9 months of age. The Mann–Whitney test and two-tailed Student’s t-test gave p<0.01 (*) for comparisons between the A/WySnJ and the A/J or AW.Baffr+/+ mice.
In human SLE, T and B cell-mediated autoimmune responses combine to yield IgG IC, which cause damage to the skin, kidneys, cardiovascular system, lung, musculoskeletal system, and other tissues [1]. Therefore, we also analyzed the serum samples for IgG antibodies to dsDNA. The IgG antibodies to dsDNA (all isotypes) were readily apparent in A/WySnJ serum up to dilutions of 1:1600 (and sometimes higher), and these mice produced significantly more IgG to dsDNA than A/J mice (p<0.01) at all dilutions tested (Fig. 1B). Most A/WySnJ mice (11/15; 73%) had these IgG autoanti-bodies by 9 months of age (ELISA absorbance 2 SD greater than the A/J mean value). The IgG autoanti-bodies were mainly of the IgG1 isotype, although other isotypes were also present (data not shown). The A/WySnJ mice that had high levels of autoantibodies to dsDNA also produced IgM and IgG autoantibodies to histone proteins (data not shown). Collectively, these data indicate that aging A/WySnJ mice develop systemic autoimmunity to nuclear antigens.
Elevated BAFF levels are thought to contribute to the subversion of B lymphocyte self tolerance and emergence of a lupus-like syndrome in MRL-lpr/lpr and NZBWF1 mice, which have serum BAFF levels elevated ≥ six fold over baseline [7]. To determine whether elevated serum BAFF levels might be driving the lupus-like syndrome in A/WySnJ mice, we performed an ELISA for BAFF on serum samples from A/WySnJ mice with high levels of dsDNA-specific antibodies (n=5; age 7–9 months) and age-matched A/J control mice (n=3). The BAFF concentration was not significantly different in these samples (A/WySnJ, 4.3 ± 2.8 ng/mL; A/J, 3.6 ± 0.5 ng/mL). Consequently, we conclude that dramatically elevated BAFF production is probably not driving the lupus-like syndrome in A/WySnJ mice as it is in MRL-lpr/lpr and NZBWF1 mice.
The high level of serum IgM and IgG antibodies to dsDNA in A/WySnJ mice was puzzling, because these mice are B lymphopenic [21, 22]. We considered two possibilities: a few auto-reactive B cells may have become hyperactivated to secrete very large amounts of dsDNA-specific autoantibodies, or a large number of auto-reactive B cells might be present and secreting a normal amount of Ig/cell. To distinguish these possibilities, an ELISPOT analysis was performed to assess the frequency of dsDNA-specific antibody-forming cells (AFC) in the spleen. The results showed that the A/J spleens had only 114 ± 49 IgM-secreting, dsDNA-specific B cells per million B cells, whereas A/WySnJ spleens had 2028 ± 405 such B cells per million B cells, an 18-fold increase in frequency (Fig. 1C). This result ruled out the possibility of relatively few auto-reactive dsDNA-specific B cells secreting large amounts of autoantibodies. The ELISPOT assay did not detect significant numbers of IgG-secreting, dsDNA-specific B cells in either the A/WySnJ or A/J spleens, or IgM- or IgG-secreting AFC in the bone marrow of these two strains (data not shown). We conclude that the dsDNA-specific B cells are not purged from the peripheral B cell repertoire in A/WySnJ mice, but instead appear to undergo enrichment and possibly expansion.
In addition to conventional B-2 B cells, there is a distinct lineage of self-renewing B-1 B cells [23]. The B-1 cells populate the peritoneal and pleural cavities and produce natural IgM antibodies that are important in the early defense against viral and bacterial infections. Some of these natural IgM antibodies cross-react with self antigens like dsDNA. We previously reported that the A/WySnJ and A/J mice do not differ significantly in the absolute number of B-1 cells in the spleen (about 0.4 million), or the rate at which the splenic B-1 cells turn over (4%/day), indicating that the Bcmd-1 mutation did not disrupt B-1 cell development [24]. To explore a possible role of B-1 cells in the A/WySnJ autoimmune phenotype, we examined the peritoneal cavity for auto-reactive AFC. If B-1 cells are major contributors to the autoimmune phenotype, then the peritoneal cavity would be predicted to have a higher frequency of auto-reactive AFC than the spleen.
A pilot study indicated that the A/WySnJ peritoneum did indeed have a higher B-1:B-2 cell ratio than the spleen (peritoneum 1:0.6; spleen 1:23), as determined by flow cytometry (B-1 cells, IgM+B220lowCD23−CD11b+; B-2 cells, IgM+B220+CD23+CD11b−) but did not show a proportionally higher ELISPOT frequency (peritoneum, 1303 AFC/million B cells; spleen, 1073 AFC/million B cells) compared to the spleen. The control A/J strain had negligible numbers of AFC in the peritoneum. Although these pilot data do not rule out a B-1 cell contribution to the autoimmune phenotype, they suggest that B-2 cells are the major contributors of auto-reactive IgM antibodies.
Bcmd-1 controls systemic autoimmunity in A/WySnJ mice
We next sought to determine whether the systemic autoimmune phenotypes in A/WySnJ mice were attributable to Bcmd-1 (Fig. 2A). We previously mapped Bcmd-1 between D15Mit211 and D15Mit76 on chromosome 15 through (A/WySnJ×CAST/Ei)F2 progeny analysis [14], and refined the mapping to a 1-cM interval bounded by D15Mit259 and D15Mit118 through (A/WySnJ×CAST/Ei)F1 × A/WySnJ N2 progeny analysis [15]. Based on synteny between the human and mouse genomes, the similar functions of Baffr (also known as Tnfrsf13c) and Bcmd-1 in controlling B cell survival, and an aberrant Baffr transcript in A/WySnJ mice, Thompson et al. [17] concluded that Bcmd-1 was a mutant allele of the Baffr gene. To determine whether the systemic autoimmunity in A/WySnJ mice was attributable to Bcmd-1, we evaluated Bcmd-1-congenic AW.Baffr+/+ mice from backcross generation 13 (N13) for autoimmunity. The SSLP genotyping showed that these mice have 7.8 Mb of CAST/Ei-derived DNA bounded by D15Mit68 and D15Mit72. This interval encompasses 177 identified genes and includes wild-type Baffr and Bik loci (Fig. 2B).
Figure 2.

