

Summary
Background
During meiosis, recombination between homologous chromosomes promotes their proper segregation. In budding yeast, programmed double-strand breaks (DSBs) promote recombination between homologs versus sister chromatids by dimerizing and activating Mek1, a chromosome axis-associated kinase. Mek1 is also a proposed effector kinase in the recombination checkpoint that arrests exit from pachytene in response to aberrant DNA/axis structures. Elucidating a role for Mek1 in the recombination checkpoint has been difficult since in mek1 loss-of-function mutants DSBs are rapidly repaired using a sister chromatid thereby bypassing formation of checkpoint-activating lesions. Here we tested the hypothesis that a MEK1 gain-of-function allele would enhance interhomolog bias and the recombination checkpoint response.
Results
When Mek1 activation was artificially maintained through GST-mediated dimerization, there was an enhanced skew toward interhomolog recombination and reduction of intersister events including multi-chromatid joint molecules. Increased interhomolog events were specifically repaired as noncrossovers rather than crossovers. Ectopic Mek1 dimerization was also sufficient to impose interhomolog bias in the absence of recombination checkpoint functions, thereby uncoupling these two processes. Finally, the stringency of the recombination checkpoint was enhanced in weak meiotic recombination mutants by blocking prophase exit in a subset of cells where arrest is not absolute.
Conclusions
We propose that Mek1 plays dual roles during meiotic prophase I by phosphorylating targets directly involved in the recombination checkpoint as well as targets involved in sister chromatid recombination. We discuss how regulation of pachytene exit by Mek1 or similar kinases could influence checkpoint stringency, which may differ among species and between sexes.
Keywords: meiosis, recombination checkpoint, homologous chromosome, sister chromatid
Introduction
Meiosis is a specialized process of cell division whereby one round of DNA replication is followed by two rounds of chromosome segregation to produce gametes for sexual reproduction. During meiotic prophase I, programmed recombination and synapsis facilitate the formation of interhomolog crossovers that promote proper disjunction of homologous chromosomes at the first meiotic division (MI; [1-3]). Recombination and synapsis culminate in the late prophase stage known as pachytene, which is defined cytologically by the side-by-side alignment of condensed homologous chromosomes connected by the synaptonemal complex (SC) [4, 5]. In budding yeast the structural axis of each homolog is formed by Red1, Hop1 and Mek1 and the central region of the synaptonemal complex is made of Zip1 [6-11]. Notably, pachytene is the last stage of meiotic prophase before cells become committed to undergo MI [12, 13].
Meiotic recombination is a step-wise process initiated by Spo11-induced double-strand breaks (DSBs) and implemented using DNA repair factors within the context of meiotic chromosome architecture [14-16]. A subset of DSBs is repaired by a designated interhomolog crossover pathway that transits through single-end invasion (SEI) and double-Holliday Junction (dHJ) intermediates [17-21]. Remaining breaks are repaired using a sister chromatid as a template for repair or by interhomolog repair resulting in noncrossovers (i.e. events detected as a gene conversion without exchange of flanking markers) that arise from a synthesis-dependent strand annealing mechanism [18, 22-24]. Finally, there is a minor class of crossovers that are dependent on Mus81/Mms4 [25]. Physical associations between sister chromatids can be detected as well. At least a subset of intersister recombination events involves a dHJ intermediate [17]; multi-chromatid joint molecules, (mcJMs) arise during normal meiosis when two ends of a DSB independently invade different chromatids and/or sequentially invade multiple templates, however they can lead to nonregulated crossing over and missegregation if not processed [26].
The presence of unrepaired DSBs and/or incomplete synapsis activates a recombination checkpoint response that leads to inhibition of Ndt80, a transcription factor required for the exit from pachytene [27, 28]. Cells arrest at a pachytene-like stage with compacted chromatin [29]. Repair of DSBs leads to the activation of Ndt80, which then allows for the resolution of dHJs to form crossover products, and finally the completion of MI [18, 20]. Mek1 is a proposed downstream effector kinase of the Mec1/Tell (ATR/ATM) signaling that is activated in response to Spo11-induced DSBs [30, 31]. Activation of Mek1 requires the C-terminal domain of Hop1 which itself is a target of Mec1/Tel1 [31, 32]. Unlike other checkpoint mutations that allow division in the presence of unrepaired DSBs [33-41], the mek1Δ mutation acts as a bypass suppressor of the prophase I arrest phenotype conferred by dmc1Δ by allowing repair of Spo11 1-induced breaks using the sister chromatid as a substrate [42]. For this reason, the role of Mek1 as a bona fide checkpoint protein has comes into question. The role for Mek1 in modulating interhomolog bias is well substantiated; in wild-type cells, activation of Mek1 leads to the phosphorylation and inactivation of Rad54, which is required for sister chromatid recombination [43]
Here we describe a semi-dominant, gain-of-function allele, MEK1-GST (similar to that described by Niu et al., 2005 [32]) that confers phenotypes not previously described: i) A net gain of interhomolog events that is coupled to a net loss of intersister events, including intersister-dHJ and multi-chromatid joint molecules; ii) increased levels of interhomolog-noncrossover recombination products that are not associated with either increased DSBs or a change in interhomolog-crossover products; iii) a hyper-barrier to intersister repair; and iv) increased stringency of the output of the recombination checkpoint pathway. Our data support a model in which Mek1 plays dual roles during meiosis I prophase: one is to promote interhomolog bias and second is to act as a checkpoint effector that controls exit from pachytene.
Results
MEK1-GST is a gain-of function-allele
A conserved sequence at the C-terminus of the Mek1 is implicated in the homodimerization/activation of Mek1 [44]. Here we analyzed the effect of artificial dimerization at the C-terminus of Mek1 in meiotic recombination and checkpoint control via a fused GST moiety similar to that described for an N-terminal GST-MEK1 fusion created by Niu et al., 2005 [32]. As a control, we created an allele MEK1-GST(nd) that is mutated at two residues to prevent GST dimerization. In this case, the dimerization of Mek1-GST(nd) is presumably under the control of Hop1, just as for wild-type Mek1 [32].
