

Abstract
Current drugs used in the treatment of Parkinson's disease (PD), for example, L-DOPA and dopamine agonists, are very effective at reversing the motor symptoms of the disease. However, they do little to combat the underlying degeneration of dopaminergic neurones in the substantia nigra pars compacta (SNc) and their long-term use is associated with the appearance of adverse effects such as L-DOPA-induced dyskinesia. Much emphasis has therefore been placed on finding alternative non-dopaminergic drugs that may circumvent some or all of these problems. Group III metabotropic glutamate (mGlu) receptors were first identified in the basal ganglia a decade ago. One or more of these receptors (mGlu4, mGlu7 or mGlu8) is found on pre-synaptic terminals of basal ganglia pathways whose overactivity is implicated not only in the generation of motor symptoms in PD, but also in driving the progressive SNc degeneration. The finding that drugs which activate group III mGlu receptors can inhibit transmission across these overactive synapses has lead to the proposal that group III mGlu receptors are promising targets for drug discovery in PD. This paper provides a comprehensive review of the role and target potential of group III mGlu receptors in the basal ganglia. Overwhelming evidence obtained from in vitro studies and animal models of PD supports group III mGlu receptors as potentially important drug targets for providing both symptom relief and neuroprotection in PD.
Keywords: basal ganglia, group III mGlu receptors, metabotropic glutamate receptors, motor symptoms, neurodegeneration, Parkinson's disease, substantia nigra
Introduction: unmet therapeutic needs in Parkinson's disease
Parkinson's disease (PD) is the second most common neurological disorder after Alzheimer's and is characterized by motor disturbances such as a resting tremor, slowness of movement (bradykinesia), difficulty in initiating movements (akinesia), rigidity and postural instability. Alongside these classical motor symptoms, associated problems such as depression, bladder dysfunction, gastrointestinal disturbances and pain may further impair the quality of life of individuals with PD. Although these non-motor symptoms are now starting to receive the recognition and attention they deserve (e.g. Chaudhuri and Schapira, 2009), this review will explore recent advances in targeting group III metabotropic glutamate (mGlu) receptors for the treatment of the classical motor symptoms.
The motor symptoms largely arise through the progressive degeneration of dopaminergic neurones in the substantia nigra pars compacta (SNc). These midbrain neurones project to forebrain regions, most notably the striatum, where the release of dopamine serves to regulate cortically driven firing within the basal ganglia thalamocortical motor circuits to ensure proper planning and execution of movement (Lang and Lozano, 1998). Current treatments for PD focus on reinstating striatal dopaminergic transmission through the use of the dopamine precursor, L-DOPA or dopamine agonists such as ropinirole or bromocriptine. The introduction of L-DOPA in the mid-1960s was undoubtedly a significant step forward, bringing effective relief of early stage motor symptoms to millions of PD patients worldwide. Similar symptom relief is now also provided by the range of available dopamine agonists. However, these drugs do little to combat the progressive degeneration of dopaminergic neurones in the nigrostriatal tract that characterizes the pathology of PD. This failure affects patients' long-term health since the increasing doses of drug required to stabilize worsening symptoms evoke disabling adverse effects including: L-DOPA-induced dyskinesia (LID) which are involuntary hyperkinetic movements of a choreic, dystonic or ballistic nature (Marsden and Parkes, 1977; Fabbrini et al., 2007; Jenner, 2008), psychosis (Zahodne and Fernandez, 2008) and aberrant reward-seeking behaviour (Antonini and Cilia, 2009).
The prevalence of PD increases with age. In a European survey of nearly 15 000 participants aged 65 years and over, the prevalence of PD (per 100 population of age-matched individuals) increased with age from 0.6 (65–69 years) up to 3.6 (80–84 years) (de Rijk et al., 1997). With the current global demographic revolution, the proportion of persons aged 60 years or over is expected to double to 22% by 2050 (Department of Economic and Social Affairs, 2009). The associated predicted increase in the incidence of PD has led the World Health Organization to identify this condition as an important public health issue that will place significant burden on health care resources worldwide (Janca, 2002). Given the failings of current treatments for PD, it is not surprising that much emphasis has been placed on finding alternative, non-dopaminergic treatments that may offer symptom relief while also slowing the progressive degeneration and possibly helping to stave off dyskinesia.
Abnormal firing of basal ganglia circuits in PD: contribution to clinical signs and progressive nigral degeneration
The loss of striatal dopamine innervation in PD produces opposing downstream changes in firing of the two main basal ganglia motor pathways by virtue of their distinct dopamine receptor populations (Smith et al., 1998; Blandini et al., 2000; Figure 1). In the direct pathway, loss of dopamine D1-like receptor stimulation leads to reduced firing of GABAergic striatal efferents to the basal ganglia output regions, the substantia nigra pars reticulata (SNr) and the internal globus pallidus (GPi). In contrast, in the indirect pathway, loss of the inhibitory drive normally maintained through dopamine D2-like receptor stimulation leads to increased firing in the GABAergic striatopallidal pathway, which in turn inhibits GABAergic drive to the subthalamic nucleus (STN), leading to increased firing of the glutamatergic STN efferents to the SNr/GPi (Hirsch et al., 2000). This abnormal firing of direct and indirect circuits culminates in a net increase in prevailing glutamate levels in SNr/GPi which drives inhibition of thalamocortical feedback, resulting in the motor deficits seen in PD (Obeso et al., 2000, 2008). Compensating for overactivity of these pathways may therefore combat the motor symptoms in PD. Indeed, the positive clinical outcomes including alleviation of motor deficits and reduction in LIDs seen with surgical procedures such as subthalamotomy, pallidotomy or deep brain stimulation of the STN or GPi to effectively switch off the basal ganglia output regions (Lozano, 2003; Rodriguez et al., 2007; Benabid et al., 2009) strengthen the premise that correcting abnormal firing, particularly in the indirect pathway, holds great promise. However, a pharmacological means of normalizing firing of these pathways would have far more widespread application than surgical procedures.
Figure 1.
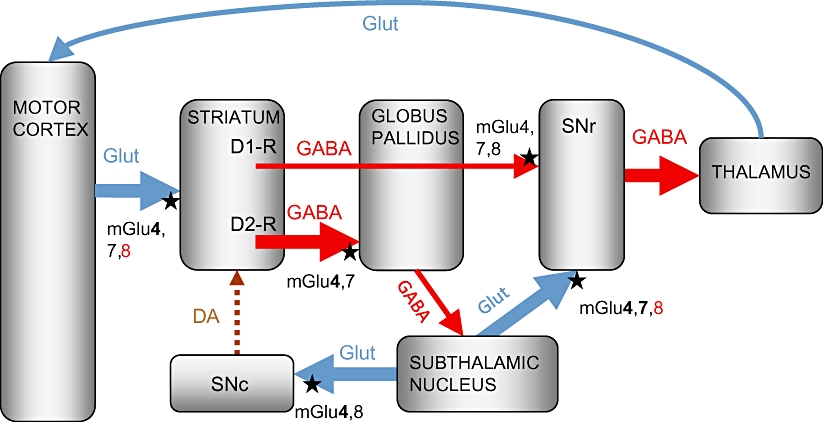
Schematic representation of the parkinsonian basal ganglia motor loop, showing the location of group III metabotropic glutamate (mGlu) receptors. The loss of striatal dopamine innervation following substantia nigra pars compacta (SNc) degeneration in Parkinson's disease (PD) produces opposing downstream changes in firing of the two main basal ganglia pathways. Thick lines indicate overactive pathways; thin lines indicate underactive pathways; dotted lines indicate degenerated pathways. In the direct pathway, loss of dopamine D1-like receptor stimulation reduces firing of GABAergic striatal efferents to the substantia nigra pars reticulata (SNr), while in the indirect pathway, loss of inhibitory D2-like receptor stimulation leads to increased firing in the GABAergic striatopallidal pathway, which in turn inhibits GABAergic drive to the subthalamic nucleus (STN), leading to increased firing of the glutamatergic STN efferents to the SNr and SNc. The increased glutamatergic innervation of the SNr ultimately drives inhibition of thalamocortical feedback, resulting in the motor deficits seen in PD, while the increased glutamatergic innervation in the SNc may contribute to the progressive SNc degeneration. Group III mGlu receptors (mGlu4, 7 or 8), indicated by black stars, are found on pre-synaptic terminals of intrinsic basal ganglia pathways where their activation may serve to restrict transmitter release. Receptor subtypes indicated in bold have been confirmed as functional, while those in red have been shown to lack function. DA, dopamine; Glut, glutamate.
The STN not only projects to the basal ganglia output regions but also sends direct projections to the SNc (Smith et al. 1990; Iribe et al., 1999; Figure 1). Therefore, increased firing of the STN under parkinsonian conditions may lead to a parallel increase in glutamate release in the SNc. Although not thought to be a primary trigger for the degeneration of dopaminergic neurones in PD, which is probably a multifactorial process involving mitochondrial dysfunction, inflammation, altered protein handling and oxidative stress (Schulz, 2007; Banerjee et al., 2009), this predicted rise in glutamate release in the SNc is implicated in the subsequent progression of cell death in PD (Blandini et al., 1996; Rodriguez et al., 1998). Compensating pharmacologically for overactivity of pathways within the indirect basal ganglia circuit may therefore slow progression of degeneration in PD in addition to combating the symptoms as outlined above. Preclinical studies in rats showing that inactivation of the STN, through chemical (kainic acid) lesioning, reduces nigrostriatal tract damage induced by the dopaminergic toxin, 6-hydroxydopamine (6-OHDA) (Piallat et al., 1996) support this suggestion. By virtue of their distribution, function and successful targeting so far in preclinical studies, group III mGlu receptors are looking like promising candidates to meet these expectations.