A/WySnJ mice are B lymphopenic, whereas A/J and congenic AW.Baffr+/+ mice are not. (A) Schematic representation of Baffr cDNA. Gray shading represents the retrotransposon insertion. (B) AW.Baffr+/+ chromosome 15. Gray shading shows the congenic interval derived from the CAST/Ei strain. (C) Splenocytes were stained with fluorescent antibodies to IgM and IgD, and analyzed for the T1 (IgMhighIgDlow), MZ/T1 (IgMhighIgDint), T2 (IgMhighIgDhigh), and mature B cell subsets (IgMlowIgDhigh). (D) Splenocytes were stained with fluorescent antibodies to IgM, C1qRp, and CD23. The IgM-gated cells were analyzed for the mature B cell subset (CD23highC1qRp−). The C1qRp-gated cells were analyzed for T1 (IgMhighCD23−), T2 (IgMhighCD23+), and T3 (IgMlowCD23+) B cell subsets. The data are representative of six mice per strain.
We first characterized peripheral B cell subsets in the AW.Baffr+/+ mice through flow cytometric analyses. The relative expression of IgM and IgD (Fig. 2C) or IgM, C1qRp, and CD23 (Fig. 2D) defined the peripheral B cell subsets. The A/WySnJ spleen had 4–5 million transitional B cells and about 5–7 million mature B cells, whereas AW.Baffr+/+ and A/J spleens had 13–25 million transitional B cells and 30–50 million mature B cells (p<0.01; Table 1). In the occasionally enlarged A/WySnJ spleens, the proportions of B cells, T cells, and myeloid lineage cells were the same as in the conventionally sized A/WySnJ spleens (data not shown). Since BAFF-R expression did not differ between A/J and A/WySnJ (data not shown), the Bcmd-1 mutation must change BAFF-R function. Consistent with data showing the BAFF-R controls optimal CD21 expression [25], we found that A/WySnJ mature follicular (FO) B cells had suboptimal CD21 expression compared to AW.Baffr+/+ and A/J B cells, but normal CD23 density (Fig. 2C). Additionally, defining marginal zone (MZ) B cells by IgM, CD21, and CD23 expression, the A/WySnJ spleens lacked MZ B cells, whereas AW.Baffr+/+ and A/J spleens had MZ B cells (data not shown). Thus, introduction of the congenic segment in AW.Baffr+/+ mice complemented all of the disrupted B cell developmental phenotypes in A/WySnJ mice.
Table 1.
Splenic B cell development in A/J, A/WySnJ, and AW.Baffr+/+ micea)
| Strain |
||||||
|---|---|---|---|---|---|---|
| A/J | A/WySnJ | AW.Baffr+/+ | ||||
| % of IgM+ | 106 | % of IgM+ | 106 | % of IgM+ | 106 | |
| IgM and IgD staining | ||||||
| T1 | 3.0 ± 0.7 | 1.7 ± 0.2 | 13.9 ± 1.0** | 1.5 ± 0.2 | 5.1 ± 0.4 | 2.5 ± 0.4 |
| MZ/T1 | 10.1 ± 1.5 | 5.9 ± 0.6 | 18.3 ± 2.8 | 2.0 ± 0.2* | 13.4 ± 0.2 | 6.4 ± 0.4 |
| T2 | 17.4 ± 3.3 | 11.2 ± 3.5 | 17.3 ± 3.2 | 2.0 ± 0.5 | 22.8 ± 0.9 | 10.9 ± 0.4 |
| Mature B | 69.5 ± 1.5 | 42.6 ± 9.2 | 50.5 ± 0.9** | 5.6 ± 0.8* | 58.7 ± 0.8 | 28.1 ± 2.6 |
| CD21 MFI | 73.9 ± 5.9 | 34.6 ± 4.0** | 83.6 ± 8.4 | |||
| CD23 MFI | 231.0 ± 50.1 | 216.2 ± 47.1 | 334.0 ± 2.5 | |||
| IgM, CD23, C1qRp staining | ||||||
| % of IgM+ | 106 | % of IgM+ | 106 | % of IgM+ | 106 | |
| Mature B | 72.7 ± 2.2 (% of C1qRp+) | 53.6 ± 10.1 | 46.0 ± 0.6** (% of C1qRp+) | 7.1 ± 0.7* | 68.5 ± 0.3 (% of C1qRp+) | 40.9 ± 2.6 |
| T1 | 19.7 ± 4.8 | 2.3 ± 0.3 | 45.5 ± 5.5* | 2.0 ± 0.2 | 45.8 ± 1.1 | 5.1 ± 0.3 |
| T2 | 29.9 ± 2.9 | 3.7 ± 0.4 | 25.5 ± 2.4 | 1.2 ± 0.2** | 27.9 ± 1.1 | 3.1 ± 0.1 |
| T3 | 50.4 ± 7.0 | 6.9 ± 2.4 | 29.0 ± 3.1 | 1.4 ± 0.3 | 26.4 ± 0.1 | 2.9 ± 0.1 |
B cells were divided into subpopulations using the IgM/IgD gating scheme of Cariappa et al. [48] as shown in Fig. 2C, or the IgM/CD23/C1qRp gating scheme of Allman et al. [47] as shown in Fig. 2D. The percentage and absolute number (mean ± SEM) of each subpopulation were measured for three mice per group. A two-tailed Student’s t-test gave p ≤ 0.05 (*) or p ≤ 0.01 (**) for comparisons between A/WySnJ and A/J B cell subsets.
A/WySnJ B cells express an abnormally high level of Bik transcripts [16], and Bik is within the congenic segment. To rule out an alteration of the Bik gene as an explanation for the short lifespan of A/WySnJ B cells and unequivocally implicate Bcmd-1, we performed Baffr complementation studies using retroviral-mediated gene transfer. The wild-type Baffr cDNA was cloned from an A/J splenocyte cDNA library into the MigRI retroviral vector, and the resulting MigRI.BR vector was used to transduce the Baffr cDNA into A/WySnJ bone marrow stem cells. The empty MigRI vector served as a negative control. A flow cytometric analysis was performed on splenocytes from A/WySnJ mice reconstituted with MigRI.BR-transduced stem cells. The GFP+ splenocytes displayed the BAFF-R protein in proportion to GFP, indicating successful Baffr gene transfer and expression (data not shown). Importantly, the GFP− splenic B cells from the Mi-gRI.BR-transduced mice, and the GFP− and GFP+ splenic B cells from the empty vector-transduced control mice had equivalent subset proportions, ruling out spurious effects of retroviral gene transfer on B cell subsets (data not shown).