We analyzed Mek1-GST and Mek1-GST(nd) protein at various time points during meiotic progression in a synchronized cell culture. Both fusion proteins appeared at about the same time (3 hours after transfer of cells to sporulation medium) by immuno-blot using an antibody to GST (Figure 1A). Mek1-GST protein levels were greater and persisted longer compared to Mek1-GST(nd). Similar results were found using two different antibodies to GST including one that recognizes an epitope outside of the dimerization domain (data not shown). The Clb5 cyclin, which is normally degraded prior to the meiosis division (MI), also persisted in the MEK1-GST background. MI timing was delayed in MEK1-GST compared to wild type MEK1 and MEK1-GST(nd) strains (Figure 1B and 1C). The MI delay was semi-dominant since MEK1-GST/MEK1 heterozygous strain exhibited an intermediate delay. The MEK1-GST induced delay requires SPO11 and RED1, suggesting that Mek1-GST kinase activities require an intact chromosome axis structure (Figure S1A). Spore viability of MEK1-GST and MEK1-GST(nd) strains was similar to wild type (Table S1). Taken together, MEK1-GST exhibits phenotypes consistent with it being a semi-dominant gain-of-function allele of MEK1.
Figure 1. MEK1-GSTis a gain of function allele.
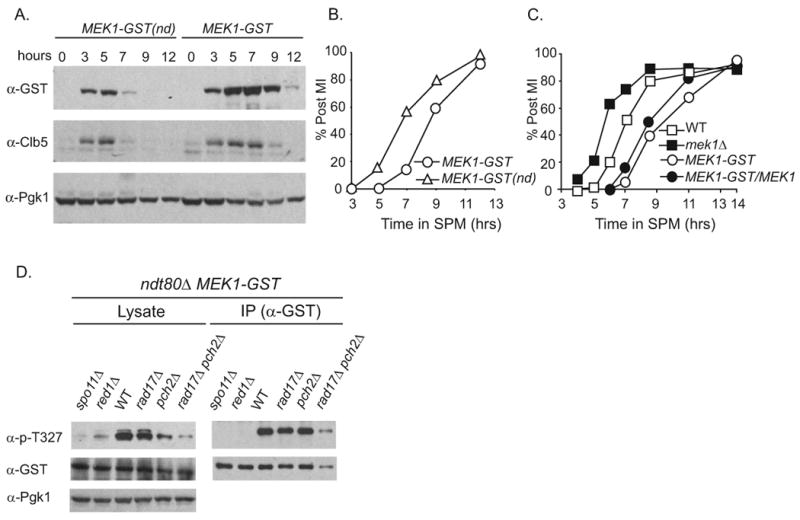
(A) Synchronous meiotic cultures of MEK1-GST and MEK1-GST(nd) were analyzed by immuno-blot at the indicated times using antibodies to GST (top), Clb5 (middle), and Pgk1 (bottom). (B) Percentage of cells in the culture at each time point that have undergone the first meiotic division as indicated by DAPI staining. (C) Same analysis as in B except for the indicated strains. (D) Western blot of cell lysates or Mek1-GST following immunoprecipitation with antibodies to p-T327, GST or Pgk1 for the indicated strains 4 hours after transfer to SPM. The levels of T-327 detection for immunoprecipitated Mek1-GST normalized to immunoprecipitated -GST for each mutant was compared to Mek1-GST alone. These values are as follows for two independent experiments (the first value corresponds to the figure shown in panel D. The second value shown is from cells collected at 4.5 hours after transfer to SPM from an independent time course): MEK1-GST spo11Δ 0.02, not determined; MEK1-GST red1Δ < 0.01, not determined; MEK1-GST, 1.00, 1.00; MEK1-GST rad17Δ, 0.94, 1.22; MEK1-GST pch2Δ, 0.88, 1.3 MEK1-GST rad17Δ pch2Δ 0.60, 0.78. See also Figure S1
The MEK1-GST allele behaves similarly to GST-MEK1 described by Niu et al., 2005 and Niu et al., 2007: Both alleles give nearly wild-type levels of sporulation and spore viability (Table S1) [32, 44]. Moreover, phosphorylation of the T327 residue for both GST-Mek1 and Mek1-GST is dependent on genes required for DSB formation including RED1 and SPO11 (Figure 1D). Notably, we are not able to distinguish whether the phenotypes associated with MEK1-GST are due to an increase in the persistence of a signal that is lower or more transient in wild-type cells or if it creates a toxic gain-of-function effect. We favor the former since MEK1-GST gives wild-type levels of spore viability (96.1% versus 96.5%; Table S1).
Repair of DSBs is delayed in MEK1-GST
To explore the effect of the MEK1-GST allele on modifying outcomes of meiotic recombination, we analyzed the physical intermediates and products of recombination at the HIS4LEU2 hot spot locus (Figure 2A; [19]). We found that steady-state levels of DSBs and slower migrating joint molecule species were greater in the MEK1-GST strain compared to MEK1-GST(nd) (Figure 2B). Consistent with this finding was the observation that crossover formation, one outcome of DSB repair, was delayed in MEK1-GST compared with MEK1-GST(nd) (Figure 2C). We next wanted whether or not the increase in steady-state levels of DSBs corresponded to an increase in their formation. We used a sae2Δ mutant background, in which DSBs are not turned over since their resection and repair is blocked. We found that DSB levels were similar in MEK1-GST sae2Δ and sae2Δ mutants suggesting that the higher levels of breaks are due to their slower turnover (Figure S2A, S2B). We also measured break levels in a dmc1 mutant background, in which breaks form and are resected but are unable to use a homolog as a substrate for repair [1-3]. DSB levels in MEK1-GST dmc1Δ strains were slightly reduced compared to the dmc1Δ single mutant, perhaps due to the inability to detect hyper-resected products by this assay (Figure S2A). Overall these data show that while DSBs form at wild type levels in MEK1-GST, their turnover is delayed.
Figure 2. Meiotic recombination analysis at the HIS4∷LEU2 hotspot in MEK1-GST compared to MEK1-GST(nd).
