

Abstract
Motivation: In the pursuits of mechanistic understanding of cell differentiation, it is often necessary to compare multiple differentiation processes triggered by different external stimuli and internal perturbations. Available methods for comparing temporal gene expression patterns are limited to a gene-by-gene approach, which ignores co-expression information and thus is sensitive to measurement noise.
Methods: We present a method for co-expression network based comparison of temporal expression patterns (NACEP). NACEP compares the temporal patterns of a gene between two experimental conditions, taking into consideration all of the possible co-expression modules that this gene may participate in. The NACEP program is available at http://biocomp.bioen.uiuc.edu/nacep.
Results: We applied NACEP to analyze retinoid acid (RA)-induced differentiation of embryonic stem (ES) cells. The analysis suggests that RA may facilitate neural differentiation by inducing the shh and insulin receptor pathways. NACEP was also applied to compare the temporal responses of seven RNA inhibition (RNAi) experiments. As proof of concept, we demonstrate that the difference in the temporal responses to RNAi treatments can be used to derive interaction relationships of transcription factors (TFs), and therefore infer regulatory modules within a transcription network. In particular, the analysis suggested a novel regulatory relationship between two pluripotency regulators, Esrrb and Tbx3, which was supported by in vivo binding of Esrrb to the promoter of Tbx3.
Availability: The NACEP program and the supplementary documents are available at http://biocomp.bioen.uiuc.edu/nacep.
Contact: szhong@illinois.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Embryonic stem (ES) cells are capable of differentiating into all cell types in an adult body, and can be triggered by different external and internal signals (Ivanova et al., 2006). One of the major themes in ES cell research is to find efficient ways of guiding ES cells to differentiate into a desired cell type. We chose to approach this theme by comparing the differentiation processes triggered by different external stimuli or internal perturbations.
Differentiating mouse ES cells by withdrawing certain growth factors [usually leukemia inhibitory factor (LIF)] leads to generating a mixture of all cell types (spontaneous differentiation) (Hong et al., 2009). Alternatively, exposing ES cells to certain growth factors can lead to enrichment of certain cell lineages during differentiation. Examples include retinoid acid (RA)-induced differentiation that enriches neuronal cells (Jones-Villeneuve et al., 1983), and activin-induced differentiation that enriches mesendoderm cells (Sulzbacher et al., 2009). Besides growth factors, repressing individual regulatory proteins can also induce differentiation. Although we previously showed that the repression of a chromatin modeling factor may encourage ES cells to differentiate and express neural markers (Hong et al., 2009), in general, it is not clear whether the repression of ES cell regulators may encourage differentiation toward any specific cell lineages (Lu et al., 2009).
We hypothesized that if two ES cell regulators are in the same regulatory pathway, the temporal transcriptional responses to repressing them should be similar, as opposed to the temporal responses of repressing a third ES regulator that is not in this regulatory pathway or acting to inhibit this pathway. We also hypothesized that if a transcription factor (TF) inhibits a signaling pathway, the temporal responses to repressing this TF may be closer to the transcriptional responses to adding a growth factor that induces this signaling pathway as compared with the temporal responses to repressing another TF. With these hypotheses in mind, we applied the new method called NACEP to compare spontaneous differentiation against eight types of induced differentiation, including one external stimulation (RA induced) and seven internal perturbations (repressing ES cell regulators). The goals of these comparisons are as follows. First, we wish to gain mechanistic insights into how RA treatment leads to differentiation toward the neural lineage. Second, we wish to test whether any of these induced differentiation processes resemble one another, and thus to infer the relative proximities of these regulators in an ES cell regulatory network. These questions inspired us to revisit analytical methods for comparing time-course gene expression data.
To date, there are very few methods for comparing time-course gene expression data. One method suppresses the temporal information and compares the ‘neighborhood’ genes between two conditions (Cheng et al., 2006). Other methods explicitly model the temporal information, but treat every gene independently (Storey et al., 2005; Telesca et al., 2009). For example, one of these methods fits a spline to the time-course data of a gene in each of two experimental conditions, and then it compares the fitted splines. This statistical approach enjoys at least two clear advantages, in that it takes full advantage of the time-course data structure, and it implements human intuition in comparing temporal patterns. The limitation is that every gene is modeled independently, and information such as co-expression is ignored. Since the two splines of a gene have to be fitted with typically a dozen data points or even fewer, the fitted splines are sensitive to biological and technical fluctuations. Compared with traditional two-sample comparison procedures that ignore the time information, the spline method can be even more prone to false positives because a random fluctuation on a data point can have a larger chance of inducing a detectable difference to the splines.
Intuitively, utilizing co-expression information, for example, by using co-clustered genes to stabilize the spline fit of a gene, might largely enhance the fit and thus enable much more accurate identification of temporal differences. We wish to formalize this intuition as a co-expression network based comparison of temporal expression patterns (NACEP).