Comparing the BAFF-R-expressing (GFP+) to the non-expressing (GFP−) splenic B cells from these mice showed that Baffr gene expression increased the FO B cell proportion from 41.6 to 61.7% (p=0.015), and decreased the NF B cell proportion from 50.3 to 23.1% (p=0.006; Fig. 3A). Baffr gene expression also significantly increased MZ B cells (p<0.05). The IgM+GFP+ mature FO B cells from the reconstituted mice showed significantly higher CD21/35 density (MFI: 197 ± 92) than the IgM+GFP− FO B cells (MFI: 110 ± 45; p=0.018). Other flow cytometry staining schemes showed similar results (Fig. 3B–D; Table 2). Thus, enforced expression of the wild-type Baffr gene fully compensated Bcmd-1, restoring MZ and FO B cell development and CD21/35 expression. Therefore, we conclude that of the 177 genes in the congenic interval, it is the Bcmd-1 allele of the Baffr gene that unequivocally controls the B cell maturation defects in A/WySnJ mice.
Figure 3.

Retroviral-mediated gene transfer of the wild-type Baffr gene into A/WySnJ HSC-restored B cell development. Flow cytometry was performed on individual splenocyte samples gated according to their GFP expression. (A) The cells were stained with antibodies to IgM, CD21 and CD23, and gated for NF, MZ and FO B cell subsets. (B) The cells were stained with antibodies to IgM and to IgD; the IgM+IgD+ cells were gated as shown to evaluate B cell subsets. (C) The cells were stained with antibodies to IgM, C1qRp, and CD23; the IgM+ cells were gated for the C1qRp−CD23+ mature B cells [47]. (D) The cells were stained with antibodies to IgM, C1qRp, and CD23; the C1qRp+ cells were subdivided into T1, T2 and T3 B cells. The data shown are representative of five mice per strain.
Table 2.
Retroviral-mediated expression of Baffr restored transitional B cell development in A/WySnJ mice
| B cell subseta) | Percentage of IgM+ splenic B cellsb) |
|||
|---|---|---|---|---|
| AW.MigRI | AW.Baffr | |||
| GFP− | GFP+ | GFP− | GFP+ | |
| IgM, CD23, C1qRp staining | ||||
| T1 | 12.3 ± 3.2 | 11.0 ± 3.6 | 14.6 ± 2.0 | 6.8 ± 2.3** |
| T2 | 11.8 ± 1.8 | 12.8 ± 5.2 | 8.8 ± 3.0 | 7.9 ± 4.0 |
| T3 | 7.9 ± 1.4 | 8.3 ± 2.0 | 8.9 ± 2.4 | 9.7 ± 4.1 |
| Mature B | 37.5 ± 6.0 | 44.1 ± 4.5 | 36.8 ± 4.3 | 62.2 ± 8.2** |
| IgM, IgD staining | ||||
| IgMhighIgDlow | 21.6 ± 4.5 | 18 ± 3.7 | 17.6 ± 1.3 | 5.8 ± 2.3** |
| IgMhighIgDhigh | 31.4 ± 2.9 | 30.2 ± 3.3 | 27.6 ± 5.3 | 22.5 ± 5.5 |
| IgMlowIgDhigh | 47.0 ± 3.6 | 52.0 ± 3.3 | 54.8 ± 6.0 | 71.7 ± 6.0** |
IgM+ B cells were divided into subsets using either the IgM/CD23/C1qRp or the IgM/IgD gating scheme.
The values represent the mean percentage (± SD) of the IgM+ B cells (four to five mice per group). A paired, two-tailed Student’s t-test gave p<0.05 (*) or p<0.01 (**) for comparisons between the GFP− and GFP+ B cell subsets.
Having constructed and characterized the AW.Baffr+/+ N13 congenic strain, and proven unequivocally that the Baffr mutation controls the B cell maturation defects in A/WySnJ mice, we evaluated the AW.Baffr+/+ congenic mice for signs of systemic autoimmunity. These mice did not develop weight loss, skin lesions, a hunched posture, or splenomegaly as they aged. Moreover, the AW.Baffr+/+ congenic mice did not produce circulating dsDNA-specific IgM antibodies (Fig. 1A), dsDNA-specific IgG antibodies (Fig. 1B), or a high frequency of IgM-secreting, dsDNA-specific B cells (Fig. 1C). We conclude that a gene in the congenic interval, most likely Bcmd-1, is responsible for the large amounts of IgM and IgG to dsDNA, and the increased frequency of dsDNA-specific B cells in A/WySnJ mice.
A/WySnJ mice develop renal pathology
Lupus patients with abundant circulating IgG antibodies to nuclear components often develop renal pathology resulting from IC deposition in the glomeruli [1]. To determine whether A/WySnJ mice developed renal dysfunction, we analyzed fresh urine samples for protein. A/WySnJ mice had significantly higher mean levels of protein in their urine than A/J and AW.Baffr+/+ mice at 11–14 months of age (p<0.05; Fig. 4A). We defined urinary protein >300 mg/dL as the threshold for evidence of severe renal damage (Fig. 4B). Using this threshold, we first detected severe proteinuria in 7–8 months-old A/WySnJ mice. This renal dysfunction increased in mice 11–14 months of age (Fig. 4B). In contrast, severe proteinuria was absent in A/J and AW.Baffr+/+ congenic mice (p<0.05; Fig. 4B).
Figure 4.

A/WySnJ mice develop proteinuria, whereas A/J and AW.Baffr+/+ mice do not. (A) Mean proteinuria score. (B) Incidence of severe proteinuria (≥300 mg/dL urinary protein). Scores were assigned as follows: score 0 = 0-trace, score 30 = 30–99 mg/dL, score 100 = 100–299 mg/dL, score 300 = ≥300 mg/dL. The data shown are from 13–22 female and male mice per age group. A two-tailed Student’s t-test for proteinuria scores and χ2 test of incidence gave p<0.05 (*) for comparisons between A/WySnJ and A/J or AW.Baffr+/+ mice.
To examine renal pathology in more detail, the kidneys from A/WySnJ mice with proteinuria and age-matched A/J and AW.Baffr+/+ mice were sectioned, periodic acid-Schiff (PAS)-stained, and examined by two veterinary pathologists. A/WySnJ kidney sections had evidence of renal pathology, whereas the A/J and AW.Baffr+/+ kidney sections did not (Fig. 5A, B; Supporting Information Fig. 1). The A/J and AW.Baffr+/+ glomeruli were of normal size (neither hyper- nor hypo-cellular), had widely patent capillary loops and Bowman’s space, and had no or minimal PAS-staining deposits in the mesangium. In contrast, the A/WySnJ glomeruli were occasionally enlarged, and often had obliterated capillary loops, PAS-staining deposits in the mesangium or glomerular tuft, and the Bowman’s space was often obstructed by adhesion to Bowman’s capsule (Fig. 5B). We did not find evidence of infiltrating mononuclear cells in any of the glomeruli. The mice used in these studies were from two different specific pathogen-free colonies, ruling out a colony-specific, infectious agent as the cause of the kidney damage and supporting the conclusion that the pathology has an autoimmune basis.