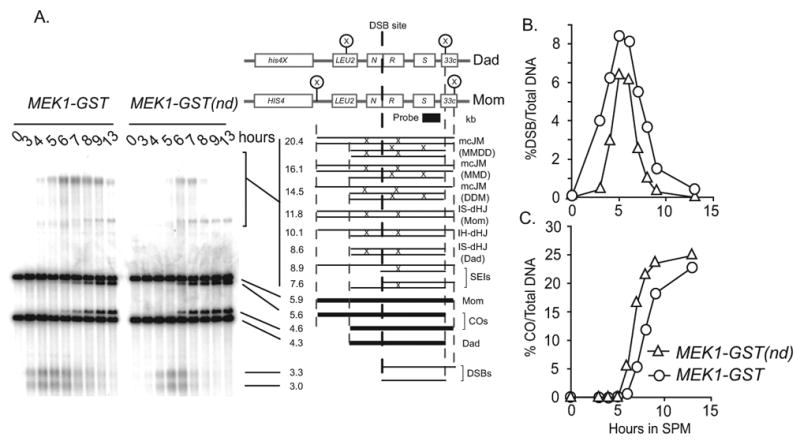
(A) Physical analysis of recombination by Southern blot to measure the formation and turnover of recombination intermediates at the indicated time points following transfer of synchronized cells to SPM in MEK1-GST and MEK1-GST(nd) strains. DNA samples in this experiment are psoralen-crosslinked. The physical structure and molecular weights corresponding to each band of a XhoI digest of the HIS4∷LEU2-(NBam)/his4-X∷LEU2-(NBam)-URA3 recombination hot spot in the Southern blot is diagramed on the right [19]. The slowly migrating species is labeled with an asterisk. (B and C) Quantification of the formation and turnover of DSB products (%DSBs/Total DNA) and crossover products (%COs/Total DNA) from the Southern blot in part A, respectively. See also Figure S2.
MEK1-GST gives a net gain of interhomolog events that is coupled with a net loss of intersister events compared to WT
Since loss of MEK1 function is associated with loss of interhomolog bias, we reasoned that the gain of function mutation, MEK1-GST might exhibit delayed turnover of DSBs due to a prolonged period of interhomolog recombination. Since crossover levels in MEK1-GST are not increased compared to wild type, we tested the possibility that excess interhomolog events are repaired as noncrossovers. For this test we used a strain carrying a HIS4LEU2 allele variant in which both crossover and noncrossover levels could be measured in the same population of cells [26, 45]. Interestingly, we found that noncrossover levels in MEK1-GST were two-fold greater than in wild type, while crossover levels were unchanged (Figure 3A-D).
Figure 3. Noncrossover recombination products are increased in MEK1-GST compared to WT while crossover levels are unchanged.
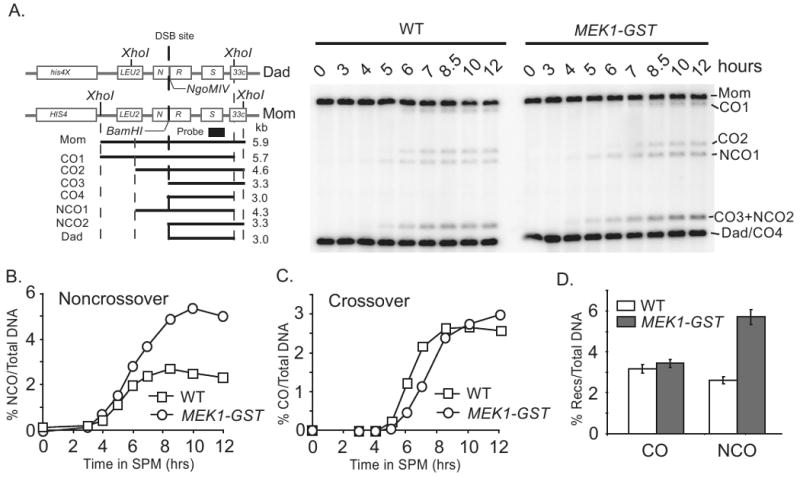
(A) Schematic of the HIS4∷LEU2-(BamHI)/his4-X∷LEU2-(NgoMIV) hotspot (left) and the timing of noncrossover and crossover formation in WT and MEK1-GST. DNA was isolated from synchronous meiotic cultures and digested with XhoI and NgoMIV prior to Southern analysis. At this hot spot, the DSB can occur at either the BamHI chromosome or the NgoMIV chromosome (the chance is equal). By digesting with XhoI and NgoMIV, there are four species of crossover products. CO4 is the same size as the DAD band (3.0kb). Further details of this hotspot are described in Martini et al. 2006 and Oh et al., 2007 [26, 45]. (B and C) Quantification of noncrossover and crossover levels at indicated times, respectively. (D) Quantification of crossover (CO) and noncrossover (NCO) levels at t = 12 hours after transfer to SPM. For each strain, four independent meiosis cultures from the same time course were analyzed.
We reasoned that the increase in noncrossover products could arise from breaks that would otherwise have been repaired using a sister chromatid as a template. We tested whether or not intersister events, including both intersister- dHJ and multi-chromatid joint molecules (mcJM), were reduced in MEK1-GST compared to wild type by using two-dimensional gel electrophoresis (Figure 4A) [26, 46]. This analysis was carried out in an ndt80Δ mutant background, which arrests prior to the exit from pachytene and prevents turnover of the majority of joint-molecule species to final products. In this way, the absolute levels of these intermediates can be directly compared [18, 26, 47]. Indeed, we observed that intermediates of DNA repair events involving sister chromatids were reduced in MEK1-GST compared to wild type, with a greater loss of mcJMs compared to IS-dHJ. By contrast, the total levels of intermediates that give rise to interhomolog crossover products, including SEIs and dHJs were the same in MEK1-GST compared to wild type (Figure 4B, 4C, 4D). Together, these results suggest that MEK1-GST promotes interhomolog recombination events at the expense of intersister recombination. Moreover, the increased level of interhomolog interactions is specifically directed to noncrossover products, suggesting that double-strand breaks are designated for repair as crossovers prior to the activation of MEK1-GST by ectopic dimerization. These data also indicate that the increased steady-state levels of JM seen by 1-D Southern blot analysis (Figure 2A) is due to slow turnover of JM into downstream products.
Figure 4. The levels of intersister-dHJs and mc-JMs are decreased in MEK1-GST compared to WT.