However, clustering time-course gene expression data by itself is a challenging problem. One group used predetermined gene expression patterns as cluster centers to cluster genes (Schulz et al., 2009). Other teams approached this problem by using a finite mixture model for the clusters and then implementing a spline fit within each cluster (Luan and Li, 2003; Ma et al., 2006). These methods are prominent advances in co-expression analysis of time-course data, but they appear to be far from what is necessary for a co-expression network-based comparison of temporal expression patterns. The outstanding challenges include but are not limited to the following. First, the cluster number has to be preset for these clustering algorithms. Although model-selection criteria such as akaike information criterion (AIC) or Bayesian information criterion (BIC) can in theory be used to judge cluster numbers, in practice the AIC or BIC curves usually do not show clear peaks or charges that are needed to make a decision (Qin, 2006). Second, clustering results are often unstable, in the sense that slight changes to the clustering algorithm or the dataset may generate vastly different clustering results. It is difficult to trust or interpret results that are sensitive to the analytical methods or noise in the data (Quackenbush, 2003). Third, a gene may not have only one function or it may not only participate in one module in the gene regulatory networks (GRNs). Forcing a gene into one cluster makes a strong assumption about the underlying GRNs, and making inferences using such an assumption may defeat the purpose of using network information to improve temporal comparisons. In our opinion, these outstanding difficulties prohibited the invention of a statistical method that explicitly utilizes network information in the identification of genes with different temporal expression patterns. The NACEP model in this article attempts to address these challenges.
2 METHODS
2.1 The NACEP model
NACEP explicitly uses co-expression network information to compare temporal gene expression data. To overcome the difficulties discussed above, NACEP first implements an infinite-mixture model for clustering time-course data. The number of clusters is automatically decided by the data and a Dirichlet process (Antoniak, 1974; MacEachern and Müller, 1998; Neal, 2000; Qin, 2006). Instead of forcing every gene into a cluster, NACEP passes the probabilities of every gene belonging to every cluster into the next step of analysis. In the second step, NACEP infers the temporal pattern of a gene as a weighted average of the temporal patterns of all the clusters, using the probability of assigning this gene to each cluster as the weight of that cluster. Finally, NACEP compares the temporal patterns of a gene between two experimental conditions with a non-parametric test, correcting for multiple hypothesis testing (Fig. 1).
Fig. 1.

NACEP method. NACEP starts with a Dirichlet process-driven clustering of time-course data. Instead of assigning each gene into a particular cluster, NACEP retains the probabilities of this gene to belong to every cluster. These probabilities and the mean expression patterns of every cluster are used in the next step of comparing the temporal expression patterns of a gene.
2.2 An infinite-mixture model for clustering time-course data
NACEP implements an infinite-mixture model for clustering time-course data. The cluster memberships are treated as missing data and are assumed to be generated from a Chinese Restaurant Process (CRP; Qin, 2006). Let C = (C(1),…, C(N)) be the cluster indicator variable, where C(i) = c, 1 ≤c ≤ C denotes that the i-th gene is assigned to the c-th cluster, 1 ≤ i ≤ N. We use |C| to denote the number of clusters present. |C| is unknown. C is treated as missing data in the model.
Given the missing data, the expression levels of a gene are modeled with a mixed-effects model (Luan and Li, 2003). In this mixed-effects model, the cluster mean is modeled as a B-spline. The measured expression level of a gene in this cluster at a time point is modeled as the sum of the cluster mean, a random gene effect and a noise term representing the overall effect of the biological and the technical fluctuations. Let Yijkl be the measured expression level for gene i, under experimental condition j, at time-point tk, from biological or technical replicate l, where i = 1,…, N; j = 1,…, J; k = 1,…, K; l = 1,…, Lk. Following Luan and Li (2003), the expression levels of the c-th cluster are modeled as:
| (1) |
where fcj (tk) is the mean profile of the c-th cluster in the j-th experimental condition and bi ∼ N(0, ϕ2) is the gene effect, which is independent from the measurement error εijkl ∼ N(0, σ2). The smooth function fcj(tk) is modeled as a B-spline, with its basis denoted as X, and
| (2) |
where βcj is the parameter set of the B-spline.
Thus, a generative probabilistic model for all the time-course gene expression data has been completely specified, with a CRP for generating the cluster indicators and a mixed-effects model for generating expression levels under given cluster indicators.
2.3 The Bayesian formulation and a Gibbs sampling algorithm for model inference
To fit the model parameters from data, we rewrote the NACEP model into a Bayesian form and then developed a Gibbs sampling algorithm to estimate the model parameters. To put NACEP into a Bayesian form, we used the theoretical developments of Dirichlet processes (Ferguson, 1973). Generally, if (Θ, B) is a measurable space, on which G0 is a probability measure and α is a positive real number, a stochastic process G is a Dirichlet process with base distribution G0 and concentration parameter α if and only if for any finite partitions (A1, A2,…, Ar) on Θ, (G(A1), G(A2),…, G(Ar)) ∼ DP(αG0(A1), αG0(A2),…, αG0(Ar)). CRP is a special form of Dirichlet process, and a CRP is often used as prior distribution for a Dirichlet process. Neal formulated a model for generating data from a Dirichlet process (Neal, 2000). In Neal's formulation, the data distribution is a mixture of distributions of form F(θ), with the mixing distribution over θ being G. Thus,
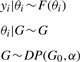 |
(3) |
where yi, i = 1,…, N are the data points and G is a Dirichlet process prior, with concentration parameter α and base distribution G0.