Localization and functional effects of group III mGlu receptors in basal ganglia circuitry
Group III mGlu receptors are one of three groups of G-protein-coupled mGlu receptor, all of which are found within the basal ganglia circuitry and have been shown to hold promise as targets for treating PD (reviewed in Conn et al., 2005). Herein, the nomenclature used for these receptors conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2008). These three groups differ in their sequence homology, agonist and antagonist pharmacology and in their signal transduction. Group I mGlu receptors, which include mGlu1 and mGlu5, couple through Gq/11, leading to phosphoinositide hydrolysis and enhanced neuronal excitation (Conn and Pin, 1997). Blockade of these receptors, either by the use of antagonists or negative allosteric modulators, has shown great promise in the treatment of PD and related dyskinetic side effects in rodent and primate models of PD (Rylander et al., 2009; Yamamoto and Soghomonian, 2009; Johnston et al., 2010). On the other hand, activation of group II mGlu receptors (comprising mGlu2 and mGlu3), which couple through Gi/Go leading to inhibition of neuronal transmission, has also proven effective in some experimental models of PD (Dawson et al., 2000; Murray et al., 2002). However, in light of recent advances in available compounds targeting group III mGlu receptors (e.g. Niswender et al., 2008b; Engers et al., 2009; Williams et al., 2009), these receptors are starting to attract increased attention. This review will therefore provide a timely, comprehensive account of current evidence surrounding the target potential of group III mGlu receptors in PD.
Group III mGlu receptors comprise four subtypes, mGlu4, mGlu6, mGlu7 and mGlu8, with all but mGlu6 playing important neuromodulatory roles in the brain (Conn and Pin, 1997). These Gi/Go-coupled receptors are found predominantly on pre-synaptic terminals of GABAergic and glutamatergic neurones where they are involved in regulating synaptic transmission, most likely through inhibition of voltage-gated calcium entry required for triggering transmitter release (Trombley and Westbrook, 1992; Conn and Pin, 1997). These receptors have also been demonstrated at post-synaptic sites in some brain regions where their signalling through activation of G-protein coupled inwardly rectifying potassium channels is expected to produce membrane hyperpolarization (Saugstad et al., 1996).
Localization of group III mGlu receptors within the basal ganglia has been unravelled to some extent through a combination of in situ hybridization, immunohistochemistry and electron microscopy studies, with each of the three receptors of interest (mGlu4, 7 and 8) exhibiting a predominantly pre-synaptic distribution. The most accepted distribution to date is summarized in Figure 1. In fact, our understanding of the distribution of mGlu4 and mGlu7 receptors has not changed significantly in the last decade (refer to Rouse et al., 2000 for a summary at that time). Both mGlu4 and mGlu7 receptors have been found on terminals of the glutamatergic corticostriatal pathway which, although not discussed above, is considered to be overactive in PD and thereby contribute to increased firing of the indirect BG pathway (Obeso et al., 2008). mGlu4 and mGlu7 receptors are also found on terminals of the GABAergic striatopallidal and striatonigral pathways (Bradley et al., 1999; Kosinski et al., 1999; Corti et al., 2002) as well as on excitatory (presumed glutamatergic) terminals in the SNr (Kosinski et al., 1999; Corti et al., 2002). While the SNr receives excitatory inputs not only from the STN but also from the pedunculopontine nucleus and frontal cortex (Carter, 1982; Kita and Kitai, 1987; Di Loreto et al., 1992), the presence of mGlu4 and mGlu7 mRNA in the STN (Testa et al., 1994; Kosinski et al., 1999; Messenger et al., 2002) strongly supports their expression on terminals of STN efferents in the so-called subthalamonigral pathway. The distribution of mGlu8 receptors in the basal ganglia is the least well characterized of all. Expression of mRNA encoding mGlu8 receptors has been reported in the cortex, striatum and STN (Messenger et al., 2002), while our unpublished findings reveal moderate to high levels of corresponding mGlu8 immunoreactivity in the striatum and SNr. Therefore, it remains a possibility that mGlu8 receptors are found on pre-synaptic terminals of corticostriatal, striatonigral or subthalamonigral pathways.
Although group III mGlu receptors are considered to be predominantly pre-synaptic within the basal ganglia, very low levels of mGlu7 immunoreactivity have been found at post-synaptic dendritic sites in both the GP (Bradley et al., 1999; Kosinski et al., 1999) and striatum (Kosinski et al., 1999). However, electrophysiological studies found no evidence for a post-synaptic action of group III mGlu receptors in the GP (Valenti et al., 2003) and any post-synaptic action in the striatum remains to be determined. At the present time, therefore, focus remains fixed on the potential of the pre-synaptic receptors.
While the existence of group III mGlu receptors on pre-synaptic terminals of pathways that are overactive in PD (corticostriatal, striatopallidal and subthalamonigral) is promising, demonstrating a functional role for each receptor in regulating transmission across these synapses is essential to support their potential as therapeutic targets for normalizing the activity of basal ganglia pathways in PD. With the lack of subtype-selective antagonists available, the only avenue open has been to explore the actions of subtype-selective agonists. That said, the task of developing subtype-selective agonists in the group III mGlu field has proven difficult and it was as recent as 2003 when the first subtype-selective agent, in this case a positive allosteric modulator (PAM) of mGlu4 receptors, N-phenyl-7-(hydroxylimino) cyclopropa[b]chromen-1a-carboxamide (PHCCC), was identified (Maj et al., 2003; Marino et al., 2003). The selectivity of PHCCC along with that of the other compounds discussed below that have been used to pharmacologically probe the actions of group III mGlu receptors in the basal ganglia are documented in Table 1.
Table 1.
Potencies (EC50 in µM) of agonists or positive allosteric modulators at indicated group III mGlu receptors in cell expression systems
| mGlu4 | mGlu6 | mGlu7 | mGlu8 | Other mGlu targets | Reference | |
|---|---|---|---|---|---|---|
| AGONISTS | ||||||
| L-AP4 | 0.2–1 | 0.2–0.9 | >100 | 0.06–0.9 | >1000 (mGlu1,5,2 & 3) | Cartmell and Schoepp, 2000 |
| ACPT-I | 1.7 | 10.6 | 281 | 5 | – | Beurrier et al., 2009 |
| LSP1-2111 | 2.2 | 1.7 | 53 | 66 | >100 (mGlu1,5,2) | Beurrier et al., 2009 |
| LSP1-3081 | 0.16 | 3.3 | 419 | 0.5 | n.e | Cuomo et al., 2009 |
| AMN082# | >10 (<20%) | >10 (none) | 0.06–0.3 (90–140%) | >10 (<20%) | >10 (mGlu1,5,2 & 3) | Mitsukawa et al., 2005 |
| (S)-DCPG | 8.8 | 3.6 | >100 | 0.03 | >100 (mGlu2,3,5) 32 (IC50 at mGlu1) | Thomas et al., 2001 |
| POSITIVE ALLOSTERIC MODULATORS | ||||||
| PHCCC (with 5 µM l-glutamate) | 2.8 | >10 | >40 | >10 | 3.4 (IC50 at Glu1) 30% max inhibition | Maj et al., 2003 |
| VU0155041* (with EC20 of l-glutamate) | 0.7–0.8 | n.e | n.e | n.e | n.e | Niswender et al., 2008a |
The main group III target for each agent is highlighted in bold while values for known activities at other mGlu receptors are shown for comparison.
VU0155041 also displays partial agonist activity with an EC50 2.5 µM at mGlu4 receptors. n.e. = no effect observed with LSP1-3081 or racemate of VU0155041 (VU0003423).
AMN082 is an allosteric site agonist. Values in parentheses indicate % increase in GTP γ-S binding produced by AMN082 in different cell lines.
ACPT-I, (1S,3R,4S)-1aminocyclopentane-1,3,4-tricarboxylic acid; AMN082, N,N′-dibenzhydryl-ethane-1,2-diamine dihydrochloride; (S)-DCPG, (S)-3,4-dicarboxyphenylglycine; L-SOP, L-serine-O-phosphate; LSP1-3081, (3S)-3-[(3-amino-3-carboxypropyl(hydroxyl)-phosphinyl)-hydroxymethyl]-5-itrothiophene; PHCCC, N-phenyl-7-(hydroxylimino) cyclopropa[b]chromen-1a-carboxamide; VU0155041, (+/−)-cis-2-(3,5,dichlorphenylcarbamoyl)cyclo-hexanecarboxylic acid.
There is now a wealth of electrophysiological evidence supporting a functional role for group III mGlu receptors in general, in each of the target pathways. In Figure 1, receptors for which functionality has been demonstrated are emphasized in bold. Considering the corticostriatal pathway first of all, in a corticostriatal slice preparation, activation of group III mGlu receptors with the broad spectrum agonists, L-serine-O-phosphate (L-SOP) and L-2-amino-4-phosphonobutyrate (L-AP4), has been shown to mediate depression of striatal excitatory post-synaptic potentials (EPSPs) evoked by cortical stimulation (Pisani et al., 1997; Cuomo et al., 2009). This depression of EPSPs is taken to reflect inhibition of glutamate release from corticostriatal terminals. These effects were lost in slices from mGlu4 knockout mice, but were replicated in normal slices by the novel mGlu4 orthosteric agonist (3S)-3-[(3-amino-3-carboxypropyl(hydroxyl)-phosphinyl)-hydroxymethyl]-5-nitrothiophene (LSP1-3081) (Cuomo et al., 2009), indicating that mGlu4 receptors play a significant role in mediating this effect. Conversely, the mGlu8-selective agonist (S)-3,4-dicarboxyphenylglycine ((S)-DCPG) did not inhibit cortically evoked EPSPs in the striatum, suggesting that mGlu8 receptors, although possibly present, are not functional at this synapse (Cuomo et al., 2009). The role of putative mGlu7 receptors in the corticostriatal pathway remains to be investigated.