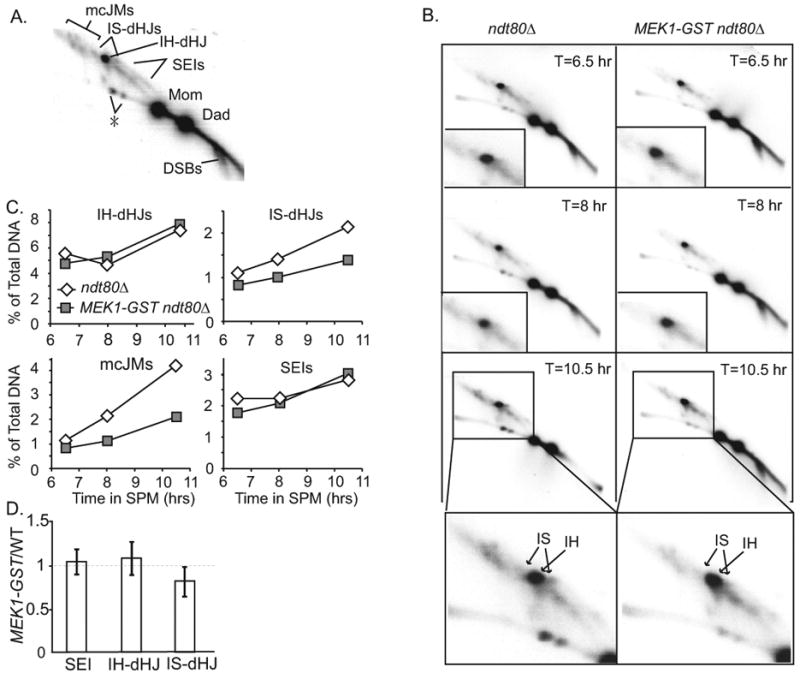
(A) Position of DNA joint molecules on Southern blot of a 2-D gel from synchronized cultures of ndt80Δ after transfer to SPM. The identity of the spot denoted by the asterisk is not known. (B) Southern blots of 2-D gels from a representative time course of ndt80Δ and ndt80 Δ MEK1-GST. DNA samples in this experiment are psoralen-crosslinked. The relative area used for quantification of intersister- and interhomolog-dHJs are as indicated in the lower panel. (C) Quantification of JM structures from time course shown in B. (D) Quantification of JMs from two independent time-courses including time points ranging from 6.5 hours to 11.5 hours. Mean +/- SD of the value of the ratio of SEI, interhomolog- dHJ and intersister-dHJ joint molecules in ndt80Δ MEK1-GST strain compared to WT. In ndt80Δ MEK1-GST, intersister-dHJs are decreased to 82% of WT levels (p = 0.035; paired t-test). Interhomolog-dHJ and SEI in MEK1-GST are not significantly different from WT (p = 0.283 and p = 0.619, respectively). The authors were able to visually assign all relevant blots with the correct strains in a blind analysis.
The MI delay conferred by rad17Δ or pch2Δ single mutants is exacerbated when MEK1-GST is present
Both Rad17, a component of mitotic DNA damage checkpoint, and Pch2, an AAA-ATPase like protein, are involved in checkpoint surveillance during meiosis [33-35, 39, 40]. Single mutants of rad17Δ and pch2Δ exhibit an MI delay, presumably because each mutant generates a lesion that activates the other's checkpoint function [39]. We tested the epistatic relationship between the delay conferred by MEK1-GST and that of either rad17Δ or pch2Δ. We found that MEK1-GST rad17Δ and MEK1 pch2Δ strains were further delayed compared to any of the three single mutants (Figure S1B). Spore viability of MEK1-GST rad17Δ was increased compared to rad17Δ (63% versus 34%) as were crossover levels measured at a late time point, albeit not to wild-type levels (Table S1, Figure S1C). Spore viability of the MEK1-GST pch2Δ double mutant remained high, just as in the pch2Δ single mutant (Table S1) even though fewer cells of the MEK1-GST pch2Δ genotype sporulated. Crossover levels were substantially reduced but this is likely due to the severe MI delay observed for the double mutant (Figure S1C). T327 phosphorylation of Mek1-GST occurs in both rad17Δ and pch2Δ single mutant strains (Figure 1D). The MI delays conferred by either rad17Δ or pch2Δ are bypassed by mek1Δ (Figure S1D, S1E). Since rad17Δ and pch2Δ function in different processes associated with recombination as well as checkpoint signaling, interpretation of the double-mutant phenotypes must be made cautiously [33, 34, 39, 40, 48-51]. Minimally, these data indicate that the delay conferred by MEK1-GST is not simply due to the activation of either the Rad17 or Pch2 dependent checkpoint pathways. Below we address the interaction of MEK1-GST with the rad17Δ pch2Δ double mutant where both recombination checkpoint functions are absent.
MEK1-GST promotes interhomolog bias in the absence of both RAD17- and PCH2-dependent checkpoint functions
We showed previsously that the rad17Δ pch2Δ double mutant progresses rapidly through MI, produces reduced levels of crossover products and gives < 1% spore viability. The double mutant is also defective in checkpoint signaling since MI divisions still occur in mutant strain backgrounds that accumulate unrepaired DSBs [39]. We reasoned that if the MEK1-GST imposed delay was due the activation of the recombination checkpoint then the MEK1-GST rad17Δ pch2Δ triple mutant would divide with the same kinetics as the rad17Δ pch2Δ double mutant. To our surprise, the MEK1-GST rad17 pch2Δ triple mutant gave a delayed MI phenotype compared to rad17Δ pch2Δ (Figure 5A) and spore viability was increased from < 1% to 47% (Table S1). This suppression of spore inviability in the rad17Δ pch2Δ was semi-dominant since spore viability in MEK1-GST/MEK1 heterozygous background was ∼29% (Table S1). Physical analysis of DSB repair in the MEK1-GST rad17Δ pch2Δ triple mutant indicated that DSB turnover was slower, yet crossover products were elevated (Figure 5B, 5C and 5D). These results indicate that MEK1-GST can uphold interhomolog bias even in the absence of the recombination checkpoint.
Figure 5. MEK1-GSTrestores interhomolog bias in the absence ofRAD17andPCH2-dependent surveillance/signaling functions.