Inspired by Neal's formulation, we rewrite the NACEP model as:
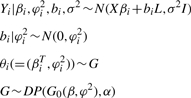 |
(4) |
where L is a column vector of 1s: (1,…, 1)T. We use conjugate priors for α, β and φ
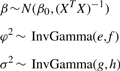 |
(5) |
where e, f, g and h are hyperparameters. Thus, we provided a Bayesian formulation for the NACEP model. Based on this formulation, we developed a Gibbs sampling algorithm to make our model inference (Bush and MacEachern, 1996) (Fig. 2; Supplementary Material).
Fig. 2.

The Gibbs sampler algorithm. The details of the updating strategies and the forms of conditional probabilities are provided in the Supplementary Material.
2.4 Comparison between experimental conditions
How different are the temporal patterns of a gene in two conditions? NACEP quantifies this difference as a weighted average of the differences between the temporal patterns of every cluster, with the posterior probabilities of the gene belonging to every cluster as the weights. Let di be the difference of gene i between two conditions, then
| (6) |
This difference can be efficiently estimated by the Gibbs sampler algorithm using the following procedure. Let s be the index of sampling iterations after burn-in, and s = 1,…, S. di can be estimated by
| (7) |
This estimation procedure saves the step of computing the posterior probabilities of each gene belonging to every cluster.
2.5 Assessing statistical significance
To assess the statistical significance of the difference of temporal patterns of a gene, NACEP obtains the distribution of 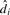 under the null hypothesis by permuting the expression data of matched time points under the two conditions. Following Storey et al. (2005), NACEP uses the permutation to compute the false discovery rate (FDR) for every gene that takes multiple hypothesis testing into consideration. NACEP ranks the genes with their FDR.
under the null hypothesis by permuting the expression data of matched time points under the two conditions. Following Storey et al. (2005), NACEP uses the permutation to compute the false discovery rate (FDR) for every gene that takes multiple hypothesis testing into consideration. NACEP ranks the genes with their FDR.
2.6 Clustering time-course gene expression data
Although NACEP was designed for utilizing co-expression information to enhance the comparison of temporal gene expression patterns, as a byproduct, NACEP provides a handy and potentially powerful tool for clustering time-course gene expression data. The major improvements of NACEP from other time-course data clustering approaches (Luan and Li, 2003; Ma et al., 2006) are 2-fold. First, NACEP employs an infinite-mixture model and thus the cluster number is judged automatically by the Gibbs sampler algorithm. Second, NACEP enables clustering data from more than one experimental condition, by simultaneously fitting temporal profiles within every experimental condition.
To use NACEP as a clustering algorithm, after running the Gibbs sampler, the clustering inference can be made by:
| (8) |
where C is a configuration of gene clustering and F is the set of all such configurations; 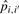 is the estimated probability for genes i and i′ being in the same cluster, which can be obtained by counting the fraction of times c(i) = c(i′) in the iterations; in any given clustering configuration C0, δi,i′(C0) = 1, if genes i and i′ are in the same cluster, otherwise δi,i′(C0) = 0.
is the estimated probability for genes i and i′ being in the same cluster, which can be obtained by counting the fraction of times c(i) = c(i′) in the iterations; in any given clustering configuration C0, δi,i′(C0) = 1, if genes i and i′ are in the same cluster, otherwise δi,i′(C0) = 0.
It should be noted that in the comparison of temporal patterns, NACEP does not force a gene to belong to one cluster; instead all the posterior probabilities of a gene belonging to all clusters are used. Thus, the temporal comparison results are not sensitive to the clustering performance.
2.7 Correlation between TFs
The time-course data of each RNAi experiment was compared with that of the control experiment by NACEP. The NACEP distance of every gene was computed according to Equation (6). Thus, each RNAi experiment produces a NACEP distance vector {d1,…, dN}, where N is the number of genes. The correlation between two TFs is the Pearson correlation between the two NACEP distance vectors of the two RNAi experiments.
3 RESULTS
3.1 Analysis of synthetic data
NACEP was evaluated on synthetic datasets for clustering performance and for comparison of temporal patterns. Although the results of the two tests are presented sequentially below, it is worth noting that NACEP's temporal comparison does not rely on a pre-fixed clustering result (see Discussion).
3.2 Clustering
We compared NACEP with three other clustering methods: K-means, MClust (Fraley and Raftery, 2006) and smoothing spline clustering (SSC) (Ma et al., 2006). Both MClust and SSC use a finite-mixture model for clustering. While MClust assumes that the samples are independent, SSC uses splines to model the time-course data structure. As a clustering method, NACEP can be regarded as an extension of SSC to an infinite-mixture model.