Turning next to the striatopallidal pathway, where the presence of mGlu4 and mGlu7 receptors was suggested from the distribution studies, L-AP4 has been shown to mediate inhibition of striatal-evoked inhibitory post-synaptic currents (IPSCs) recorded in the GP (Marino et al., 2003; Valenti et al., 2003), an effect likely explained by inhibition of GABA release from striatopallidal terminals. In support of this, our in vivo microdialysis studies indicate that local infusion of L-SOP or L-AP4 inhibits GABA release in the rat GP (MacInnes and Duty, 2008). The mGlu4 receptor appears crucial in the striatopallidal pathway, since the L-AP4-mediated inhibition of IPSCs was lost in slices from mGlu4 knockout mice (Valenti et al., 2003), but was enhanced in normal slices by the mGlu4 PAM PHCCC (Marino et al., 2003) and mimicked by the novel mGlu4 selective agonist LSP1-2111 (Beurrier et al., 2009). Although an involvement of mGlu7 receptors seems unlikely, given the loss of L-AP4's efficacy in slices from mGlu4 knockout animals (Valenti et al., 2003), until the effects of mGlu7 selective agents have been examined, a functional role for these receptors cannot be ruled out at this stage.
Reaffirming what was suspected from the distributional data, electrophysiological evidence also indicates the presence of functional group III mGlu receptors on terminals of the subthalamonigral pathway. Thus, L-AP4 depresses STN-evoked EPSCs in the SNr (Wittmann et al., 2001; 2002), and inhibits EPSPs in dopaminergic neurones of the SNc (Wigmore and Lacey, 1998), most likely reflecting inhibition of glutamate release from STN terminals in these regions. In support of this, we found that L-SOP and L-AP4 inhibited depolarization-evoked release of [3H]-D-aspartate from slices of rat SN in vitro and that local intranigral infusion of L-SOP reduced glutamate release in the SNr in vivo (Austin et al., 2010). Once again, mGlu4 receptors have been identified as one of the functional subtypes, since STN-evoked EPSCs in dopaminergic neurones of the SNc were inhibited by the mGlu4 PAM PHCCC (Valenti et al., 2005). This is consistent with our findings that PHCCC potentiates L-AP4-mediated inhibition of [3H]-D-aspartate release in SN slices (Broadstock et al. unpublished). Although no published reports have examined the effect of stimulating mGlu7 receptors on STN terminals, our preliminary data indicate that these receptors act to inhibit glutamate release in the SN since the mGlu7 allosteric agonist N,N′-dibenzhydryl-ethane-1,2-diamine dihydrochloride (AMN082; Mitsukawa et al., 2005) inhibits [3H]-D-aspartate release in SN slices. In contrast, a role for pre-synaptic mGlu8 receptors looks unlikely given that activation of mGlu8 receptors with (S)-DCPG neither inhibited [3H]-D-aspartate release in our preliminary studies (Broadstock et al. unpublished) nor reduced STN-evoked EPSCs in the SNc (Valenti et al., 2005).
The presence of group III mGlu receptors on GABAergic striatonigral terminals raises a potential cause for concern. When activated by L-AP4, these receptors have been shown to mediate inhibition of striatal-evoked IPSCs in the SNr (Wittmann et al., 2002). No subtype-specific studies have been undertaken to identify which group III receptors are the culprits. From a therapeutic standpoint, however, these receptors are a potential stumbling block since restricting GABA release in the SNr would exacerbate an already compromised striatonigral transmission and potentially undo the benefits of inhibiting glutamate release in this region from STN terminals. Fortunately, current evidence seems to rule out any likely detrimental effect of activating these receptors in PD. Electrophysiological studies have shown that under conditions of dopamine depletion, that is, in slices taken from animals treated with reserpine to deplete all catecholamines, while the ability of L-AP4 to inhibit STN-evoked EPSCs in the SNr is maintained, its ability to inhibit striatal-evoked IPSCs in the SNr is lost (Wittmann et al., 2002). These findings indicate that under parkinsonian conditions, the pharmacological effects of group III mGlu receptors on the desired subthalamonigral terminals most likely override those of the receptors on striatonigral terminals. The behavioural studies described below in animal models of PD certainly appear to back up this supposition.
Given that one or more functional group III mGlu receptor subtype has been identified at several desirable locations in the basal ganglia circuitry, activation of these receptors has been predicted to bring about relief of parkinsonian symptoms, by normalizing firing within the basal ganglia circuitry, and to offer protection against SNc degeneration, by reducing glutamate-mediated excitotoxicity (Rouse et al., 2000; Conn et al., 2005). The remainder of this review will focus on current evidence obtained in support of these predictions.
Symptomatic relief following group III mGlu receptor activation
Targeting of group III mGlu receptors to bring about symptomatic relief has been studied in a number of different rodent models of PD. In the reserpine-treated rat, systemic injection of reserpine depletes the brain of all monoamines, including dopamine, and induces akinesia within 12–18 h that is maintained for up to 48 h. Drug-induced reversal of akinesia is monitored as an index of symptomatic efficacy. In the haloperidol-treated rat, systemic injection of haloperidol produces a temporary state of catalepsy, the reversal of which is also taken to indicate symptomatic efficacy of test drugs. Finally, in the unilateral 6-OHDA-lesioned rat, akinesia in the forelimb contralateral to the lesion is manifest as reduced reaching in cylinder reaching tests, and the ability of drugs to reverse this akinesia is taken as a measure of symptomatic efficacy. In support of the validity of all three models, neurochemical or electrophysiological findings support features of increased STN firing and increased glutamate release in the basal ganglia output regions, the SNr or entopeduncular nucleus (the rat homologue of GPi) (You et al., 1996; Biggs et al., 1997; Vila et al., 1999).
Because of the limited availability of systemically active group III mGlu receptor agonists or PAMs, the majority of studies have examined the efficacy of centrally administered agents. Although of limited clinical relevance, site-directed injections have been very useful in teasing out which regions of the basal ganglia circuitry can be targeted to mediate beneficial outcomes. Given the activation of pre-synaptic group III mGlu receptors in the striatum, GP or SNr could potentially restrict transmitter release from overactive pathways to normalize firing of the basal ganglia circuits and thereby restore movement, these regions have all been subject to some degree of investigation.
Targeting the striatum
Intrastriatal injections of the broad spectrum group III mGlu agonists (1S,3R,4S)-1aminocyclopentane-1,3,4-tricarboxylic acid (ACPT-I) and L-AP4 have been shown to produce relief of haloperidol-induced catalepsy (Konieczny et al., 2007) or to improve akinesia in the 6-OHDA lesioned rat (Cuomo et al., 2009). These data are consistent with the L-AP4-mediated depression of cortical-evoked EPSPs in the striatum (Pisani et al., 1997), indicating that restriction of glutamate release from overactive cortical inputs likely underlies this symptom relief. Consistent with the electrophysiological findings, the involvement of mGlu4 receptors is favoured in this action, since intrastriatal injection of the orthosteric mGlu4 agonist, LSP1-3081, reduces akinesia in the 6-OHDA lesioned rat (Cuomo et al., 2009). While mGlu8 receptors are unlikely to be involved in this region, given their lack of functional effects noted above (Cuomo et al., 2009), any positive contribution from mGlu7 receptors remains to be examined.
Targeting the GP
The GP was one of the first regions implicated in mediating the anti-parkinsonian responses of broad spectrum group III mGlu agonists. Studies from our laboratory first demonstrated that direct injection of L-SOP into the GP reversed akinesia in the reserpine-treated rat (MacInnes et al., 2004). Others have since confirmed that direct intrapallidal injections of the broad spectrum agonists, L-AP4 or ACPT-I reversed reserpine-induced akinesia as well as reversing haloperidol-induced catalepsy and 6-OHDA-induced forelimb akinesia (Konieczny et al., 2007; Lopez et al., 2007). These effects which, when tested, were blocked by treatment with group III mGlu receptor antagonists, most likely reflect inhibition of GABA release within the GP (Marino et al., 2003; Valenti et al., 2003; MacInnes and Duty, 2008) which is predicted to normalize firing within the indirect basal ganglia pathway and hence restore motor activity. The most recent findings concur with earlier electrophysiological data (Marino et al., 2003; Valenti et al., 2003) indicating that mGlu4 receptors play a key role in the GP. Thus, Beurrier et al. (2009) demonstrated that injection of the mGlu4 agonist, LSP1-2111 reversed akinesia in the 6-OHDA lesion rat model of PD. In contrast, consistent with the lack of clear evidence favouring the existence of mGlu8 receptors on striatopallidal terminals, intrapallidal injection of the mGlu8 agonist (S)-DCPG was ineffective (Lopez et al., 2007; Beurrier et al., 2009), ruling out any target potential of mGlu8 receptors here. Again, a role for mGlu7 receptors in the GP awaits verification.