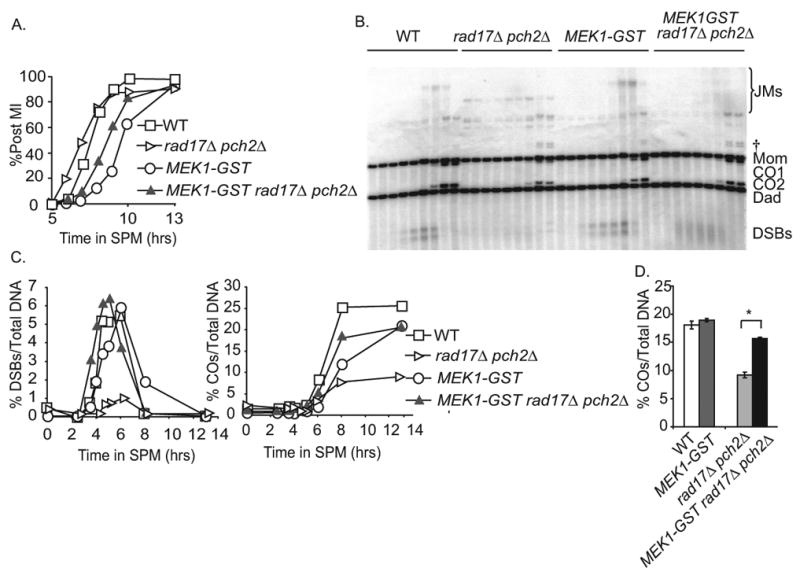
(A) Percentage of cells in a synchronized meiotic cultures that have undergone the first meiotic division at the indicated time points in WT, rad17Δ pch2Δ, MEK1-GST, MEK1-GST rad17Δ pch2Δ. (B) Southern blot and quantification of recombination intermediates and crossovers at the HIS4LEU2 hotspot in WT, rad17Δ pch2Δ, MEK1-GST and MEK1-GST rad17Δ pch2Δ. For each strain, the 0, 2.5, 3.5 4, 4.5, 5, 6, 8 and 13 hour time points were analyzed. The bands immediately above the Mom fragment in the rad17Δ pch2Δ– containing strains are ectopic recombination species [34]. DNA samples in this experiment are Psoralen-crosslinked. †, DNA fragments arising from ectopic recombination between HIS4∷LEU2 and the linked, endogenous leu2 locus [34]. (C) Quantification of the formation and turnover of DSB products (%DSBs/Total DNA), crossover products (%COs/Total DNA) and %post MI cells from the Southern blot experiment is shown in part B, (D) Final crossover levels/total DNA in WT and checkpoint mutants used in parts B and C. Mean +/- SD of three independent cultures is shown. The asterisks indicate the differences were statistically significant (p < 0.05) by paired t-test. Results presented in (A) to (C) are from the same time course.
We ruled out possibility that another DNA damage checkpoint was activated in the MEK1-GST rad17Δ pch2 Δ triple mutant. First, we found that the addition of the dmc1Δ mutation to the triple mutant background did not lead to MI arrest, as would be expected if another checkpoint function were activated. In fact, dmc 1Δ MEK1-GST rad17 Apch2Δ MI kinetics were indistinguishable from the MEK1-GST rad17Δ pch2Δ triple mutant (Figure 6A, 6B). Second, we showed that these cells divided even though DSBs were not fully repaired as indicated by physical analysis at the HIS4LEU2 hot spot (Figure 6C). In addition, DAPI stained chromosomes in the quadruple mutant background were fragmented (data not shown), consistent with the presence of unrepaired breaks.
Figure 6. MEK1-GSTimposes a delay independent of meiotic checkpoint.
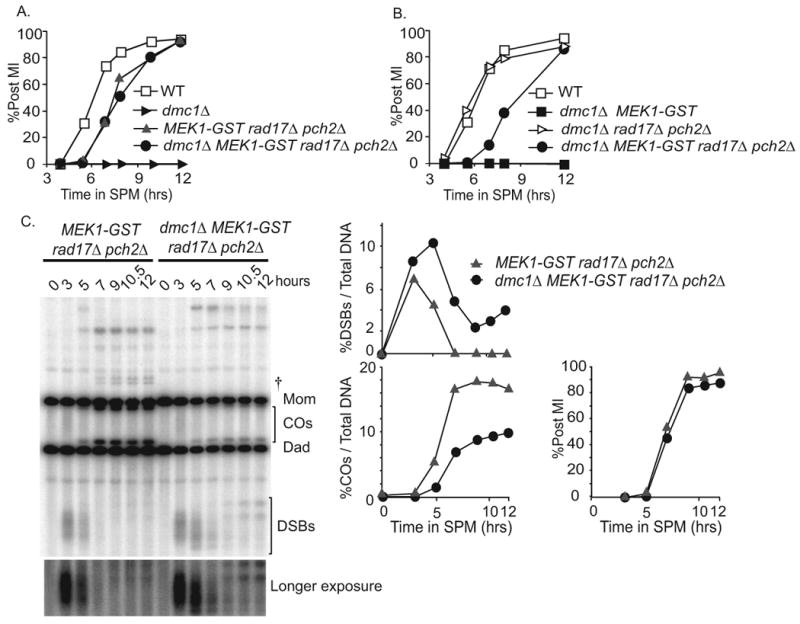
(A-B) Percentage of cells in synchronized meiotic cultures that have undergone the first meiotic division. Strains used include WT, MEK1-GST, dmc1Δ, MEK1-GST rad17Δ pch2Δ, dmc 1Δ MEK1-GST and dmc1Δ MEK1-GST rad17Δ pch2Δ (C) Southern blot analysis of MEK1-GST rad17Δ pch2Δ versus dmc1Δ MEK1-GST rad17Δ pch2Δ DSB formation and turnover (%DSBs/Total DNA), CO formation (%COs/Total DNA) and post MI kinetics (%Post MI/Total DNA) are shown. DNA samples in this experiment are not Psoralen-crosslinked. Two slower migrating bands appeared in the quadruple mutant at late time points, after the majority of cells had already undergone MI division. The migration pattern of these bands is reminiscent of hairpin structures that form from hyper-extended 3′ ends of DSBs at HIS4LEU2 locus in the rad52 mutants [47]. †, meiosis-specific hybridization bands, probably ectopic recombination resulted from HIS4∷LEU2 recombining with the leu2 locus [34].
MEK1-GST increases the stringency of the meiotic recombination checkpoint response
The delay phenotype conferred by MEK1-GST suggests that it may be influencing the regulation of MI division, independent of upstream signaling functions. We next wanted to see what effect MEK1-GST would have in mutants that exhibit checkpoint mediated delay, but eventually go on to divide. As shown previously, deletion of ZIP1, NDJ1 and CSM4 results in checkpoint-mediated MI delay yet the majority of cells ultimately divide, albeit with low spore viability (Figure 7 A, 7B and 7C; Table S1) [52-54]. Interestingly, the MEK1-GST zip1Δ double mutant exhibited near complete MI arrest, unlike the single mutants (Figure 7A). For the MEK1-GST ndj1Δ and MEK1-GST csm4Δ double mutants, MI division timing was further delayed compared to either single mutant and a greater fraction of cells failed to undergo the MI division, however crossover levels were not affected (Figure 7B and 7C; Figure S3A and S3B).