We simulated 100 datasets. Each simulation was composed of four clusters, containing 30, 40, 50 and 30 genes in each cluster. Every gene was measured at 10 time points. Following Ma et al. (2006), the mean expression patterns were simulated with the following functions:
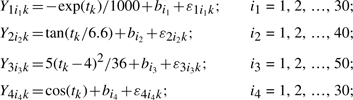 |
where k is the index of time points; i is the index of genes; bi is the random gene effect; εijk is the random measurement error.
Two comparisons were made. First, we compared how often a method incorrectly identifies the cluster number by ‘cluster number prediction error’ (CNPE; Fig. 3B). The BIC was used for MClust and SSC to choose the cluster number. Since K-means cannot automatically determine the cluster number (unless assuming some parametric form of data distribution), we assigned the correct cluster number to K-means and exempted it from the first comparison. Using BIC with SSC (18% CNPE) improved the chances of correctly identifying cluster numbers compared with using BIC with MClust (43% CNPE).
Fig. 3.

Clustering performance. (A) One hundred datasets were simulated. Each dataset contained four clusters. Representative expression values of three genes for each cluster are shown, in plus, open circle and open triangle. (B) Clustering performance was evaluted. CNPE, the proportion of predictions with incorrect cluster numbers, among all simulated datasets. A higher CNPE correlates with worse performance. AMR, the average proportion of genes being misclassified. A higher AMR correlates with worse performance.
Moreover, the results of NACEP (0% CNPE) were further improved from the results of using BIC with SSC (18% CNPE).
Second, when the cluster number was correct, we compared how often a gene is incorrectly clustered [average misclassification rate (AMR), Fig. 3B]. In this comparison, we assigned the correct cluster number to K-means, Mclust and SCC. By doing so, we gave the other algorithms an advantage over NACEP. In this comparison, SSC and NACEP largely outperformed K-means and MClust, consistent with the expectation that explicitly modeling the time-course data structure might boost clustering performance. NACEP further exhibited a 22-fold (1.54/0.0733) improvement of clustering accuracy compared with SSC.
3.3 Comparison of temporal patterns
We compared NACEP with a single-gene-based time-course comparison method called EDGE (Storey et al., 2005).
We did four simulations. In each simulation, time-course gene expression data from two experimental conditions were generated, with 10 time points in each condition. In the first simulation, a total of 1000 genes were simulated. Among these genes, 600 genes were simulated to have the same temporal pattern in the two conditions (Group f, Fig. 4A). The remaining 400 genes had different expression patterns. They were separated into four groups (Groups a–d, Fig. 4A). Their expression patterns differ between the two conditions as follows. Groups a and b had different trends. Group c was generated from gamma functions with different parameters. Group d was generated from sine functions with different phases. The gene groups were assumed to have different variances on their gene effects (bi) and the same variance on the measurement errors (Var(εijk) = 1). We made Var(bi) increase from Group a to Group d, as reflected by the increasing confidence intervals of the mean trajectories in Figure 4A.
Fig. 4.

Cross-condition comparison. (A) Four synthetic datasets. Five to seven groups of genes were simulated in each synthetic dataset, with each group exhibiting either a different pattern (Groups a–e), or a similar pattern (Groups f and g) between the two experimental conditions. Genes in Groups e and g do not form any clusters. Each gene was generated from its own temporal pattern. Group d differs in the two conditions by a phase shift. The cluster averages and their confidence intervals are shown in solid curves and shaded regions, respectively. Expression values of 10 representative genes are shown for each gene group in dots. The standard deviation of the expression values of all genes in a group is shown as a green band. (B) ROC curves. Panels 1–4 correspond to synthetic datasets 1–4. The ROC curves of the best, median and worst performance on 50 simulations are plotted in solid (NACEP) and dashed (EDGE) lines.
To simulate more realistic data, in the second simulation, we increased the variances of the gene effect and the measurement error. To simulate the situations in which some genes cannot be easily clustered (scatter genes), we added a new gene group to the third simulation. This new group (Group g, Fig. 4A) contained 100 genes, each independently generated to have its own temporal pattern, which is the same in the two conditions. Group g genes differ in the two conditions by the average expression level (gene effect bi) and measurement variation (εijk). In order to challenge NACEP even further, in the fourth simulation, we added yet another group of scatter genes (Group e) with different temporal patterns in the two conditions.
Each simulation was independently repeated 50 times. Both NACEP and EDGE were applied to these datasets to detect genes with different trajectories. True and false positives of these predictions are summarized as receiver operating characteristic (ROC) curves in Figure 4B. NACEP out-performed EDGE in all four simulations. Notably, when the false positive rates are small (using high thresholds), EDGE performed not much better than a random decision, by producing a small number of true positives (Fig. 4B). NACEP largely increased the sensitivities under the same specificities of EDGE, causing the ROC curves to shoot up almost vertically to 20% at a 0% false positive rate in all simulations. This indicates a particularly useful feature of NACEP, in that its top predictions are likely to be reliable.