Targeting the substantia nigra pars reticulata
As discussed above, within the SNr, group III mGlu receptors are found not only on the desired glutamatergic subthalamonigral terminals but also on GABAergic striatonigral terminals. This raises the possibility that activation of these receptors could produce a number of different outcomes – a net inhibition of GABAergic transmission (i.e. inhibition of striatal-evoked IPSCs), a net inhibition of glutamatergic transmission (i.e. inhibition of glutamate-evoked EPSCs) or functional stale-mate with neither effect dominating. In vitro, Wittmann et al. (2002) had shown that under conditions of dopamine depletion, the beneficial action of group III mGlu receptors to inhibit STN-evoked EPSCs in the SNr predominated over the potentially detrimental action to inhibit striatal-evoked IPSCs. Fortunately, this phenomenon appears to hold true in vivo. Under conditions of marked dopamine depletion or dopamine receptor blockade, injections of broad spectrum group III agonists L-SOP, L-AP4 or ACPT-I into the SNr brought about beneficial anti-parkinsonian actions including reversal of reserpine-induced akinesia (MacInnes et al., 2004; Austin et al., 2010) and haloperidol-induced catalepsy (Konieczny et al., 2007). In light of the above electrophysiological, release and microdialysis data, these responses most likely reflect receptor-mediated inhibition of glutamate release from overactive subthalamonigral neurones. However, in normal animals, injection of ACPT-I into the SNr induced catalepsy (Lopez et al., 2007), probably reflecting a predominant effect to inhibit GABA release from the striatonigral pathway, as noted in vitro. Of some concern, intranigral injections of ACPT-I also worsened akinesia in a partial 6-OHDA lesioned rat model of PD displaying (58%) reduction in striatal dopamine innervation (Lopez et al., 2007), implying that even with this level of dopamine loss, the detrimental effects on GABAergic transmission still dominate. Estimations of the threshold level of nigrsotriatal tract degeneration required for symptoms to appear in patients vary widely. Some have estimated a 68% loss of cells in the lateral ventral tier of the SNc (average 52% throughout the SNc) at the time of symptom onset (Fearnley and Lees, 1991). Others have estimated, using positron emission tomography or single photon emission tomography, that striatal dopamine transporters are reduced by between 25 and 64% at the point of symptom onset (Morrish et al., 1998; Booji et al., 2001; Hilker et al., 2005). This variability makes it difficult to predict the likely outcome of targeting group III mGlu receptors in the SNr in the early stages of PD. Clearly, it will be essential to establish at what level of lesion development the detrimental effects of targeting group III mGlu receptors in the SNr are replaced by the beneficial ones, or indeed which group III mGlu receptor subtype might be responsible for the detrimental effects, before the full impact of this finding is realized.
The subtype of group III mGlu receptor mediating these anti-parkinsonian effects in the SNr is unlikely to be mGlu8, since intranigral injection of the mGlu8-selective agonist (S)-DCPG failed to reverse haloperidol-induced catalepsy (Lopez et al., 2007), in agreement with the lack of in vitro efficacy with (S)-DCPG in the SN (Valenti et al., 2005). However, both mGlu4 and mGlu7 receptors remain as possible targets in the SNr and our preliminary findings showing relief of reserpine-induced akinesia following intranigral injection of both the mGlu4 PAM, PHCCC and the mGlu7 allosteric agonist AMN082 (Broadstock et al. unpublished) support further investigation of their potential as drug targets.
Targeting multiple sites simultaneously
While site-directed injections have revealed much about the basal ganglia regions in which activating group III mGlu receptors could offer symptomatic potential in PD, routes of administration that can target multiple sites simultaneously give a better indication of the likely translation of these findings into therapeutic strategies. So far, positive outcomes have been demonstrated in a number of animal models of PD, following intracerebroventricular (i.c.v.) administration of group III agents. The first studies of this kind demonstrated the efficacy of i.c.v. injections of the broad spectrum agonists L-AP4 or L-SOP against reserpine-induced akinesia (Valenti et al., 2003; MacInnes et al., 2004), against haloperidol-induced catalepsy and against forelimb akinesia in the 6-OHDA lesioned rat (Valenti et al., 2003). Of importance, the degree of reversal of forelimb asymmetry produced by L-AP4 following this route of administration was similar to that produced by the gold-standard drug for treating PD, L-DOPA, adding strength to the potential for a useful clinical outcome in the future with this approach. The identity of the receptor subtype(s) mediating the responses following i.c.v. injection has so far only been partly addressed. Following i.c.v. injection, PHCCC was found to reverse reserpine-induced akinesia (Marino et al., 2003) indicating that mGlu4 receptors were useful targets for providing anti-parkinsonian relief. Further support for targeting mGlu4 receptors has come from studies showing a similar reversal of reserpine-induced akinesia and haloperidol-induced catalepsy following i.c.v. injection of the novel mGlu4 PAM (+/−)-cis-2-(3,5,dichlorphenylcarbamoyl)cyclo-hexanecarboxylic acid (VU0155041) (Niswender et al., 2008a). Offering most promise from a therapeutic perspective are the outcomes of studies utilizing systemic routes of administration. Battaglia et al. (2006) first showed that systemic administration of the mGlu4 PAM PHCCC could reverse reserpine-induced akinesia. Since then, both the broad spectrum agonist ACPT-I (Lopez et al., 2008) and the novel mGlu4 orthosteric agonist LSP1-2111 (Beurrier et al., 2009) have been shown to reverse haloperidol-induced catalepsy following systemic injection. The ability of selective mGlu7 or 8 agonists (or PAMs) to relieve symptoms in animal models of PD following i.c.v. injection has not yet been examined, although systemic administration of a mixed mGlu8 agonist/AMPA antagonist (R,S)-3,4-DCPG increased haloperidol-induced catalepsy (Ossowska et al., 2004). While this finding throws doubt on the anti-parkinsonian potential of mGlu8 receptors, judgement must be delayed until studies with ‘cleaner’ mGlu8 agonists have been undertaken.
Neuroprotection afforded by group III mGlu receptor activation
The finding that broad spectrum group III mGlu agonists like L-AP4 could inhibit glutamatergic drive to the SNc (Wigmore and Lacey, 1998), coupled with the proposed contribution of glutamate-mediated excitotoxicity in the progressive SNc degeneration in PD (Blandini et al., 1996; Rodriguez et al., 1998), make a clear case for considering the therapeutic potential of targeting group III mGlu receptors to bring about neuroprotection in PD.
Few groups have so far examined the potential protective effects of group III mGlu receptor activation in animal models of PD, but all have demonstrated favourable outcomes. Vernon et al. (2005, 2007) found that injections of the broad spectrum group III mGlu agonist L-AP4 into the rat SNc 1 h before toxin and for 3 or 7 days thereafter can protect against 6-OHDA-induced nigrostriatal tract degeneration. The beneficial effects – including preservation of striatal dopaminergic neurone markers, for example, tyrosine hydroxylase (TH), protection of TH-positive cells in the SNc and preservation of striatal dopamine content – were lost with concomitant administration of a group III mGlu receptor antagonist, confirming they were receptor mediated. Studies in this laboratory have recently confirmed this protective effect of sub-chronic treatment with L-AP4 against 6-OHDA-induced nigrostriatal tract degeneration and have additionally revealed that the motor behaviour of animals is also preserved under this treatment regimen (Austin et al., 2010). Although the identity of the receptor subtype mediating this protection was not examined in these reports, our preliminary data support a role for mGlu4 receptors, since the mGlu4 PAM, VU0155041, provides protection against a similar 6-OHDA lesion, preserving histological and neurochemical markers of dopaminergic neurones as well as protecting against the loss of motor function (Betts et al., 2009). This finding is consistent with those of Battaglia et al. (2006) who found that systemic injections of the mGlu4 PAM PHCCC, given 30 min before toxin injection, offered around 50% protection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced loss of striatal dopamine content and reduction in TH-positive cell numbers in the SNc in mice. In this MPTP study, a similar protection against MPTP-induced degeneration was seen following injection of PHCCC directly into the GP. This finding implies that normalization of glutamatergic drive to the SNc, either directly by actions in the SNc itself or indirectly via correction of GP firing and subsequent downstream reductions in STN glutamatergic drive to the SNc, can produce therapeutic relief in these animal models of PD. Studies investigating the neuroprotective potential of activating mGlu7 or mGlu8 receptors are now eagerly awaited as are studies involving the protective potential following systemic administration of group III modulators.
Potential mechanisms underlying neuroprotective effects
On the question of what events might underpin group III mGlu receptor-mediated neuroprotection, a number of mechanisms are proposed on the basis of current evidence (Figure 2). The electrophysiological and transmitter release data certainly point towards direct or indirect inhibition of glutamate release in the SNc being a key influence. However, there are a number of other tangible explanations that have arisen out of in vitro studies designed to examine the mechanisms underlying group III mGlu receptor-mediated protection. Although many early clues were gained from studies performed in other cell types, such as cortical or hippocampal neurones, discussion here will focus mainly on the studies performed in primary ventral mesencephalic (VM) cultures encompassing TH-positive, dopaminergic neurones. From these studies, the two most compelling alternative explanations involve activation of group III mGlu receptors either on glial cells supporting the SNc neurones or on the SNc neurones themselves.
Figure 2.