Figure 7. MEK1-GST exhibits a hyper-checkpoint function.
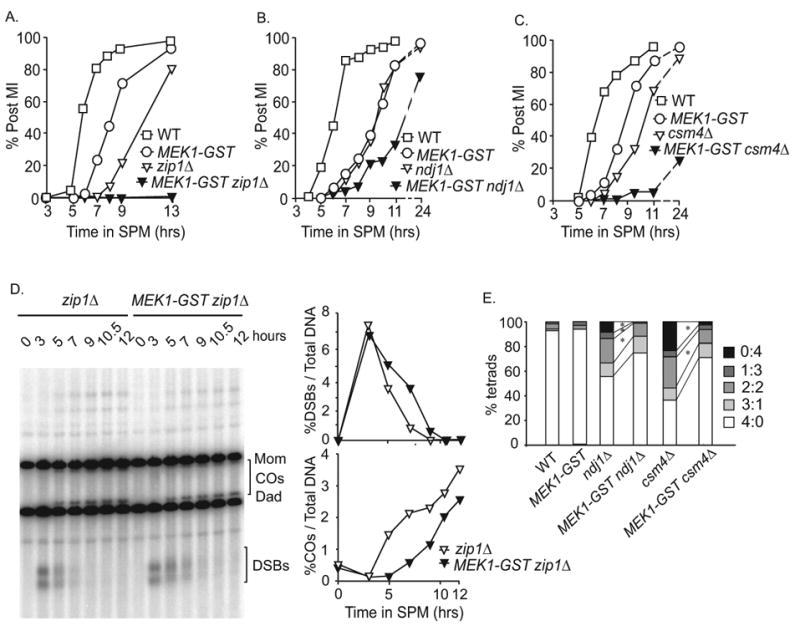
(A-C) Percentage of cells in a synchronized meiotic cultures that have undergone the first meiotic division at the indicated time points in WT, MEK1-GST, zip1Δ, MEK1-GST zip1Δ, ndj1Δ,MEK1-GST ndj1Δ, csm4Δ, and MEK1-GST csm4Δ. (D) Southern blot and quantification of zip1Δ compared to MEK1-GST zip 1Δ (E) Categories of tetrads from ndj1Δ and csm4Δ mutants (viable: inviable). A 2:2 pattern of viable to nonviable spores is expected from a MI non-disjunction event. A 3:1 pattern might arise by non-disjunction in MII or premature sister chromatid segregation in MI. The 0:4 and 1:3 classes indicate multiple meiosis I and/or meiosis II non-disjunction events so were not further considered in this analysis. Only four spore tetrads were analyzed. The asterisks indicate the differences were statistically significant (p < 0.05) by Fisher's exact test. Note: the strains used in A do not contain the HIS4LEU2 hotspot and are described in the strain Table S2 See also Figure S3.
We next asked if the effect of MEK1-GST on MI delay/arrest in these double mutant situations was due to a synergistic defect causing an accumulation of recombination intermediates that in turn led to a persistent checkpoint response. We measured DSB formation and repair at the HIS4LEU2 locus in zip1Δ and MEK1-GST zip 1Δ mutants and found that DSBs were efficiently repaired in both situations (Figure 7D). We ruled out the possibility that the failure to detect breaks is due to hyper-resection of 5′ ends by analyzing full-length chromosome fragments using clamped homogenous electric field (CHEF) gel analysis. There was no smear that would indicate unrepaired, hyper-resected DSBS in MEK1-GST zip1Δ (Figure S3C). arrest conferred by MEK1-GST is not due to the failure to repair DSBs, as far as our limits of detection can discern. Together, these results suggest that once Mek1-GST is dimerized/activated, it is less able than wild type to deactivate and shut off an inhibitory affect on MI division, even when checkpoint-activating lesions have been repaired.
Strikingly, compared to each single mutant, MEK1-GST rescued the spore inviability patterns conferred by ndj1 Δ (75% versus 90%) and csm4Δ (64% versus 87%; Table S1). The zip1 Δ MEK1-GST double mutant gave no spore products for analysis. For ndj1 Δ and csm4Δ mutants, we classified tetrads into four categories according to the number of viable spores per tetrad (Figure 7E). While all aberrant classes were suppressed to some extent in the presence of the MEK1-GST allele, 2:2 and 0:4 classes were significantly reduced in the ndj1Δ and csm4Δ backgrounds. Combined, these results suggest that MEK1-GST selectively suppresses MI division in ndj1Δ or csm4Δ cells that were unsuccessful in the maturation of chromosome structures that facilitate meiosis I disjunction.
Discussion
Our findings provide insight into the roles of the chromosome axis associated protein Mek1 in meiotic recombination and in regulating the exit from meiosis I in budding yeast. We show evidence that Mek1 kinase has multiple targets that include proteins involved in suppressing sister chromatid recombination as well as those that function to regulate the exit from pachytene. We found that ectopic Mek1 dimerization impacted events of meiosis I prophase in two ways: (i) by enhancing interhomolog bias, perhaps through the prolonged or premature activation of a Mek1 target substrate such as Rad54 [43] and (ii) by inhibiting the re-entry into the cell cycle following checkpoint-mediated arrest in certain mutant situations, perhaps through the persistent phosphorylation of a protein that regulates the exit from pachytene.
Crossover/noncrossover designation may be imposed prior to or concomitant with Mek1 dimerization
An outstanding problem in the field of meiosis has been to define the point among the myriad chromosomal processes when DSBs are designated for crossover repair versus noncrossover repair. Crossover designation is thought to occur at early stages of meiosis I prophase prior to formation of SEI joint molecule formation [17-21], however there is evidence that single-stranded 3′ ends of DSBs may sample a number of substrates, including sister chromatids, prior to being committed for repair as a crossover [26]. It is conceivable then that crossover designation occurs prior to or concomitant with Mek1 dimerization, after which point the remaining breaks would be repaired as either noncrossovers or using sister chromatids, with the former being favored due to Mek1 dimerization and/or activation. This model would predict that artificially prolonging the period of interhomolog bias or premature activation of Mek1 might result in an increase in noncrossover products and a decrease in the levels of intersister joint molecules, with no change in crossover levels; the MEK1-GST allele confers this same constellation of phenotypes.