Figure 5.
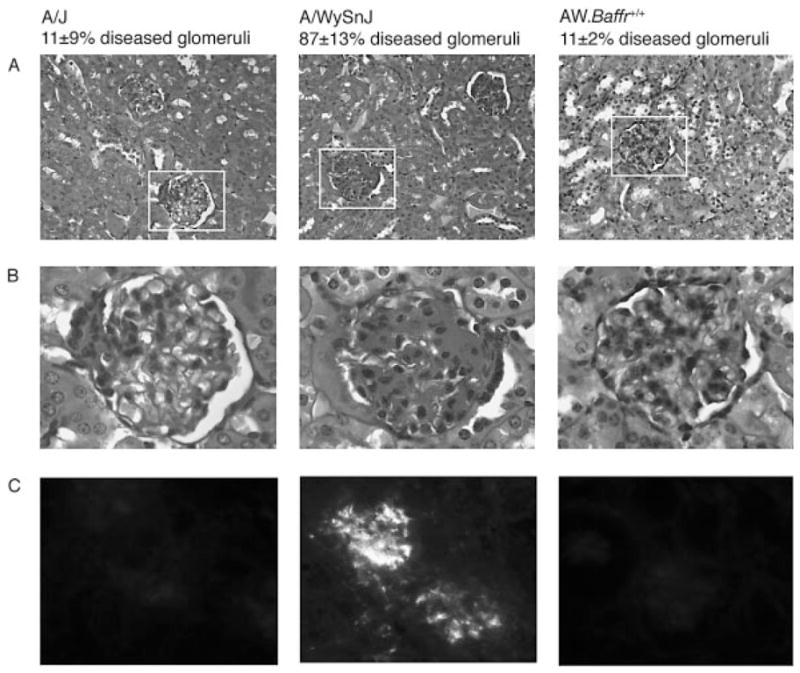
A/WySnJ mice develop renal pathology and accumulate IgG deposits in the glomeruli, whereas A/J and AW.Baffr+/+ mice do not. (A) Kidney sections stained with PAS reagent. (B) Kidney sections within the white boxes in panel (A) were enlarged to show renal pathology. (C) Kidney sections stained with FITC-coupled antibodies to mouse IgG. Histopathological and immunohistochemical analyses were performed on kidneys from mice aged 11–12 months.
Application of a blinded scoring method yielded results showing pathology in 11% of the A/J and AW.Baffr+/+ glomeruli and 87% of the A/WySnJ glomeruli (p<0.01; Table 3). Additionally, of the glomeruli showing pathology, A/WySnJ kidneys showed higher levels of damage as evidenced by disease scores significantly higher than A/J or AW.Baffr+/+ (p<0.01, Table 3). There was a 100% incidence of severe renal pathology in A/WySnJ mice with proteinuria, suggesting that proteinuria was a reliable indicator of renal damage. Immunofluorescence staining identified IgG IC deposited in the glomeruli but not the tubular basement membrane (Fig. 5C). Together, the proteinuria, histopathology, and immunofluorescence data indicate that A/WySnJ mice with circulating IgG autoantibodies and proteinuria developed renal pathology attributable to IC deposition, whereas A/J and AW.Baffr+/+ mice did not. We conclude that the Bcmd-1 allele of Baffr, or a closely linked locus, is responsible for the systemic autoimmune phenotypes in A/WySnJ mice.
Table 3.
Renal pathology in A/WySnJ mice
| Strain | Number of mice examined | Glomeruli with lesions (%)a) | Cumulative glomerular pathology scoreb) | Incidence of renal pathologyc) |
|---|---|---|---|---|
| A/J | 3 | 11 ± 9 | 11 ± 1 | 0/3 (0%) |
| A/WySnJ | 8 | 87 ± 13** | 62 ± 6** | 8/8 (100%) |
| AW.Baffr+/+ | 4 | 11 ± 2 | 18 ± 2 | 0/4 (0%) |
Kidney samples from male and female mice (age 12 months) were prepared, PAS-stained, and scored in blinded fashion. At least 25 glomeruli were examined per section. A pathology score was given to each glomerulus as described in the Materials and methods. A χ2 test gave p<0.01 (**) for comparisons between A/WySnJ and A/J or AW.Baffr+/+ glomeruli.
The severity of the renal pathology was quantified by summing the pathology scores for 25 blindly scored glomeruli. A two-tailed Student’s t-test gave p<0.01 (**) for comparisons between A/WySnJ and A/J or AW.Baffr+/+ scored glomeruli.
Discussion
We have presented evidence linking disrupted BAFF-R function to a systemic autoimmune syndrome in B lymphopenic A/WySnJ mice. These mice developed outward signs of a lupus-like disease as they aged. They also showed the immunological hallmarks and the characteristic renal pathology of lupus. The retroviral gene complementation data establish that a mutation in Bcmd-1 rather than Bik accounts for the B lymphocyte abnormalities in A/WySnJ mice. The genetic studies also implicated Bcmd-1 in the regulation of the lupus-like syndrome, because congenic AW.Baffr+/+ mice did not develop autoimmunity. To our knowledge, this is the first report of a single gene mutation in the Baffr locus that has been linked to lupus susceptibility [1].
Current models to explain the loss of tolerance in B lymphopenic animals would not predict development of systemic autoimmunity in A/WySnJ mice [4]. Chronic BCR stimulation of self-reactive B cells is thought to induce anergy and strong BAFF dependence leading to the deletion of these cells at the T2 stage in a diverse pool of B cells [4, 11, 12]. To explain why B lymphopenic mice and humans commonly develop systemic auto-immunity, others have suggested that an excess of BAFF per B cell could rescue anergic, self-reactive B cells, predisposing to systemic autoimmune disease [1, 4]. However, this excess-BAFF-per-B cell model requires a competent BAFF-R through which excess BAFF could signal survival. The A/WySnJ B cells appeared not to respond to BAFF in vivo [19], so Bcmd-1 is widely considered to be a complete loss-of-function mutation [5]. If Bcmd-1 is a complete loss-of-function mutation, then excessive survival signaling through the BAFF-R in a B lymphopenic environment cannot explain the lupus-like disease in A/WySnJ mice.