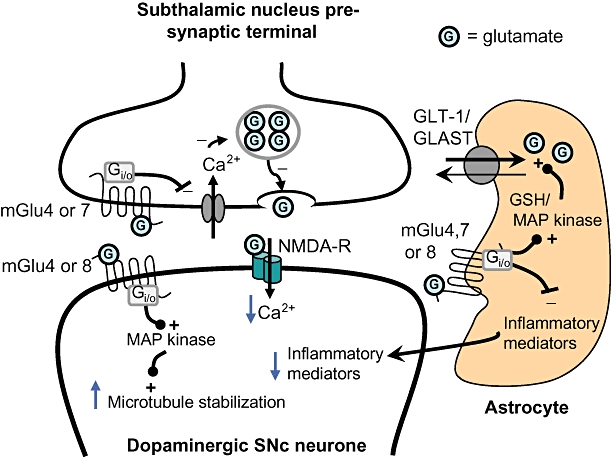
Putative mechanisms underlying the protection of dopaminergic substantia nigra pars compacta (SNc) neurones following group III metabotropic glutamate (mGlu) receptor activation. Activation of group III mGlu receptors located at three different sites in the glutamatergic subthalamonigral synapse may underlie the protection of dopaminergic SNc neurones. (i) Activation of pre-synaptic mGlu4 or mGlu7 receptors will restrict the pre-synaptic Ca2+ entry required for glutamate release leading in turn to reduced synaptic glutamate levels and reduced stimulation of post-synaptic NMDA receptors. The resultant reduction in Ca2+ entry into SNc neurones is proposed to reduce excitotoxic damage. (ii) Activation of glial mGlu4, 7 or 8 receptors may lead, via activation of MAP kinase or elevation of the antioxidant reduced glutathione (GSH) levels, to increased glutamate uptake via GLT-1 or GLAST glutamate transporters, further reducing synaptic glutamate levels. Activation of glial receptors may also reduce the production of inflammatory mediators, such as chemokines, that in turn will reduce the inflammatory stress upon SNc neurones. (iii) Activation of mGlu4 or mGlu8 receptors on SNc neurones directly may lead to increased microtubule stabilization through a MAP kinase-dependent pathway. One or more of these events may contribute to the overall neuroprotective effects of group III mGlu receptor activation. G, glutamate; MAP kinase, mitogen-activated protein kinase.
So far, mGlu4 and mGlu7 receptors have been identified on primary cultured rat astrocytes (Besong et al., 2002; Yao et al., 2005) and mGlu4 and mGlu8 receptors on rat brain microglia, which were devoid of mGlu7 receptor expression (Taylor et al., 2003). Exposure of astrocytes to activators such as lipopolysaccharide (LPS) is known to trigger the release of glial mediators such as glutamate, nitric oxide and hydrogen peroxide that are toxic to dopaminergic neurones (McNaught and Jenner, 2000). Indeed, conditioned medium from LPS-treated astrocytes has been shown to cause neuronal cell death or apoptosis in primary VM cultures (McNaught and Jenner, 1999; Zhou et al., 2006). Of great significance, Zhou et al. (2006) found that concomitant exposure of the LPS-treated astrocytes to the broad spectrum group III mGlu receptor agonist L-AP4 protects VM cultures from LPS-conditioned medium-induced apoptosis. L-AP4 was further found to enhance glutamate re-uptake into the treated astrocytes, secondary to producing recovery of the astrocyte levels of the antioxidant, reduced glutathione (Zhou et al., 2006). Astrocyte activation is also implicated in MPTP-induced degeneration of dopaminergic neurones. Thus, conditioned medium from astrocytes exposed to the toxic pyridinium ion 1-methyl-4-phenylpyridinium (MPP+) was toxic to midbrain neuronal cultures (Yao et al., 2005). Like LPS, MPP+ was found to reduce glutamate uptake into astrocytes and concomitant exposure to L-AP4 was again able to restore astrocyte glutamate uptake and reduce the toxicity of the conditioned medium (Yao et al., 2005). Reducing prevailing glutamate levels in the SNc by enhancing reuptake of glutamate into surrounding astrocytes may therefore be a key mechanism underpinning the neuroprotective potential of group III mGlu receptor activation. It is noteworthy that the related group II mGlu receptors, which also couple through Gi/o have been shown to produce an up-regulation of the glial glutamate transporters GLT-1 and GLAST through a mitogen-activated protein (MAP) kinase and phosphatidyl-inositol-3 (PI-3) kinase-dependent pathway (Aronica et al., 2003). L-AP4-mediated stimulation of group III mGlu receptors has similarly been shown to stimulate MAP kinase and PI-3 kinase pathways in cultured cerebellar granule cells (Iacovelli et al., 2002), so it remains a possibility that a similar up-regulation of glial glutamate transporters may underlie the enhanced astrocyte glutamate uptake reported in the above studies. L-AP4 treatment of astrocytes has also been shown to reduce the production of pro-inflammatory chemokines (Besong et al., 2002), an effect abolished in astrocyte cultures from mGlu4 -/- mice, indicating that activation of astoglial mGlu4 receptors may offer additional anti-inflammatory actions. Although these mechanisms have yet to be explored in animal models of PD, given that gliosis and inflammation are increasingly recognized as features of PD pathology (Whitton, 2007), it is very likely that some component derived from activation of astroglial group III mGlu receptors contributes to the overall protective potential of targeting these receptors in PD.
The remaining and undoubtedly more controversial piece of the mechanistic jigsaw stems from a study which showed that L-AP4 could protect TH-positive neurones in rat embryonic midbrain cultures against the toxicity of rotenone (Jiang et al., 2006). Rotenone is known to inhibit mitochondrial complex I (like MPTP) and to cause depolymerization of microtubules, the latter effect having been critically linked to the toxic capacity of rotenone on dopaminergic neurones (Ren et al., 2005). Although not as widely used as MPTP or 6-OHDA, rotenone has recently been used to trigger dopaminergic neurone death both in vitro (Falk et al., 2009) and in vivo (Panov et al., 2005). The protective effects of L-AP4 against rotenone toxicity again appeared to involve activation of the MAP kinase pathway since L-AP4 increased activation of the MAP kinase extracellular signal-regulated kinase in these midbrain TH-positive neurone cultures and the protection against rotenone toxicity was blocked by pharmacological inhibition of MAP kinase kinase (Jiang et al., 2006). Inhibition of MAP kinase kinase also prevented L-AP4-induced attenuation of rotenone-induced microtubule depolymerization, leading the authors to conclude that L-AP4 provided protection of midbrain TH-positive neurones through activation of the MAP kinase pathway to stabilize microtubules (Jiang et al., 2006). What is particularly interesting about this study is that, given that the cells were incubated in a cytosine arabinoside-supplemented medium that inhibits glial cell growth, the findings highlight for the first time that targeting group III mGlu receptors on the dopaminergic neurones themselves, rather than on surrounding glial cells or indeed on the pre-synaptic terminals of incoming glutamatergic neurones, can offer neuroprotection. To date, very little is known about a likely post-synaptic action of group III mGlu receptors in the SNc. While current evidence rules out the presence of mGlu7 receptors in the SNc [with negligible mRNA or mGlu7 immunoreactivity detected in the SNc (Kosinski et al., 1999; Messenger et al., 2002) ], a post-synaptic presence for mGlu4 or mGlu8 receptors cannot be entirely ruled out since mRNA (Messenger et al., 2002) and immunoreactivity (Gu et al., 2003) for each has been reported in this region. However, Wigmore and Lacey (1998) failed to detect any direct post-synaptic effects of L-AP4 on SNc neurones, despite observing the aforementioned inhibition of STN-evoked EPSCs in the SNc, reflective of L-AP4's pre-synaptic action. Subsequent electrophysiological studies of Valenti et al. (2005) also ruled out any post-synaptic action of L-AP4 in regulating SNc neuronal excitability. Future studies are therefore required to clarify whether mGlu4 or mGlu8 receptors might mediate any post-synaptic action, distinct from modulation of neuronal excitability, which might relate to this potential neuroprotective effect.
Future directions
One of the main drawbacks of long-term treatment with L-DOPA is the onset of disabling, excessive involuntary movements or LIDs. Future studies should aim to examine whether, when given alone, group III mGlu agents evoke unwanted dyskinetic effects, akin to LIDs or whether, as seen recently with group I mGlu5 receptor antagonists in rodents (Rylander et al., 2009; Yamamoto and Soghomonian, 2009) and primates (Johnston et al., 2010), these agents may actually be helpful in reducing LIDs when given in combination with low-dose L-DOPA. It is still too early to say whether activation of group III mGlu receptors offer any degree of long-term potential in the treatment of PD; however, the advent of improved systemically active drugs that are more amenable for long-term administration to animals should enable due attention to be paid to these issues in the future. Further studies examining the long-term effectiveness of targeting group III mGlu receptors will also clarify whether receptor desensitization will have any bearing on the long-term utility of this approach. Certainly, many of the responses noted above with the broad spectrum agonists, ranging from electrophysiological responses in vitro through to behavioural responses and neuroprotection in vivo, show that responses tail off at the higher concentrations or doses tested (e.g. Valenti et al., 2003; MacInnes et al., 2004; Vernon et al. 2007). These observations may be explained by receptor desensitization since the broad spectrum agonist L-AP4 has been shown to induce rapid internalization of at least two of the group III mGlu receptors in vitro, mGlu4 (Iacovelli et al., 2004) and mGlu7 (Pelkey et al., 2005). Targeting the allosteric sites of receptors is likely to reduce the chance of desensitization becoming a problem (Kew, 2004) and may explain the recent emphasis on developing mGlu4 receptor PAMs (Niswender et al., 2008b; Engers et al., 2009; Williams et al., 2009).
To date, investigations in the group III mGlu receptor field have still not progressed into the final preclinical model of PD, the MPTP-treated primate. This model is the most clinically relevant model in which to monitor the long-term effectiveness of agents given alone or in combination with low-dose L-DOPA. As such, studies in the MPTP-treated primate will provide invaluable insight into the likely clinical potential of targeting group III mGlu receptors in PD as well as revealing any unforeseen side effects that may arise from this strategy. Given that combating excessive glutamatergic transmission with the use of ionotropic glutamate receptor antagonists is predicted to lead to unacceptable cognitive and psychomimetic adverse effects (Kornhuber and Weller, 1997), it will be essential to establish whether targeting group III mGlu receptors to achieve, among other things, such inhibition of glutamatergic transmission can happen without the occurrence of similar adverse effects. Despite the lack of current data for group III mGlu agonists, recent clinical trials conducted with agonists of the related class of group II (mGlu2 and mGlu3) receptors in patients with schizophrenia or generalized anxiety disorder have reported good tolerance, improvements in symptoms and, most importantly here, no major treatment-related adverse events (Patil et al., 2007; Dunayevich et al., 2008). While the outcome of longer-duration clinical trials is still awaited, these earliest observations of targeting group II mGlu receptors in humans do offer hope that drugs targeting group III mGlu receptors might prove to be equally well tolerated and devoid of unwanted adverse effects. However, the challenge of producing ‘hit-to-lead’ compounds for group III mGlu receptors with agreeable systemic activity (Williams et al., 2009) may still hamper translational progress in this area.