3.4 Analysis of ES cell differentiation
3.4.1 Differential temporal responses between spontaneous and RA-induced differentiation of ES cells
We applied NACEP to identify the genes and pathways that mediate RA-induced differentiation of ES cells. RA is known to facilitate ES cell differentiation and to enrich neurogenic precursor cells among the differentiated cells, although the molecular mechanisms of such an effect remain elusive (Glaser and Brüstle, 2005). We hypothesized that the neurogenic effect of RA is mediated by a set of neurogenic regulatory genes, whose expression patterns are different between RA-induced and spontaneous differentiation. To test this hypothesis, we reanalyzed the data by Ivanova et al. (2006), who subjected mouse ES cells to spontaneous and RA-induced differentiation processes (Ivanova et al., 2006). Gene expression was measured in each differentiation condition every day over a 7-day period.
To preprocess the data, we used Gene Ontology (GO) annotations to obtain the genes involved in transcriptional regulation or signal transduction and filtered out the other genes. We also filtered out the genes with small changes of expression levels in both of the two time series and subjected the remaining 783 genes to NACEP analysis. NACEP's Gibbs sampling computation stabilized after 10 000 iterations and generated 37 clusters (Fig. 5A). Canonical ES cell regulators including Nanog, Oct4, Klf2, Esrrb and Utf1 all showed up in one cluster, lending credibility to the clustering result. Two Ets domain TFs, Etv4 and Etv5, were clustered together with the canonical ES cell regulators, suggesting that these TFs involved in organ morphogenesis might have a neglected role in ES cell regulation. Interestingly, Etv5's DNA binding motif was reported to be enriched in Nanog bound regions (Zhou et al., 2007). Another cluster contained Shh and Gli1, the key ligand and TF of the shh signaling pathway, suggesting the shh pathway might be tightly regulated in both spontaneous and RA-induced differentiation of mouse ES cells.
Fig. 5.

Comparison of RA-induced and spontaneous differentiation of ES cells. (A) Mean expression patterns of 37 clusters in Days 0–7 of two differentiation conditions. (B) A hypothetical regulatory pathway that responds to RA and induces neural differentiation of ES cells.
Comparing the two differentiation processes, NACEP reported 156 genes with different temporal patterns (FDR < 10−5). We then separated these 156 genes into two groups, i.e. the induced (134) and the repressed (22) genes, by RA as compared with spontaneous differentiation. The 22 genes repressed by RA included pluripotency and self-renewal regulators Esrrb, Utf1, Nanog, Klf2 and Oct4. These data are consistent with the notion that RA facilitates differentiation.
The top-ranked RA-induced genes included Gli3, Zic3 and others. GLI3 is one of the three Gli family proteins in mice, which serve as key TFs of the shh pathway. Consistent with this result, Zic3 is a known downstream transcriptional target of Gli family TFs (Mizugishi et al., 2001). These data, together with the result that Shh and Gli were clustered together (Fig. 5A), suggest that the shh pathway genes might be activated by RA and mediate the differentiation of ES cells into neural precursors. Consistent with this hypothesis, the induction of the shh pathway promotes neuronal differentiation from embryoid bodies that are differentiated from ES cells (Vokes et al., 2007). Moreover, in mouse brains, shh activation has been associated with neoplastic growth and development of brain tumors (Flora et al., 2009). Finally, Etv4, which inhibits Shh during limb bud development (Zhang et al., 2009), was identified by NACEP as one of the top-repressed genes by RA. These data are in-line with the hypothesis that the shh pathway is positively regulated during RA-induced differentiation.
The top RA-induced genes also included Nr2f2 and Igf1. Igf1 can serve as a ligand to the insulin receptor (INSR) pathway. Nr2f2 was shown to be recruited to the Igf1 promoter region, modulating its expression (Kim et al., 2009). These data tempted us to hypothesize that RA-induced neuronal differentiation is at least partially mediated by the INSR pathway. Coincidentally, INSR induces neuronal differentiation of neuroendocrine tumors (Dikic et al., 1994), and it is vital for keeping neural stem cells alive (Siegrist et al., 2010). Taken together, these analyses suggest that RA may induce neuronal differentiation through activating shh and INSR pathways (Fig. 5B).
3.4.2 Comparison of temporal expression responses to RNAi reveals placement of TFs in a GRN
A number of ES cell TFs that sustain self-renewal and inhibit differentiation have been identified. Less clear are the interaction relationships of these ES cell TFs, or the full picture of the ES cell GRN. In order to elucidate the GRN of ES cells, biochemical assays including sequential chromatin immunoprecipitation (ChIP) were used to directly assess the interaction of two TFs (Geisberg and Struhl, 2004); co-localization of binding regions (from ChIP-seq or ChIP-chip data) was used to infer TF interactions (Chen et al., 2008), and co-expression information together with protein–protein interaction data were used to infer GRNs (Müller et al., 2008).