Excessive BAFF-R-mediated survival signaling could contribute to the lupus-like disease if Bcmd-1 were a partial loss-of-function mutation. Others found (and we confirmed) that the mutant BAFF-R protein binds BAFF and is correctly expressed in B cell development [17, 25, 26]. Moreover, the A/WySnJ B cell deficiency is not as severe as the deficiency in mice harboring a null Baffr allele, suggesting some residual survival signaling through the mutant BAFF-R [25, 27]. The mutant BAFF-R lacks the C-terminal eight amino acids of the cytoplasmic domain and has 21 additional retrotransposon-encoded residues [16, 18]. Others have suggested that these eight truncated amino acids have no role in signaling [28]. Importantly, the mutant BAFF-R retains the putative TRAF3-binding motif implicated in BAFF-R signaling [29, 30], and we and others found that A/WySnJ B cells showed the NF-κB2 activation that is characteristic of BAFF-R signaling [31, 32]. Moreover, BAFF-R signaling enhanced pro-survival Bcl-2 and Bcl-xL gene expression [5], and we found equal Bcl-2 and greater Bcl-xL gene expression in vivo in A/WySnJ T1 B cells than in A/J T1 B cells [16].
Collectively, these data support the interpretation that the mutant BAFF-R retains some pro-survival signaling function, although it has lost the ability to up-regulate CD21 [25] and down-regulate Bik [16]. That A/WySnJ B cells did apparently not respond to BAFF in vivo [19] might be due to the fact that these cells already had an excess of BAFF due to the B lymphopenic environment, and hence could not further increase their longevity.
BAFF-mediated signaling through transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) is a second factor that may contribute to the rescue of dsDNA-specific B cells in A/WySnJ mice. The A/WySnJ mice make robust B cell responses to TI-II antigens [21, 22], and DNA has a repeating structure like a TI-II antigen. The dsDNA-specific B cells could bind the DNA released from B cells dying due to excess Bik expression [16], and undergo T cell-independent activation to secrete dsDNA-specific IgM. DNA cross-linking of the BCR on dsDNA-specific B cells could lead to DNA internalization, TLR9 binding, and B cell activation and proliferation [33]. In addition, BAFF supports IgM responses to TI-II antigens through TACI [34], and A/WySnJ mice have fully functional TACI receptors. Thus, in the B lymphopenic environment, excess BAFF might act through TACI to promote the survival of dsDNA-specific B cells with cross-linked BCR. The excess BAFF/TACI model predicts that mice harboring a null Baffr allele would develop systemic autoimmunity, a prediction that is currently being tested.
Suboptimal CD21 expression on A/WySnJ B cells is a third factor that may contribute to the failure to delete dsDNA-specific B cells. Homozygous deficiencies in complement components C4A, C4B, and C1q are strong genetic risk factors for SLE [28]. Moreover, Cr2, which encodes the complement receptors 1 (also known as CD35) and 2 (also known as CD21), is a candidate gene for the murine Sle1c lupus susceptibility locus [35]. The CD19/CD21/CD35 complex couples microbial antigen recognition to B cell activation [28]. As the BCR co-receptor, this complex may be important in establishing the threshold for BCR-mediated negative selection [28]. BAFF-R-mediated signals control optimal CD21 expression on maturing B cells, and the Bcmd-1 mutation disrupted this function [25]. In A/WySnJ T2 B cells, the combined BCR plus CD21 signal transmitted by self antigens in the context of complement components might be too weak to trigger apoptosis. B lymphoctes from Baffr-null mice also fail to up-regulate CD21 on their surface [25, 27]. Therefore, the suboptimal CD21 model also predicts that mice harboring a null Baffr allele would develop autoimmunity.
It is not clear how the dsDNA-specific B cells that are retained in the A/WySnJ B cell repertoire switch to produce IgG. SLE pathology is generally attributed to pathogenic IgG autoantibodies, and self-reactive T cells are thought to be required to promote class-switch recombination and somatic hypermutation [1]. Thus, a break in T cell tolerance may also need to occur in A/WySnJ mice. BAFF-R is expressed on a subset of T cell; so it is possible that Bcmd-1 disrupted some T cell function [36, 37]. Alternatively, T cell-independent class switching could occur, since class-switched pathogenic antibodies developed in T cell-deficient, BAFF-Tg mice [38].
Lupus-prone mouse strains have long been recognized as valuable SLE models. Genetic and biological dissection of SLE pathogenesis in these strains has yielded insight into the mechanisms that subvert peripheral B cell self tolerance in humans [1]. Most lupus-prone strains like (NZB×NZW)F1, MRL, and BXSB have B cell hyperplasia, several genetic abnormalities that together give the full lupus-prone phenotype, and no evidence for BAFF-R involvement. In sharp contrast, the A/WySnJ strain has B lymphopenia and the Bcmd-1 allele of the Baffr locus is clearly necessary for the autoimmune phenotype. Experiments are currently underway to determine whether additional A-strain genes are also required. Single gene mutations that subvert peripheral B cell self tolerance are rare. We suggest that the unique features of the A/WySnJ SLE model justify further study of these mice in search of novel insights into the genetic basis of the syndrome, the role of TACI and CD21, the B-1 vs. B-2 cell source of the pathogenic antibodies, the developmental stage when the tolerance breakdown occurs, and how the BCR and BAFF-R signals are integrated to control the process of selection against self-reactive B cells in the peripheral B cell repertoire.
Materials and methods
Mice
Male and female A/J, A/WySnJ, and congenic mice were produced in our specific pathogen-free mouse colony in the Department of Biochemistry, University of Wisconsin-Madison. The mice were maintained at 23°C with 40–60% humidity and 12-h light-dark cycles. Mice studied for B cell subset analyses were 7–10 wk of age. Mice studied for circulating autoimmune antibodies, AFC, proteinuria, and renal pathology were 7–14 months of age. The Institutional Animal Care and Use Committee approved the protocols (protocol A073000-A01149–5-01-04).