Potential added value of targeting group III mGlu receptors in PD
In closing, it is worth turning our attention back to the clinical situation. As mentioned in the Introduction, in addition to the classical motor symptoms of PD, patients also exhibit a variety of non-motor symptoms which add to their reduced quality of life. Of these, depression and pain are particularly problematic. It is therefore promising to note that group III mGlu receptor activation following i.c.v. administration of the broad spectrum agonists like ACPT-I or the mGlu4 PAM PHCCC in combination with low-dose ACPT-I has been shown to produce antidepressant effects in rats (Palucha et al., 2004; Klak et al., 2007). Equally promising are the reports that intrathecal administration of ACPT-I or PHCCC or systemic administration of the mGlu8 agonist (S)-DCPG relieved hyperalgesia in inflammatory pain models (Marabese et al., 2007; Goudet et al., 2008), most likely through inhibition of glutamate release from primary afferents in the spinal cord (Zhang et al., 2009). Finally, activation of group III receptors, especially mGlu7, has been shown to inhibit glutamate release onto dopaminergic neurones in the ventral tegmental area, and systemic injection of ACPT-I has been shown to produce antipsychotic effects in rodents (Palucha-Poniewiera et al., 2008). This portfolio of actions raises the possibility that, in addition to combating the motor symptoms and progressive degeneration of dopaminergic neurones in PD, targeting group III mGlu receptors may also provide a means of tackling some of the non-motor signs in PD and help to stave off treatment-related addictive behaviours or psychosis. These offer exciting prospects for drug discovery in the group III mGlu receptor field in relation to the treatment of PD.
Concluding remarks
In the 10 years since Bradley et al. (1999) first demonstrated immunoreactivity for group III mGlu receptors in the basal ganglia motor loop, we have seen the potential of targeting these receptor for therapeutic gain flourish from mere speculation to something tangible. Activation of pre-synaptic group III mGlu receptors has been shown to reduce transmission across synapses in the indirect pathway that are overactive in PD (i.e. corticostriatal, striatopallidal and subthalamonigral), an effect which most likely underlies the reversal of symptoms seen in rodent models of the disease following administration of agonist or PAMs of group III mGlu receptors. Group III mGlu receptor activation has also been shown to protect dopaminergic neurones both in vitro and in vivo against a range of known dopaminergic toxins. While the exact molecular mechanisms underlying these protective effects remain to be established, the likelihood is that beneficial actions are mediated through activation of both neuronal and glial group III mGlu receptors. In parallel fields, activation of group III mGlu receptors has been shown to produce actions (antidepressant, analgesic and potentially anti-addictive) that raise the possibility that, in addition to combating the motor symptoms and progressive degeneration of dopaminergic neurones in PD, targeting group III mGlu receptors may also provide a means of tackling some of the non-motor signs in PD and help to stave off treatment-related addictive behaviours or psychosis. These results all offer exciting prospects for drug discovery in the group III mGlu receptor field for the treatment of PD. Efforts should now be directed towards elucidating which of the specific group III mGlu receptors holds greatest potential (with most studies to date favouring mGlu4 receptors), unravelling the molecular mechanisms underlying neuroprotection and targeting of group III mGlu receptors in the MPTP-treated primate model of PD in order to discover the likely long-term therapeutic benefits of this strategy.
Acknowledgments
I wish to thank the BBSRC, MRC and Guy's and St Thomas' Charitable Trustees for funding the relevant group III mGlu projects in this lab.
Glossary
Abbreviations
- 6-OHDA
6-hydroxydopamine
- ACPT-I
(1S,3R,4S)-1aminocyclopentane-1,3,4-tricarboxylic acid
- AMN082
N,N′-dibenzhydryl-ethane-1,2-diamine dihydrochloride
- EPSC/P
excitatory post-synaptic current/potential
- GP(i) (internal)
globus pallidus
- i.c.v.
intracerebroventricular
- IPSC
inhibitory post-synaptic current
- L-AP4
L-2-amino-4-phosphonobutyrate
- LID
L-DOPA-induced dyskinesia
- LPS
lipopolysaccharide
- L-SOP
L-serine-O-phosphate
- LSP1-3081
(3S)-3-[(3-amino-3-carboypropyl(hydroxyl)-phosphinyl)-hydroxymethyl]-5-nitrothiophene
- MAP kinase
mitogen-activated protein kinase
- mGlu
metabotropic glutamate
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PAM
positive allosteric modulator
- PD
Parkinson's disease
- PHCCC
N-phenyl-7-(hydroxylimino) cyclopropa[b]chromen-1a-carboxamide
- (S)-DCPG
(S)-3,4-dicarboxyphenylglycine
- SNc/r
substantia nigra pars compacta/reticulata
- STN
subthalamic nucleus
- TH
tyrosine hydroxylase
- VM
ventral mesencephalic
- VU0155041
(+/−)-cis-2-(3,5,dichlorphenylcarbamoyl)cyclo-hexanecarboxylic acid
Conflict of interest
The author has no conflict of interest regarding the content of this review.
Supplemental material
Supporting Information: Teaching Materials; Figs 1–2 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini A, Cilia R. Behavioural adverse effects of dopaminergic treatments in Parkinson's disease: incidence, neurobiological basis, management and prevention. Drug Saf. 2009;32:475–488. doi: 10.2165/00002018-200932060-00004. [DOI] [PubMed] [Google Scholar]
- Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yanakaya B, Leenstra S, et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurol. 2003;17:2106–2118. doi: 10.1046/j.1460-9568.2003.02657.x. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Betts MJ, Broadstock M, O'Neill MJ, Mitchell SN, Duty S. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson's disease. Br J Pharmacol. 2010;160:1741–1753. doi: 10.1111/j.1476-5381.2010.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson's disease pathogenesis. Biochim Biophys Acta. 2009;1792:651–663. doi: 10.1016/j.bbadis.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Busceti CL, Molinaro G, Biagioni F, Traficante A, Nicoletti F, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci. 2006;26:7222–7229. doi: 10.1523/JNEUROSCI.1595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabid AL, Chabardes S, Mitrofanis J, Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson's disease. Lancet Neurol. 2009;8:67–81. doi: 10.1016/S1474-4422(08)70291-6. [DOI] [PubMed] [Google Scholar]
- Besong G, Battaglia G, D'Onofrio M, Di Marco R, Ngomba RT, Storto M, et al. Activation of group III metabotropic glutamate receptors inhibits the production of RANTES in glial cell cultures. J Neurosci. 2002;22:5403–5411. doi: 10.1523/JNEUROSCI.22-13-05403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts M, O'Neill M, Mitchell S, Duty S. Allosteric modulation of group III mGlu receptor 4: a potential neuroprotective approach for the treatment of Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 2):S121. [Google Scholar]
- Beurrier C, Lopez S, Révy D, Selvam C, Goudet C, Lhérondel M, et al. Electrophysiological and behavioural evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J. 2009;23:3619–3628. doi: 10.1096/fj.09-131789. [DOI] [PubMed] [Google Scholar]
- Biggs CS, Fowler LJ, Whitton PS, Starr MS. Extracellular levels of glutamate and aspartate in the entopeduncular nucleus of the rat determined by microdialysis: regulation by striatal dopamine D2 receptors via the indirect striatal output pathway? Brain Res. 1997;753:163–175. doi: 10.1016/s0006-8993(97)00033-4. [DOI] [PubMed] [Google Scholar]
- Blandini F, Porter RH, Greenamayre JT. Glutamate and Parkinson's disease. Mol Neurobiol. 1996;12:73–94. doi: 10.1007/BF02740748. [DOI] [PubMed] [Google Scholar]
- Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson's disease. Prog Neurobiol. 2000;62:63–88. doi: 10.1016/s0301-0082(99)00067-2. [DOI] [PubMed] [Google Scholar]
- Booji J, Bergmans P, Winogrodzka A, Speelman JD, Wolters EC. Imaging of dopamine transporters with [123I]FP-CIT SPECT does not suggest a significant effect of age on the symptomatic threshold of disease in Parkinson's disease. Synapse. 2001;39:101–108. doi: 10.1002/1098-2396(200102)39:2<101::AID-SYN1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Bradley SR, Standaert DG, Levey AI, Conn PJ. Distribution of group III mGluRs in rat basal ganglia with subtype-specific antibodies. Ann N Y Acad Sci. 1999;868:531–534. doi: 10.1111/j.1749-6632.1999.tb11322.x. [DOI] [PubMed] [Google Scholar]
- Carter CJ. Topographical distribution of possible glutamatergic pathways from the frontal cortex to the striatum and substantia nigra in rats. Neuropharmacology. 1982;21:379–383. doi: 10.1016/0028-3908(82)90019-3. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464–474. doi: 10.1016/S1474-4422(09)70068-7. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci. 2005;6:787–798. doi: 10.1038/nrn1763. [DOI] [PubMed] [Google Scholar]
- Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- Cuomo D, Martella G, Barabino E, Platania P, Vita D, Madeo G, et al. Metabotropic glutamate receptor subtype 4 selectively modulates both glutamate and GABA transmission in the striatum: implications for Parkinson's disease treatment. J Neurochem. 2009;109:1096–1105. doi: 10.1111/j.1471-4159.2009.06036.x. [DOI] [PubMed] [Google Scholar]