Interestingly, the loss of intersister interactions observed in the presence of Mek1-GST comes largely from a pool of mcJM species, which are aberrant products involving three or four chromatids that occur in an unchallenged meiosis [26]. Our findings using MEK1-GST suggest that Mek1 antagonizes mcJM formation, as is also the case for Sgs1 [26].
Sheridan and Bishop have postulated that disassembly of axial constraints at the end of prophase could activate the “clean-up” of residual DSBs via intersister recombination [55]. This constraint might be mechanistically tied to the active state of a Mek1 target substrate and the slow turnover of DSBs that we observe. If this constraint were prolonged due to an artificially maintained active state of Mek1, more breaks may end up being repaired via interhomolog noncrossover recombination rather than using a sister chromatid.
Our finding that MEK1-GST preserves crossover levels, even when total interhomolog events are increased is reminiscent of “crossover homeostasis”. Crossover homeostasis has been observed in situations when the number of crossovers per chromosome remains constant even when the total number of breaks is reduced, as in a spo11 hypomorph [45, 56]. Our results indicate that the converse is also true; when the total number of interhomolog events per chromosome is increased, as in the case of MEK1-GST, crossover levels remain unchanged. This phenomenon indicates a possible late role for Mek1-GST impacting the output of the crossover/noncrossover decision. In either case, the dimerization/activation of Mek1 may serve as a regulatory landmark to couple these two processes.
Dual roles for Mek1 in interhomolog bias and recombination checkpoint signaling
Findings to date suggest that early stages of meiotic recombination checkpoint function and interhomolog bias are inextricably linked. Both checkpoint signaling and partner choice require the activation of the ATM/ATR signaling pathways [31, 33]. It is conceivable that Mek1 acts solely to ensure that the interhomolog bias is upheld, primarily through its role in phosphorylating Rad54 or other like targets [43]. On the other hand, Mek1 is similar to the Rad53 DNA damage checkpoint kinase that acts downstream of ATM/ATR in vegetative cells and regulates a wide range of targets in response to DNA damage [57]. While Rad53 is phosphorylated in response to unrepaired DSBs during MI, it does not affect cell cycle arrest until after the MI division [42, 58]. Thus Mek1 is an attractive candidate for maintaining recombination-induced arrest during and after the formation of interhomolog connections.
It would seem reasonable to consider that by extending the period of interhomolog bias, there would be slower turnover of DSBs, which in turn would activate a checkpoint-mediated delay from pachytene exit. We were surprised, however, to find that MEK1-GST conferred an MI delay even when the Rad17 and Pch2-dependent recombination checkpoint functions were absent. We were able to rule out the possibility that MEK1-GST activated an alternative DNA damage pathway (e.g. Rad9/Rad53) in this situation since deletion of DMC1 in this strain background did not induce an additional MI delay or arrest phenotype. These data suggest that Mek1 dimerization can influence the regulation of prophase exit even in the absence of surveillance/signaling functions of Rad17 and Pch2. The increased period of interhomolog bias thus can account for the delay observed in the MI division and the turnover of DSBs in MEK1-GST cells. We suspect that Mek1-GST exhibits at least some transient association/activation within the chromosome loop-axis structure that has experienced a Spo11-induced DSB, independent of Hop1 phosphorylation by ATM/ATR (Carballo et al; Nui et al). This could be true for wild type Mek1 as well.
Checkpoint stringency can be modulated by controlling exit from pachytene arrest
Deletion of a subset of meiotic genes leads to a checkpoint induced MI delay, followed by a division that gives rise to inviable spore products. We found that MEK1-GST exacerbates the delay in which ndj1Δ and csm4Δ□ mutants progress through MI and even blocks MI division in a subset of cells. The observed increase in spore viability for the double mutants suggest that those cells arrested in the presence of MEK1-GST may represent the same pool of cells that would have otherwise gone on to form inviable spore products.
What mechanism prevents MI division in a subset of cells in ndj1Δ/csm4Δ mutants, and complete arrest in zip1Δ when MEK1-GST is present? Conversely, why do zip1Δ/ndj1Δ/csm4Δ single mutants (with wild-type MEK1) proceed through MI division, only to give rise to inviable spore products? To accommodate the delay/arrest phenotype conferred by the zip1Δ/ndj1Δ/csm4Δ mutants in combination with MEK1-GST, we propose that prolonged dimerization/activation of MEK1-GST inhibits the re-entry into the cell cycle following checkpoint-mediated arrest in these mutant backgrounds, perhaps through the persistent phosphorylation of a protein that regulates the exit from pachytene. Based on these data we suggest that Mek1 phosphorylates at least one target that is directly involved in the meiotic recombination checkpoint in a challenged meiosis in addition to its targets that are directly involved in interhomolog bias.
One possibility is that MEK1-GST inhibits the process of checkpoint recovery. Checkpoint recovery occurs when signaling through the checkpoint response has ended and cells re-enter the cell cycle [59]. To date, checkpoint recovery has been described only in nonmeiotic cells of yeast, so this would provide the first observation of the phenomenon in meiosis. Another possibility is that MEK1-GST mediated arrest in the zip1Δ/ndj1Δ/csm4Δ mutant situations is due to inhibition of the checkpoint adaptation response where divisions occur even though lesions are not repaired [38]. This seems less likely since unrepaired DSBs do not accumulate in the MEK1-GST zip1Δ mutant, as would be expected if adaptation were inhibited. On the other hand, there could be a low level of breaks that were not detected with the assays we used here.
It is curious to consider why an intact checkpoint would not function to maximize spore viability in a “challenged meiosis”. It is perhaps advantageous for yeast to progress quickly through sporulation since the resultant spore stage is resistant to myriad environmental insults. Sexually dimorphic levels of checkpoint stringency observed in animals [60] may be determined in part through the modulation of a similar checkpoint-related kinase. To date, Mek1orthologs have only been found in fungi. In S. pombe, the Mek1 ortholog phosphorylates Cdc25 phosphatase and causes arrest prior to MI [61]. Since Cdc25 is also a target of Chk1/Chk2 kinases in mitotic checkpoint pathways in metazoans [62], Chk1/Chk2 might play a role similar to Mek1 to uphold checkpoint stringency during meiosis in higher eukaryotes.