DNA isolation and polymerase chain reaction
The production of congenic A/WySnJ mice carrying a wild-type Baffr allele from strain CAST/Ei was described previously [15]. This congenic strain was previously designated AW.Bcmd-1c [15]. Here, the congenic strain is designated AW.Baffr+/+, since Baffr is the commonly recognized gene name for the gene designated Tnfrsf13c. For genotyping, tail DNA was isolated [15]. PCR amplification was performed with one forward primer specific for both Baffr alleles (5′-ACCCTC-CTCAGAAACCCCTCAT-3′), and two reverse primers, one specific for wild-type Baffr (5′-GCCTCCACTGCTGCTATTG-CTC-3′; 170-bp product) and one specific for Bcmd-1 (5′-TGGTCGTAAATACCCTTGGCTC-3′; 235-bp product) [20]. The PCR products were separated on GenePure HiRes agarose (ISC BioExpress, Kaysville, UT).
B cell staining and FACS analysis
For flow cytometric analysis, splenocytes were dissociated into ice-cold Hank’s balanced salt solution with HEPES buffer, and RBC were depleted. Duplicate samples (106 cells/sample) were stained (30–45 min on ice) with optimal amounts of the mAb conjugates in staining buffer (PBS pH 7.3 with 5% heat-inactivated FBS and 0.1% NaN3). Reference samples stained with single-color mAb served as controls to select fluorescence gates and flow cytometer compensation. Stained samples were analyzed on a FACScalibur™ using CELLQuest™ software (BD Biosciences, Franklin Lakes, NJ). The streptavidin-allophycocyanin and streptavidin-Cy-Chrome™ secondary labels, the PE- and biotin-coupled rat mAb to mouse CD23 (clone B3B4), and the FITC-coupled rat mAb to mouse CD21/35 (clone 7G6) were obtained from BD Biosciences. The allophycocyanin-goat polyclonal antibodies to mouse IgM, and the FITC-, PE-, Tricolor-™, allophycocyanin- and biotin-rat IgG2B and IgG2A isotype control mAb were obtained from Caltag Laboratories (Burlingame, CA). PE-coupled rat mAb to mouse CD21/35 was a generous gift from Dr. John Kearney (University of Alabama, Birmingham, AL).
Antibodies to nuclear antigens
An ELISA was performed to quantify serum antibodies to nuclear antigens. The ELISA was performed as described [39], except that plates were coated with 100 μL of fragmented calf thymus DNA grade I (10 μg/mL; Sigma-Aldrich, St. Louis, MO) or histone protein (50 μg/mL; Sigma-Aldrich). Plates were blocked with 2% BSA in PBS, and triplicate 100-μL aliquots of diluted serum samples were added. Plates were washed with 0.05% Tween-20 in PBS. Biotin-coupled goat antibodies to IgM or to IgG (1:500 dilution; Southern Biotechnology, Birmingham, AL), streptavidin-coupled alkaline phosphatase (1:1000 dilution; Southern Biotechnology), and p-nitrophenyl phosphate liquid substrate (Sigma-Aldrich) were used for detection of the serum autoantibodies.
Analysis of antibody-forming cells
An ELISPOT assay was performed to quantify AFC specific for dsDNA as described [40], except that splenocytes were added to triplicate wells at 2.5×105–1.0×106 per well and incubated at 37°C overnight. Biotin-conjugated rat anti-mouse IgM (1:500 dilution; clone 1B4B1; Southern Biotechnology), streptavidin-conjugated HRP (1:2000 dilution; Southern Biotechnology), and 3-amino-9-ethylcarbazole (Sigma-Aldrich) were used for detection. DNA-specific spots from triplicate wells were counted twice on a dissecting microscope, and reported as mean number of AFC per 106 B cells. To determine B cell percentages by flow cytometry, 106 splenocytes from each mouse were stained as above with APC-conjugated goat polyclonal antibodies to mouse IgM (Caltag), PE-coupled rat mAb to mouse IgD (clone 11–26; Southern Biotechnology), and FITC-coupled rat mAb to mouse CD4 (clone GK1.5; Southern Biotechnology).
Assessment of renal pathology
For histopathological evaluation, excised kidneys were formalin-fixed, paraffin-embedded, and sectioned at 5 μm. Kidney sections were stained with PAS stain (StatLab) and counterstained in hematoxylin (Sigma-Aldrich). The sections were examined using a Zeiss Axioskop microscope equipped with a Plan-Neofluar 20×/0.5 objective. Bright field images were acquired with AxioVision 3.0 software controlling an Axiocam digital camera.
The scoring of glomerular lesions was done in blinded fashion. A glomerulus was scored 0 if its size was normal (neither hyper- nor hypo-cellularity), the capillary loops were widely patent, the mesangium had no or minimal PAS-staining deposits, the Bowman’s space was not occluded by PAS-staining deposits or adhesion to Bowman’s capsule or crescent formation, and infiltrating mononuclear cells were not present. A glomerulus was given 1 point for each of these abnormalities: an enlarged size (hyper-cellularity), capillary loops >50% obliterated, PAS-staining deposits in >50% of the mesangium or glomerular tuft, Bowman’s space obstructed by adhesion to Bowman’s capsule or PAS-staining deposits or crescent formation, and infiltrating mononuclear cells [41, 42]. A mouse was considered to have evidence of renal pathology if ≥33% of the glomeruli scored ≥2 [43, 44]. The severity of the renal pathology was quantified by summing the lesion scores for 25 glomeruli per section.
A published immunofluorescence method was employed for the assessment of IgG deposition in the kidney [45]. In brief, kidneys were snap-frozen in OCT compound, cryostat-sectioned at 10 μm, fixed with acetone, and blocked with 2% normal rabbit serum (Jackson Immunoresearch) in 0.5% Triton X-100/PBS. The sections were then incubated overnight at 4°C with FITC-conjugated rabbit antibodies to mouse IgG (Jackson Immunoresearch) diluted 1:100 in blocking solution. Sections were examined using an Eclipse E600 microscope (Nikon) under a 40× objective. Fluorescent images were acquired using a SPOT RTKE camera and software (Diagnostic Instruments).
Proteinuria was quantified by placing a fresh urine sample on an Albustix Reagent Strip for Urinalysis (Bayer Corp., Elkhart, IN). The resulting color was compared to the manufacturer’s calibration chart.
MigRI retroviral vectors
The wild-type Baffr gene was PCR-amplified from an A/J mouse splenic cDNA library. Deep Vent DNA polymerase (New England Biolabs, Beverly, MA) was used to amplify base pairs 8–589 of the BAFF-R mRNA sequence (GenBank ID 15208476; forward primer 5′-GCCCAGACTCGGAACTGT-3′ and reverse primer 5′-AGCCTCCACTGCTGCTATTG-3′). This fragment was cloned into the pCRII-TOPO vector using the TOPO TA PCR Cloning Kit for Sequencing (Invitrogen, Carlsbad, CA). Plasmid DNA was isolated and sequenced at the University of Wisconsin-Madison Biotechnology Center’s DNA Sequence Laboratory. Inserts from error-free clones were excised with EcoRI and subcloned into the EcoRI site of the MigRI vector’s multiple cloning site [46].