- Dawson L, Chadha A, Megalou M, Duty S. The group II metabotropic glutamate receptor agonist, DCG-IV, alleviates akinesia following intranigral or intraventricular administration in the reserpine-treated rat. Br J Pharmacol. 2000;129:541–546. doi: 10.1038/sj.bjp.0703105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Department of Economic and Social Affairs. World Population Ageing 2009. New York: United Nations; 2009. Available at: http://www.un.org/esa/population/publications/WPA2009/WPA2009_WorkingPaper.pdf (accessed 12 June 2010) [Google Scholar]
- de Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, et al. Prevalence of parkinsonism and Parkinson's disease in Europe: The EUROPARKINSON collaborative study. European community concerted action on the epidemiology of Parkinson's disease. J Neurol Neurosurg Psychiatry. 1997;62:10–15. doi: 10.1136/jnnp.62.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Loreto S, Florio T, Scarnati E. Evidence that non-NMDA receptors are involved in the excitatory pathway from the pedunculopontine region to nigrostriatal dopaminergic neurons. Exp Brain Res. 1992;89:79–86. doi: 10.1007/BF00229003. [DOI] [PubMed] [Google Scholar]
- Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology. 2008;33:1603–1610. doi: 10.1038/sj.npp.1301531. [DOI] [PubMed] [Google Scholar]
- Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, et al. Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs) J Med Chem. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goedt CG. Levodopa-induced dyskinesia. Mov Disord. 2007;22:1379–1389. doi: 10.1002/mds.21475. [DOI] [PubMed] [Google Scholar]
- Falk T, Zhang S, Sherman SJ. Pigment epithelium derived factor (PEDF) is neuroprotective in two in vitro models of Parkinson's disease. Neurosci Lett. 2009;458:49–52. doi: 10.1016/j.neulet.2009.04.018. [DOI] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Goudet C, Chapuy E, Alloui A, Acher F, Pinn J-P, Eschalier A. Group III metabotropic glutamate receptors inhibit hyperalgesia in animal models of inflammation and neuropathic pain. Pain. 2008;137:112–124. doi: 10.1016/j.pain.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Gu B, Zhang Y-D, Hu G. A unilateral 6-hydroxydopamine lesion decreases the expression of metabotropic glutamate receptors in rat substantia nigra. Neurosci Lett. 2003;351:186–190. doi: 10.1016/j.neulet.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, et al. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol. 2005;62:378–382. doi: 10.1001/archneur.62.3.378. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Périer C, Orieux G, François C, Féger J, Yelnik J, et al. Metabolic effects of nigrostriatal denervation in basal ganglia. Trends Neurosci. 2000;23:S78–S85. doi: 10.1016/s1471-1931(00)00021-5. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, et al. Native group III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phopshatidylinositol-3-kinase pathways. J Neurochem. 2002;82:216–223. doi: 10.1046/j.1471-4159.2002.00929.x. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Capobianco L, Iula M, Di Giorgi Gerevini V, Picascia A, Blahos J, et al. Regulation of mGlu4 metabotropic glutamate receptor signaling by type-2 G-protein coupled receptor kinase (GRK2) Mol Pharmacol. 2004;65:1103–1110. doi: 10.1124/mol.65.5.1103. [DOI] [PubMed] [Google Scholar]
- Iribe Y, Moore K, Pang KC, Tepper JM. Subthalamic stimulation-induced synaptic responses in substantia nigra pars compacta dopaminergic neurones in vitro. J Neurophysiol. 1999;82:925–933. doi: 10.1152/jn.1999.82.2.925. [DOI] [PubMed] [Google Scholar]
- Janca A. Parkinson's disease from WHO perspective and a public health point of view: short survey. Parkinsonism Relat Disord. 2002;9:3–6. doi: 10.1016/s1353-8020(02)00038-x. [DOI] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Yan Z, Feng J. Activation of group III metabotropic glutamate receptors attenuates rotenone toxicity on dopaminergic neurons through a microtubule-dependent mechanism. J Neurosci. 2006;26:4318–4328. doi: 10.1523/JNEUROSCI.0118-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston T, Fox SH, McIldowie MJ, Piggott MJ, Brotchie JM. Reduction of L-DOPA-induced dyskinesia by the selective metabotropic glutamate receptor 5 (mGlu5) antagonist MTEP in the MPTP-lesioned macaque model of Parkinson's disease. J Pharmacol Exp Ther. 2010;333:865–873. doi: 10.1124/jpet.110.166629. [DOI] [PubMed] [Google Scholar]
- Kew JN. Postiive and negative allosteric modulation of metabotropic glutamate receptors: emerging therapeutic potential. Pharmacol Ther. 2004;104:233–244. doi: 10.1016/j.pharmthera.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol. 1987;260:435–452. doi: 10.1002/cne.902600309. [DOI] [PubMed] [Google Scholar]
- Klak K, Palucha A, Brański P, Sowa M, Pilc A. Combined administration of PHCCC, a positive allosteric modulator of mGlu4 receptors and ACPT-I, mGlu III receptor agonist evokes antidepressant-like effects in rats. Amino Acids. 2007;32:169–172. doi: 10.1007/s00726-006-0316-z. [DOI] [PubMed] [Google Scholar]
- Konieczny J, Wardas J, Kuter K, Pilc A, Ossowska K. The influence of group III metabotropic glutamate receptor stimulation by (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid on the parkinsonian like akinesia and striatal proenkephalin and prodynorphin expression in rats. Neuropharmacology. 2007;145:611–620. doi: 10.1016/j.neuroscience.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Weller M. Psychotogenicity and N-methyl-D-aspartate receptor antagonism: implications for neuroprotective pharmacotherapy. Biol Psychiatry. 1997;41:135–144. doi: 10.1016/S0006-3223(96)00047-9. [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Bradley SR, Conn PJ, Levey AI, Landwehrmeyer GB, Penney JB, Jr, et al. Localisation of metabotropic glutamate 7 mRNA and mGluR7a protein in the rat basal ganglia. J Comp Neurol. 1999;415:266–284. [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Acher F, De Leonibus E, Mele A, Amalric M. Targeting group III metabotropic glutamate receptors produces complex behavioural effects in rodent models of Parkinson's disease. J Neurosci. 2007;27:6701–6711. doi: 10.1523/JNEUROSCI.0299-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Johnston TH, Brotchie JM, Schann S, Neuville P, et al. Functional interaction between adenosine A2A and group III metabotropic glutamate receptors to reduce parkinsonian symptoms in rats. Neuropharmacology. 2008;55:483–490. doi: 10.1016/j.neuropharm.2008.06.038. [DOI] [PubMed] [Google Scholar]
- Lozano AM. Surgery for Parkinson's disease, the five W's: why, who, what, where, and when. Adv Neurol. 2003;91:303–307. [PubMed] [Google Scholar]
- MacInnes N, Duty S. Group III metabotropic glutamate receptors act as hetero-receptors modulating GABA release in the globus pallidus in vivo. Eur J Pharmacol. 2008;580:95–99. doi: 10.1016/j.ejphar.2007.10.030. [DOI] [PubMed] [Google Scholar]
- MacInnes N, Messenger MJ, Duty S. Activation of group III metaobotropic glutamate receptors in selected regions of the basal ganglia alleviates akinesia in the reserpine-treated rat. Br J Pharmacol. 2004;141:15–22. doi: 10.1038/sj.bjp.0705566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, et al. (-)-PHCCC, a positive allosteric modulator of mGlu receptor4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- Marabese I, de Novellis V, Palazzo E, Scafuro MA, Vita D, Rossi F, et al. Effects of (S)-3,4-DCPG, an mGlu8 receptor agonist, on inflammatory and neuropathic pain in mice. Neuropharmacology. 2007;52:253–262. doi: 10.1016/j.neuropharm.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O'Brien JA, Valenti O, McDonald TP, Clements MK, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc Natl Acad Sci U S A. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD, Parkes JD. Success and problems of long-term levodopa therapy in Parkinson's disease. Lancet. 1977;1:345–349. doi: 10.1016/s0140-6736(77)91146-1. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Altered glial function causes neuronal death and increases neuronal susceptibility to 1-methyl-4-phenylpyridinium- and 6-hydroxydopamine-induced toxicity in astrocytic/ventral mesencephalic co-cultures. J Neurochem. 1999;73:2469–2476. doi: 10.1046/j.1471-4159.1999.0732469.x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Extracellular accumulation of nitric oxide, hydrogen peroxide, and glutamate in astrocytic cultures following glutathione depletion, complex I inhibition, and/or lipopolysaccharide-induced activation. Biochem Pharmacol. 2000;60:979–988. doi: 10.1016/s0006-2952(00)00415-9. [DOI] [PubMed] [Google Scholar]
- Messenger MJ, Dawson LG, Duty S. Changes in metabotropic glutamate receptor 1-8 gene expression in the rodent basal ganglia motor loop following lesion of the nigrostriatal tract. Neuropharmacology. 2002;43:261–271. doi: 10.1016/s0028-3908(02)00090-4. [DOI] [PubMed] [Google Scholar]