We hypothesized that the transcriptomic responses to knockdown of TFs should also contain useful information on the relative placement of these TFs in a GRN. To test this hypothesis, we started by considering a hypothetical situation, wherein there are three TFs (A, B and C), with A and B ‘closer’ in the GRN as compared with C (Fig. 6A). The relative proximity of A and B can be substantiated in the following examples: A and B often interact with each other, forming a dimer to co-bind and co-regulate target genes; C can only interact with A or B through the assistance of other proteins; C is an upstream regulator of A and B; or C independently regulates a set of its own downstream genes. In these hypothetical examples, the genome-wide temporal transcriptional responses to the knockdown of A and B should be similar, as opposed to the temporal responses to C knockdown.
Fig. 6.

Comparison of time-course data of knockdown of seven TFs. (A) A hypothetical gene regulatory pathway. (B) The top 30 NACEP predicted genes with differential temporal patterns between an RNAi condition and the control. (C) Pearson correlation between TF knockdowns. The Pearson correlation was derived from the NACEP distances (di) of all genes between two TF knockdowns. (D) Predicated relative TF placement in a GRN, drawn with Cytoscape (Shannon et al., 2003). The pairwise TF correlations are visualized as the thickness of the edges. Dashed edges represent experimentally verified regulatory interactions.
Using this idea, we applied NACEP to a set of time-course gene expression data generated by seven RNAi experiments (Ivanova et al., 2006), namely the gene expression data of Days 0–7 after the knockdown of Nanog, Oct4, Sox2, Esrrb, Tbx3, Tcl1 and Ccnb1ip1. We compared the temporal responses with each RNAi experiment to the control data, i.e. the gene expression data of wild-type ES cells on matched time (Days 0–7). In every comparison (by NACEP), the genes with a different expression pattern between the RNAi and the control conditions were identified (Fig. 6B). To quantify the similarity of the genes affected by two RNAi experiments, we used the Pearson correlation of the NACEP distances of the two RNAi experiments (see Methods). The Pearson correlation was regarded as a similarity/proximity metric between the two TFs on which the RNAi were performed (Fig. 6C). To obtain a global view of the proximity of all TFs in the GRN, we clustered the TFs by the Pearson's correlations. For visualization purposes, an edge was drawn between two TFs if their correlation was beyond an ad hoc threshold of 0.54 and if the width of the edge was proportional to the correlation between the two connecting TFs (Fig. 6D). This result indicates that Nanog, Oct4, Sox2 and Esrrb may form a heavily connected regulatory module, and Tbx3, Tcf1 and Ccnb1ip1 are attached to the Nanog-containing module through a few specific links. To check whether the predicted GRN structure was sensitive to the choice of Pearson's correlation as the similarity/proximity metric or sensitive to the use of all genes, we applied another similarity metric (Supplementary Fig. S1A) and restricted the calculation to the top 5% of genes most strongly affected by all RNAi experiments (Supplementary Fig. S1B), and we found that the predicted TN structure was robust. This analysis based solely on RNAi transcriptomes identified the interactions among Nanog, Oct4, Sox2, Esrrb and Tbx3 consistent with evidence drawn from protein–protein interaction, protein–DNA binding data, and mutation analysis of TF binding sites (dashed edges, Fig. 6D) (Boyer et al., 2005; Chen et al., 2008; Han et al., 2010; Niwa et al., 2009; Pirity and Dinnyes, 2010; van den Berg et al., 2008; Zhang et al., 2008). This analysis failed to predict the transcriptional regulatory relationship between Oct4 and Tcl1 (Matoba et al., 2006), which might be explained by the hypothesis that Tcl1 only specifically regulates a much smaller subset of genes than Oct4, and thus Tcl1 RNAi only reflects a small subset of transcriptomic changes downstream to Oct4 RNAi. The strong correlation of the temporal responses to knockdowns of Esrrb and Tbx3 (Fig. 6D) predicts the proximity of these two TFs in the GRN. Further experiments are needed to test whether these TFs could directly interact with each other or if one is under the transcriptional control of the other. Interestingly, ChIP-seq data showed that there were two Esrrb binding sites near the Tbx3 gene, at 1k bp upstream and 100 bp downstream to the transcription start site of Tbx3 (Supplementary Fig. S2).
4 DISCUSSION
Comparative biology plays a central role in biological discovery. Most, if not all, principles in biology are proved with comparative experiments or in contrasts of observations. As the capacities of making genome-wide measurements increase, it becomes a typical exercise to monitor a biological process by taking genome-wide measurements at multiple time points during this process. Time-course gene expression data have become a common data type. As of April 12, 2010, Gene Expression Omnibus (GEO) has documented 1796 time-course data series on 118 measurement platforms in over 50 species. These data treasures await adequate analysis tools.