Retroviral transduction and chimeric mice
The MigRI and MigRI.BR retroviral particles were produced and A/WySnJ HSC were transduced as previously described [20, 46]. Briefly, purified vector DNA (Wizard Plus Maxipreps; Promega, Madison, WI) was CaPO4-precipitated onto 80–90% confluent monolayers of the Bosc23 packaging cells grown in IMDM supplemented with 10% heat-inactivated FBS (Hyclone, Logan, UT), 100 U/mL penicillin/streptomycin, and 2 mmol/L L-glutamine. Retroviral supernatants were collected 48–72 h later, stored at −70°C, and titered on NIH/3T3 cells.
To produce chimeric mice, the A/WySnJ donors were injected i.v. with 5-fluorouracil (5 mg in 400 μL PBS; Sigma-Aldrich). Five days later, bone marrow cells were collected and cultured (10×106–20×106 cells) in IMDM (4 mL) supplemented with 15% heat-inactivated FBS, 5% WEHI-3B conditioned medium, 100 U/mL penicillin/streptomycin, 2 mmol/L L-glutamine, recombinant mouse IL-3 (6 ng/mL), recombinant mouse IL-6 (10 ng/mL), and recombinant mouse stem cell factor (100 ng/mL; all from Peprotech, Rocky Hill, NJ). After 24 h of culture, the medium was removed, and two sequential retroviral infections separated by 24 h were performed by spinoculation as described [46]. The infected cells were collected 4 h after the second spinoculation, resuspended in PBS, and 0.5×106–1×106 cells were injected i.v. into each lethally irradiated A/WySnJ mouse. Lethal irradiation (1100 rad in two doses, separated by 4 h) was done just prior to bone marrow cell transfer. Splenocytes were collected and analyzed 11–12 wk later.
Supplementary Material
Acknowledgments
We thank Drs. John Kearney, John Cambier, Antonius Rolink, Warren Pear, and Mark Cook for reagents. We are indebted to Dr. Annette Gendron-Fitzpatrick (University of Wisconsin Research Animal Resource Center), Dr. Tom Pugh (GRECC, Madison Veteran’s Hospital), and Dr. Erik Ranheim (University of Wisconsin Medical School, Department of Pathology) for assistance with the renal pathology studies. This work was supported by an NIH Predoctoral Training Grant in Genetics (5T32GM07133) and the Lupus Foundation of America Gina Finzi Memorial Student Fellowship to C.G.M., an NIH Predoctoral Training Grant in Cell and Molecular Biology (T32GM07215) and a Petersen Fellowship from the Department of Biochemistry to I.J.A., and Wisconsin Alumni Research Foundation (WARF) Grant 135-GG99.
Abbreviations
- AFC
antibody-forming cells
- BAFF
B cell-activating factor belonging to the TNF family
- BCL-2
B cell lymphoma
- Bcmd-1
B cell maturation defect-1
- FO
follicular
- IC
immune complex
- MZ
marginal zone
- PAS
periodic acid-Schiff
- SLE
systemic lupus erythematosus
- TACI
trans-membrane activator and calcium modulator and cyclophilin ligand interactor
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
Supporting Information for this article is available at http://www.wiley-vch.de/contents/jc_2040/2008/37817_s.pdf
References
- 1.Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: Multiple immunological phenotypes in a complex genetic disease. Adv Immunol. 2006;92:1–69. doi: 10.1016/S0065-2776(06)92001-X. [DOI] [PubMed] [Google Scholar]
- 2.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 3.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 5.Mackay F, Sierro F, Grey ST, Gordon TP. The BAFF/APRIL system: An important player in systemic rheumatic diseases. Curr Dir Autoimmun. 2005;8:243–265. doi: 10.1159/000082106. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, Bastian H, et al. Cutting edge: A role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10. doi: 10.4049/jimmunol.166.1.6. [DOI] [PubMed] [Google Scholar]
- 7.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 8.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 9.Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, Harris AW. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci USA. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 11.Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, Brink R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, Cyster JG. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 13.Lentz VM, Cancro MP, Nashold FE, Hayes CE. Bcmd governs recruitment of new B cells into the stable peripheral B cell pool in the A/WySnJ mouse. J Immunol. 1996;157:598–606. [PubMed] [Google Scholar]
- 14.Hoag KA, Clise-Dwyer K, Lim YH, Nashold FE, Gestwicki J, Cancro MP, Hayes CE. A quantitative-trait locus controlling peripheral B-cell deficiency maps to mouse chromosome 15. Immunogenetics. 2000;51:924–929. doi: 10.1007/s002510000223. [DOI] [PubMed] [Google Scholar]
- 15.Clise-Dwyer K, Amanna IJ, Duzeski JL, Nashold FE, Hayes CE. Genetic studies of B-lymphocyte deficiency and mastocytosis in strain A/WySnJ mice. Immunogenetics. 2001;53:729–735. doi: 10.1007/s00251-001-0382-z. [DOI] [PubMed] [Google Scholar]
- 16.Amanna IJ, Clise-Dwyer K, Nashold FE, Hoag KA, Hayes CE. Cutting edge: A/WySnJ transitional B cells overexpress the chromosome 15 proapoptotic Blk gene and succumb to premature apoptosis. J Immunol. 2001;167:6069–6072. doi: 10.4049/jimmunol.167.11.6069. [DOI] [PubMed] [Google Scholar]
- 17.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, et al. Baff-R, a newly identified TNF receptor that specifically interacts with baff. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 18.Yan M, Brady JR, Chan B, Lee WP, Hsu B, Harless S, Cancro M, et al. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol. 2001;11:1547–1552. doi: 10.1016/s0960-9822(01)00481-x. [DOI] [PubMed] [Google Scholar]
- 19.Harless SM, Lentz VM, Sah AP, Hsu BL, Clise-Dwyer K, Hilbert DM, Hayes CE, Cancro MP. Competition for BLyS-mediated signaling through Bcmd/BR3 regulates peripheral B lymphocyte numbers. Curr Biol. 2001;11:1986–1989. doi: 10.1016/s0960-9822(01)00598-x. [DOI] [PubMed] [Google Scholar]