- Mitsukawa K, Yamamoto R, Ofner S, Nozulak J, Pescott O, Lukic S, et al. A selective metabotropic glutamate receptor 7 agonist: activation of receptor signaling via an allosteric site modulates stress parameters in vivo. Proc Natl Acad Sci U S A. 2005;102:18712–18717. doi: 10.1073/pnas.0508063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish PK, Rakshi JS, Bailey DL, Sawler GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with [18F]dopa PET. J Neurol Neurosurg Psychiatry. 1998;64:314–319. doi: 10.1136/jnnp.64.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray TK, Messenger MJ, Ward MA, Woodhouse S, Osborne DJ, Duty S, et al. Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson's disease. Pharmacol Biochem Behav. 2002;73:455–466. doi: 10.1016/s0091-3057(02)00842-0. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, et al. Discovery, characterisation and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol. 2008a;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Lebois EP, Luo Q, Kim K, Muchalski H, Yin H, et al. Positive allosteric modulators of the metabotropic receptor subtype 4 (mGluR4): Part I. Discovery of pyrazolol[3,4-d]pyrimidines as novel mGluR4 positive allosteric modulators. Bioorg Med Chem Lett. 2008b;18:5626–5630. doi: 10.1016/j.bmcl.2008.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Orez MC, Rodriguez M, Lanciego JL, Artieda J, Gonzalo N, et al. Pathophysiology of the basal ganglia in Parkinson's disease. Trends Neurosci. 2000;23:S8–S19. doi: 10.1016/s1471-1931(00)00028-8. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Orez MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, et al. Functional organization of the basal ganglia: therapeutic implications for the treatment of Parkinson's disease. Mov Disord. 2008;23:S548–S559. doi: 10.1002/mds.22062. [DOI] [PubMed] [Google Scholar]
- Ossowska K, Pietraszek M, Wardas J, Wolfarth S. Potential antipsychotic and extrapyramidal effects of (R,S)-3,4-dicarboxyphenylglycine [(R,S)-3,4-DCPG], a mixed AMPA antagonist/mGluR8 agonist. Pol J Pharmacol. 2004;56:295–304. [PubMed] [Google Scholar]
- Palucha A, Tatarczyńska E, Brański P, Szewczyk B, Wierońska JM, Klak K, et al. Group III mGlu receptor agonists produce anxiolytic- and antidepressant-like effects after central administration in rats. Neuropharmacology. 2004;46:151–159. doi: 10.1016/j.neuropharm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Palucha-Poniewiera A, Klodzińska A, Stachowicz K, Tokarski K, Hess G, Schann S, et al. Peripheral administration of group III mGlu receptor agonist ACPT-I exerts potential antipsychotic effects in rodents. Neuropharmacology. 2008;55:517–524. doi: 10.1016/j.neuropharm.2008.06.033. [DOI] [PubMed] [Google Scholar]
- Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson's disease: multiple brain mitochondria dysfunctions after short-term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–42035. doi: 10.1074/jbc.M508628200. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGlu receptor7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci. 1996;8:1408–1414. doi: 10.1111/j.1460-9568.1996.tb01603.x. [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Centonze D, Bernardi G. Activation of group III metabotropic glutamate receptors depresses glutamatergic transmission at corticostriatal synapse. Neuropharmacology. 1997;36:845–851. doi: 10.1016/s0028-3908(96)00177-3. [DOI] [PubMed] [Google Scholar]
- Ren Y, Liu W, Jiang H, Jiang Q, Feng J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J Biol Chem. 2005;280:34105–34112. doi: 10.1074/jbc.M503483200. [DOI] [PubMed] [Google Scholar]
- Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol. 1998;44:S175–S188. doi: 10.1002/ana.410440726. [DOI] [PubMed] [Google Scholar]
- Rodriguez JP, Walters SE, Watson P, Stell R, Mastaglia FL. Globus pallidus stimulation in advanced Parkinson's disease. J Clin Neurosci. 2007;14:208–215. doi: 10.1016/j.jocn.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Rouse ST, Marino MJ, Bradley SR, Awad H, Wittmann M, Conn PJ. Distribution and roles of metabotropic glutamate receptors in the basal ganglia motor circuit: implications for treatment of Parkinson's disease and related disorders. Pharmacol Ther. 2000;88:427–435. doi: 10.1016/s0163-7258(00)00098-x. [DOI] [PubMed] [Google Scholar]
- Rylander D, Recchia A, Mela F, Dekundy A, Danysz W, Cenci MA. Pharmacological modulation of glutamate transmission in a rat model of L-DOPA-induced dyskinesia: effects on motor behavior and striatal nuclear signaling. J Pharmacol Exp Ther. 2009;330:227–235. doi: 10.1124/jpet.108.150425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J Neurosci. 1996;16:5979–5985. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB. Mechanisms of neurodegeneration in idiopathic Parkinson's disease. Parkinsonism Relat Disord. 2007;13:S306–S308. doi: 10.1016/S1353-8020(08)70021-X. [DOI] [PubMed] [Google Scholar]
- Smith Y, Hazrati LN, Parent A. Efferent projections of the subthalamic nucleus in the squirrel monkey as studied by the PHA-L anterograde tracing method. J Comp Neurol. 1990;294:306–323. doi: 10.1002/cne.902940213. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998;86:353–387. doi: 10.1016/s0306-4522(98)00004-9. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci. 2003;23:2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NK, Wright RA, Howson PA, Kingston AE, Schoepp DD, Jane DE. (S)-3,4-DCPG, a potent and selective mGlu8a receptor agonist, activates metabotropic glutamate receptors on primary afferent terminals in the neonatal rat spinal cord. Neuropharmacology. 2001;40:311–318. doi: 10.1016/s0028-3908(00)00169-6. [DOI] [PubMed] [Google Scholar]
- Trombley PQ, Westbrook GL. L-AP4 inhibits calcium currents and synaptic transmission via a G-protein-coupled glutamate receptor. J Neurosci. 1992;12:2043–2050. doi: 10.1523/JNEUROSCI.12-06-02043.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG, et al. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J Neurosci. 2003;23:7218–7226. doi: 10.1523/JNEUROSCI.23-18-07218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. Group III metabotropic glutamate receptor-mediated modulation of excitatory transmission in the rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther. 2005;313:1296–1304. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Palmer S, Datla KP, Zbarsky V, Croucher MJ, Dexter DT. Neuroprotective effects of metabotropic glutamate receptor ligands in a 6-hydroxydopamine rodent model of Parkinson's disease. Eur J Neurosci. 2005;22:1799–1806. doi: 10.1111/j.1460-9568.2005.04362.x. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Zbarsky V, Datla KP, Dexter DT, Croucher MJ. Selective activation of group III metabotropic glutamate receptors by L-(+)-2-amino-4-phosphonobutyric acid protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J Pharmacol Exp Ther. 2007;320:397–409. doi: 10.1124/jpet.106.108159. [DOI] [PubMed] [Google Scholar]
- Vila M, Marin C, Ruberg M, Jimenez A, Raisman-Vozari R, Agid Y, et al. Systemic administration of NMDA and AMPA receptor antagonists reverses the neurochemical changes induced by nigrostriatal denervation in basal ganglia. J Neurochem. 1999;73:344–352. doi: 10.1046/j.1471-4159.1999.0730344.x. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigmore MA, Lacey MG. Metabotopic glutamate receptors depress glutamate-mediated synaptic input to rat midbrain dopamine neurones in vitro. Br J Pharmacol. 1998;123:667–674. doi: 10.1038/sj.bjp.0701662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R, Niswender CM, Luo Q, Le U, Conn PJ, Lindsley CW. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4). Part II: challenges in hit-to-lead. Bioorg Med Chem Lett. 2009;19:962–966. doi: 10.1016/j.bmcl.2008.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann M, Marino M, Bradley SR, Conn PJ. Activation of group III mGluRs inhibits GABA-ergic and glutamatergic transmission in the substantia nigra pars reticulata. J Neurophysiol. 2001;85:1960–1968. doi: 10.1152/jn.2001.85.5.1960. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Marino M, Conn PJ. Dopamine modulates the function of group II and group III metabotropic glutamate receptors in the substantia nigra pars reticulata. J Pharmacol Exp Ther. 2002;302:433–441. doi: 10.1124/jpet.102.033266. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Soghomonian JJ. Metabotropic glutamate mGluR5 receptor blockade opposes abnormal involuntary movements and the increases in glutamic acid decarboxylase mRNA levels induced by L-DOPA in striatal neurons of 6-hydroxydopamine-lesioned rats. Neuroscience. 2009;163:1171–1180. doi: 10.1016/j.neuroscience.2009.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HH, Ding JH, Zhou F, Wang F, Hu LF, Sun T, et al. Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes. J Neurochem. 2005;92:948–961. doi: 10.1111/j.1471-4159.2004.02937.x. [DOI] [PubMed] [Google Scholar]
- You Z-B, Herrera-Marschitz M, Pettersson E, Nylander I, Goiny M, Shou HZ, et al. Modulation of neurotransmitter release by cholecystokinin in the neostriatum and substantia nigra of the rat: regional and receptor specificity. Neuroscience. 1996;74:793–804. doi: 10.1016/0306-4522(96)00149-2. [DOI] [PubMed] [Google Scholar]
- Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson's disease: a review. Drugs Aging. 2008;25:665–682. doi: 10.2165/00002512-200825080-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HM, Chen SR, Pan HL. Effects of activation of group III metabotropic glutamate receptors on spinal synaptic transmission in a rat model of neuropathic pain. Neuroscience. 2009;158:875–884. doi: 10.1016/j.neuroscience.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Yao H-H, Wu J-Y, Yang Y-J, Ding J-H, Zhang J, et al. Activation of group II/III metabotropic glutamate receptors attenuates LPS-induced astroglial neurotoxicity via promoting glutamate uptake. J Neurosci Res. 2006;84:268–277. doi: 10.1002/jnr.20897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.