4.1 Clustering time-course data
The purpose of clustering analysis is usually to discover co-expression patterns that can be translated to biological knowledge or new hypotheses (Thalamuthu et al., 2006). However, clustering remains a difficult problem, as exemplified by ad hoc criteria for choosing optimal clusters and results being sensitive to the initial conditions. As a result, the applications of clustering analyses of expression data are limited by strong noise in the results. Some genes known to be involved in a particular pathway are invariably missed, whereas other apparently unrelated genes exhibit expression profiles that are strikingly similar to bona fide pathway components (Quackenbush, 2003). To address these issues, new methods are needed to simultaneously tackle at least two methodological challenges. First, the cluster number has to be intelligently determined. Second, the time-course nature of the data has to be explicitly utilized. The NACEP method represents an attempt toward these goals. The heart of this method is a generative probabilistic model with a Dirichlet process (Neal, 2000) generating the clusters and a mixed-effects model (Wang, 1998) with a B-spline (Luan and Li, 2003) mean generating the gene expression patterns. NACEP can potentially be generalized to handle non-time-course data by not requiring fcj(tk) in Equation (1) to be a time-dependent function.
4.2 Comparison of temporal patterns
The main function of NACEP is to compare time-course data between two experimental conditions. At least two questions have to be addressed to make an effective comparison. First, how should the time-course data structure be utilized so as to increase the sensitivity and robustness of the comparison? Second, how can we minimize detection errors introduced by noise in the measurements? The first challenge was elegantly addressed by a method called EDGE (Storey et al., 2005). EDGE models the time-course data with splines and compares the splines between two conditions one gene at a time. Since the data points for a single gene are often limited, noise in the data can strongly influence the comparison result. This imposes a pressing need to address the second question. It appears to be difficult to extend EDGE to incorporate prior information of pathways and networks to improve the comparison. A major difficulty is that the accurate and complete pathway information is typically unavailable. The NACEP method utilizes the clustering information to assist the detection of different time-course expression patterns in a soft way. The premise of this method is that the gene clusters are correlated with regulatory pathways, but that any clustering result cannot be fully trusted. To detect the differential expression of a gene, NACEP borrows information from every other gene. The amount of information borrowed is proportional to the probability that the other gene will co-cluster with the gene under comparison. Thus, the detection of differential expression does not reply on a prefixed clustering result.
4.3 GRN structure and TF knockdown
Although gene expression responses to knockdown of TFs are often measured, such data were often used to identify the transcriptional targets of the inhibited TFs. To our knowledge, there is as yet no principled approach to infer the interaction relationships among the inhibited TFs, except in some special cases in which one TF is the transcriptional target of another. As a proof of principle, this work demonstrated that the temporal transcriptional responses to the knockdown of a set of TFs could be used to identify the interaction relationships of these TFs, as well as their relative proximity in the GRN. Our data suggest that Esrrb may contribute to maintaining pluripotency through transcriptionally regulating Tbx3, a T-box transcriptional repressor. We were also interested in testing whether any of the TFs maintain pluripotency through, at least partially, inhibiting the RA pathway and its downstream genes. The differences between RA treatment and any TF knockdown are larger than the differences between any two TF knockdowns (Fig. 6C; Supplementary Fig. S1), suggesting that the seven TFs included in our analyses are likely involved in pathways independent to the RA pathway.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dan Xie, Drs Guixian Lin, Xuming He, Kimberly A. Hughes, Jianhua Guo and Ningzhong Shi for useful discussions.
Funding: NSF DEB 08-48386, NSF DBI 08-45823, NIH R01 DK082605, NIH supplement to GM058686-0851.
Conflict of Interest: none declared.
REFERENCES
- Antoniak CE. Mixtures of Dirichlet processes with applications to Bayesian nonparametric problems. Ann. Stat. 1974;2:1152–1174. [Google Scholar]
- Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush CA, MacEachern SN. A semiparametric Bayesian model for randomised block designs. Biometrika. 1996;83:275–285. [Google Scholar]
- Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cheng C, et al. MARD: a new method to detect differential gene expression in treatment-control time courses. Bioinformatics. 2006;22:2650–2657. doi: 10.1093/bioinformatics/btl451. [DOI] [PubMed] [Google Scholar]
- Dikic I, et al. PC12 cells overexpressing the insulin receptor undergo insulindependent neuronal differentiation. Curr. Biol. 1994;4:702–708. doi: 10.1016/s0960-9822(00)00155-x. [DOI] [PubMed] [Google Scholar]
- Ferguson TS. A Bayesian analysis of some nonparametric problems. Ann. Stat. 1973;1:209–230. [Google Scholar]
- Flora A, et al. Deletion of Atoh1 disrupts sonic hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science. 2009;326:1424–1427. doi: 10.1126/science.1181453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C, Raftery AE. Technical Report. Vol. 504. Department of Statistics, University of Washington; 2006. MCLUST Version 3 for R: Normal mixture modeling and model-based clustering. [Google Scholar]