- 20.Amanna IJ, Dingwall JP, Hayes CE. Enforced bcl-x(L) gene expression restored splenic B lymphocyte development in BAFF-R mutant mice. J Immunol. 2003;170:4593–4600. doi: 10.4049/jimmunol.170.9.4593. [DOI] [PubMed] [Google Scholar]
- 21.Miller DJ, Hanson KD, Carman JA, Hayes CE. A single autosomal gene defect severely limits IgG but not IgM responses in B lymphocyte-deficient A/WySnJ mice. Eur J Immunol. 1992;22:373–379. doi: 10.1002/eji.1830220213. [DOI] [PubMed] [Google Scholar]
- 22.Miller DJ, Hayes CE. Phenotypic and genetic characterization of a unique B lymphocyte deficiency in strain A/WySnJ mice. Eur J Immunol. 1991;21:1123–1130. doi: 10.1002/eji.1830210506. [DOI] [PubMed] [Google Scholar]
- 23.Hardy RR. B-1 B cell development. J Immunol. 2006;177:2749–2754. doi: 10.4049/jimmunol.177.5.2749. [DOI] [PubMed] [Google Scholar]
- 24.Lentz VM, Hayes CE, Cancro MP. Bcmd decreases the life span of B-2 but not B-1 cells in A/WySnJ mice. J Immunol. 1998;160:3743–3747. [PubMed] [Google Scholar]
- 25.Shulga-Morskaya S, Dobles M, Walsh ME, Ng LG, MacKay F, Rao SP, Kalled SL, Scott ML. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol. 2004;173:2331–2341. doi: 10.4049/jimmunol.173.4.2331. [DOI] [PubMed] [Google Scholar]
- 26.Gorelik L, Cutler AH, Thill G, Miklasz SD, Shea DE, Ambrose C, Bixler SA, et al. Cutting edge: BAFF regulates CD21/35 and CD23 expression independent of its B cell survival function. J Immunol. 2004;172:762–766. doi: 10.4049/jimmunol.172.2.762. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173:2245–2252. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 28.Carroll MC. A protective role for innate immunity in systemic lupus erythematosus. Nat Rev Immunol. 2004;4:825–831. doi: 10.1038/nri1456. [DOI] [PubMed] [Google Scholar]
- 29.Ni CZ, Oganesyan G, Welsh K, Zhu X, Reed JC, Satterthwait AC, Cheng G, Ely KR. Key molecular contacts promote recognition of the BAFF receptor by TNF receptor-associated factor 3: Implications for intracellular signaling regulation. J Immunol. 2004;173:7394–7400. doi: 10.4049/jimmunol.173.12.7394. [DOI] [PubMed] [Google Scholar]
- 30.Xu LG, Shu HB. TNFR-associated factor-3 is associated with BAFF-R and negatively regulates BAFF-R-mediated NF-kappa B activation and IL-10 production. J Immunol. 2002;169:6883–6889. doi: 10.4049/jimmunol.169.12.6883. [DOI] [PubMed] [Google Scholar]
- 31.O’Connor S, Shumway SD, Amanna IJ, Hayes CE, Miyamoto S. Regulation of constitutive p50/c-Rel activity via proteasome inhibitor-resistant IkappaBalpha degradation in B cells. Mol Cell Biol. 2004;24:4895–4908. doi: 10.1128/MCB.24.11.4895-4908.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Debnath I, Roundy KM, Weis JJ, Weis JH. Analysis of the regulatory role of BAFF in controlling the expression of CD21 and CD23. Mol Immunol. 2007;44:2388–2399. doi: 10.1016/j.molimm.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 34.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14:573–582. doi: 10.1016/s1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 35.Boackle SA, Holers VM, Chen X, Szakonyi G, Karp DR, Wakeland EK, Morel L. Cr2, a candidate gene in the murine Sle1c lupus susceptibility locus, encodes a dysfunctional protein. Immunity. 2001;15:775–785. doi: 10.1016/s1074-7613(01)00228-x. [DOI] [PubMed] [Google Scholar]
- 36.Huard B, Arlettaz L, Ambrose C, Kindler V, Mauri D, Roosnek E, Tschopp J, et al. BAFF production by antigen-presenting cells provides T cell co-stimulation. Int Immunol. 2004;16:467–475. doi: 10.1093/intimm/dxh043. [DOI] [PubMed] [Google Scholar]
- 37.Ye Q, Wang L, Wells AD, Tao R, Han R, Davidson A, Scott ML, Hancock WW. BAFF binding to T cell-expressed BAFF-R costimulates T cell proliferation and alloresponses. Eur J Immunol. 2004;34:2750–2759. doi: 10.1002/eji.200425198. [DOI] [PubMed] [Google Scholar]
- 38.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, et al. BAFF and MyD88 signals promote a lupus like disease independent of T cells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates BCL-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci USA. 2000;97:2703–2708. doi: 10.1073/pnas.040577497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitching AR, Holdsworth SR, Ploplis VA, Plow EF, Collen D, Carmeliet P, Tipping PG. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med. 1997;185:963–968. doi: 10.1084/jem.185.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenkranz AR, Mendrick DL, Cotran RS, Mayadas TN. P-selectin deficiency exacerbates experimental glomerulonephritis: A protective role for endothelial P-selectin in inflammation. J Clin Invest. 1999;103:649–659. doi: 10.1172/JCI5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohan C, Morel L, Yang P, Watanabe H, Croker B, Gilkeson G, Wakeland EK. Genetic dissection of lupus pathogenesis: A recipe for nephrophilic autoantibodies. J Clin Invest. 1999;103:1685–1695. doi: 10.1172/JCI5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morel L, Tian XH, Croker BP, Wakeland EK. Epistatic modifiers of autoimmunity in a murine model of lupus nephritis. Immunity. 1999;11:131–139. doi: 10.1016/s1074-7613(00)80088-6. [DOI] [PubMed] [Google Scholar]
- 45.Johnson BA, Ikeda S, Pinto LH, Ikeda A. Reduced synaptic vesicle density and aberrant synaptic localization caused by a splice site mutation in the Rs1h gene. Vis Neurosci. 2006;23:887–898. doi: 10.1017/S0952523806230244. [DOI] [PubMed] [Google Scholar]
- 46.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 47.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol. 2001;167:6834–6840. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 48.Cariappa A, Tang M, Parng C, Nebelitskiy E, Carroll M, Georgopoulos K, Pillai S. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. Immunity. 2001;14:603–615. doi: 10.1016/s1074-7613(01)00135-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.