- Geisberg JV, Struhl K. Quantitative sequential chromatin immunoprecipitation, a method for analyzing co-occupancy of proteins at genomic regions in vivo. Nucleic Acids Res. 2004;32:e151. doi: 10.1093/nar/gnh148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Brüstle O. Retinoic acid induction of ES-cell-derived neurons: the radial glia connection. Trends Neurosci. 2005;28:397–400. doi: 10.1016/j.tins.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Han J, et al. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature. 2010;463:1096–1100. doi: 10.1038/nature08735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, et al. Dissecting early differentially expressed genes in a mixture of differentiating embryonic stem cells. PLoS Comput. Biol. 2009;5:e1000607. doi: 10.1371/journal.pcbi.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova N, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- Jones-Villeneuve EM, et al. Retinoic acid-induced neural differentiation of embryonal carcinoma cells. Mol. Cell. Biol. 1983;3:2271–2279. doi: 10.1128/mcb.3.12.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, et al. Chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) regulates growth and patterning of the postnatal mouse cerebellum. Dev. Biol. 2009;326:378–391. doi: 10.1016/j.ydbio.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, et al. Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature. 2009;462:358–362. doi: 10.1038/nature08575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan Y, Li H. Clustering of time-course gene expression data using a mixed-effects model with B-splines. Bioinformatics. 2003;19:474–482. doi: 10.1093/bioinformatics/btg014. [DOI] [PubMed] [Google Scholar]
- Ma P, et al. A data-driven clustering method for time course gene expression data. Nucleic Acids Res. 2006;34:1261–1269. doi: 10.1093/nar/gkl013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEachern SN, Müller P. Estimating mixture of Dirichlet process models. J. Comput. Graph. Stat. 1998;7:223–238. [Google Scholar]
- Matoba R, et al. Dissecting Oct3/4-regulated gene networks in embryonic stem cells by expression profiling. PLoS ONE. 2006;1:e26. doi: 10.1371/journal.pone.0000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, et al. Molecular properties of Zic proteins as transcriptional regulators and their relationship to GLI proteins. J. Biol. Chem. 2001;276:2180–2188. doi: 10.1074/jbc.M004430200. [DOI] [PubMed] [Google Scholar]
- Müller FJ, et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature. 2008;455:401–405. doi: 10.1038/nature07213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal RM. Markov chain sampling methods for Dirichlet process mixture models. J. Comput. Graph. Stat. 2000;9:249–265. [Google Scholar]
- Niwa H, et al. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- Pirity MK, Dinnyes A. Tbx3: another important piece fitted into the pluripotent stem cell puzzle. Stem Cell Res. Ther. 2010;1:12. doi: 10.1186/scrt12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZS. Clustering microarray gene expression data using weighted Chinese restaurant process. Bioinformatics. 2006;22:1988–1997. doi: 10.1093/bioinformatics/btl284. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Genomics. Microarrays — guilt by association. Science. 2003;302:240–241. doi: 10.1126/science.1090887. [DOI] [PubMed] [Google Scholar]
- Schulz H, et al. The FunGenES database: a genomics resource for mouse embryonic stem cell differentiation. PLoS ONE. 2009;4:e6804. doi: 10.1371/journal.pone.0006804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist SE, et al. Inactivation of both foxo and reaper promotes long-term adult neurogenesis in Drosophila. Curr. Biol. 2010;20:643–648. doi: 10.1016/j.cub.2010.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, et al. Significance analysis of time course microarray experiments. Proc. Natl Acad. Sci. USA. 2005;102:12837–12842. doi: 10.1073/pnas.0504609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzbacher S, et al. Activin A-Induced differentiation of embryonic stem cells into endoderm and pancreatic progenitors - the influence of differentiation factors and culture conditions. Stem Cell Rev. Rep. 2009;5:159–173. doi: 10.1007/s12015-009-9061-5. [DOI] [PubMed] [Google Scholar]
- Telesca D, et al. Differential expression and network inferences through fun-ctional data modeling. Biometrics. 2009;65:793–804. doi: 10.1111/j.1541-0420.2008.01159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalamuthu A, et al. Evaluation and comparison of gene clustering methods in microarray analysis. Bioinformatics. 2006;22:2405–2412. doi: 10.1093/bioinformatics/btl406. [DOI] [PubMed] [Google Scholar]
- van den Berg DLC, et al. Estrogen-related receptor beta interacts with Oct4 to positively regulate Nanog gene expression. Mol. Cell. Biol. 2008;28:5986–5995. doi: 10.1128/MCB.00301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, et al. Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development. 2007;134:1977–1989. doi: 10.1242/dev.001966. [DOI] [PubMed] [Google Scholar]
- Wang Y. Mixed effects smoothing spline analysis of variance. J. R. Stat. Soc. B. 1998;60:159–174. [Google Scholar]
- Zhang X, et al. Esrrb activates Oct4 transcription and sustains self-renewal and pluripotency in embryonic stem cells. J. Biol. Chem. 2008;283:35825–35833. doi: 10.1074/jbc.M803481200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. FGF-regulated Etv genes are essential for repressing Shh expression in mouse limb buds. Dev. Cell. 2009;16:607–613. doi: 10.1016/j.devcel.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, et al. A gene regulatory network in mouse embryonic stem cells. Proc. Natl Acad. Sci. USA. 2007;104:16438–16443. doi: 10.1073/pnas.0701014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.