Experimental Procedures
Standard methods were used to construct yeast strains, synchronize meiotic cells and prepare DNA and proteins samples for analysis by Southern and western blotting, respectively. Detailed methods are described in supplementary materials.
Highlights.
MEK1 gain-of-function mutation enhances meiotic interhomolog recombination
Meiotic crossover designation occurs prior to or concomitant with Mek1 dimerization
Mek1 plays dual roles in interhomolog bias and the recombination checkpoint
Checkpoint stringency can be enhanced by regulating the exit from pachytene
Supplementary Material
Acknowledgments
We thank JoAnne Engebrecht, Joshua Chang Mell, Neil Hunter and Michael Lichten for discussions and thoughtful comments on the manuscript. We thank Michael Lichten for sharing unpublished results. We also thank the Hunter lab for strains. National Institutes of Health grant RO1GM075119 to S.M.B supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- 2.Bhalla N, Dernburg AF. Prelude to a division. Annual Review of Cell and Developmental Biology. 2008;24:397–424. doi: 10.1146/annurev.cellbio.23.090506.123245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter N. Meiotic recombination. In: Aguilera A, Rothstein R, editors. Molecular Genetics of Homologous Recombination. Heidelberg: Springer-Verlag; 2006. pp. 381–342. [Google Scholar]
- 4.Moses MJ. Synaptonemal complex. Annual Review of Genetics. 1968;2:363–412. [Google Scholar]
- 5.Page SL, Hawley RS. The genetics and molecular biology of the synaptonemal complex. Annual Review of Cell and Developmental Biology. 2004;20:525–558. doi: 10.1146/annurev.cellbio.19.111301.155141. [DOI] [PubMed] [Google Scholar]
- 6.Rockmill B, Roeder GS. RED1: a yeast gene required for the segregation of chromosomes during the reductional division of meiosis. Proc Natl Acad Sci USA. 1988;85:6057–6061. doi: 10.1073/pnas.85.16.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rockmill B, Roeder GS. A meiosis-specific protein kinase homolog required for chromosome synapsis and recombination. Genes Dev. 1991;5:2392–2404. doi: 10.1101/gad.5.12b.2392. [DOI] [PubMed] [Google Scholar]
- 8.Sym M, Engebrecht J, Roeder GS. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell. 1993;72:365. doi: 10.1016/0092-8674(93)90114-6. [DOI] [PubMed] [Google Scholar]
- 9.Hollingsworth NM, Ponte L. Genetic interactions between HOP1, RED1 and MEK1 suggest that MEK1 regulates assembly of axial element components during meiosis in the yeast Saccharomyces cerevisiae. Genetics. 1997;147:33–42. doi: 10.1093/genetics/147.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith AV, Roeder GS. The yeast Red1 protein localizes to the cores of meiotic chromosomes. J Cell Biol. 1997;136:957–967. doi: 10.1083/jcb.136.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollingsworth NM, Byers B. HOP1: a yeast meiotic pairing gene. Genetics. 1989;121:445–462. doi: 10.1093/genetics/121.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidow LS, Byers B. Enhanced gene conversion and postmeiotic segregation in pachytene-arrested Saccharomyces cerevisiae. Genetics. 1984;106:165–183. doi: 10.1093/genetics/106.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuster EO, Byers B. Pachytene arrest and other meiotic effects of the start mutations in Saccharomyces cerevisiae. Genetics. 1989;123:29–43. doi: 10.1093/genetics/123.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P. An atypical topoisomerase II from archaea with implications for meiotic recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- 15.Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 16.Blat Y, Protacio RU, Hunter N, Kleckner N. Physical and functional interactions among basic chromosome organizational features govern early steps of meiotic chiasma formation. Cell. 2002;111:791–802. doi: 10.1016/s0092-8674(02)01167-4. [DOI] [PubMed] [Google Scholar]
- 17.Schwacha A, Kleckner N. Identification of double Holliday junctions as intermediates in meiotic recombination. Cell. 1995;83:783–791. doi: 10.1016/0092-8674(95)90191-4. [DOI] [PubMed] [Google Scholar]
- 18.Allers T, Lichten M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell. 2001;106:47. doi: 10.1016/s0092-8674(01)00416-0. [DOI] [PubMed] [Google Scholar]
- 19.Hunter N, Kleckner N. The single-end invasion: An asymmetric intermediate at the double-strand break to double-Holliday junction transition of meiotic recombination. Cell. 2001;106:59–70. doi: 10.1016/s0092-8674(01)00430-5. [DOI] [PubMed] [Google Scholar]
- 20.Clyne RK, Katis VL, Jessop L, Benjamin KR, Herskowitz I, Lichten M, Nasmyth K. Polo-like kinase Cdc5 promotes chiasmata formation and cosegregation of sister centromeres at meiosis I. Nat Cell Biol. 2003;5:480–485. doi: 10.1038/ncb977. [DOI] [PubMed] [Google Scholar]
- 21.Borner GV, Kleckner N, Hunter N. Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell. 2004;117:29–45. doi: 10.1016/s0092-8674(04)00292-2. [DOI] [PubMed] [Google Scholar]
- 22.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessop L, Rockmill B, Roeder GS, Lichten M. Meiotic chromosome synapsis-promoting proteins antagonize the anti-crossover activity of sgs1. PLoS Genet. 2006;2:e155. doi: 10.1371/journal.pgen.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMahill MS, Sham CW, Bishop DK. Synthesis-dependent strand annealing in meiosis. PLoS Biol. 2007;5:e299. doi: 10.1371/journal.pbio.0050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de los Santos T, Hunter N, Lee C, Larkin B, Loidl J, Hollingsworth NM. The Mus81/Mms4 endonuclease acts independently of double-Holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics. 2003;164:81–94. doi: 10.1093/genetics/164.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh SD, Lao JP, Hwang PY, Taylor AF, Smith GR, Hunter N. BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell. 2007;130:259–272. doi: 10.1016/j.cell.2007.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chu S, Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell. 1998;1:685–696. doi: 10.1016/s1097-2765(00)80068-4. [DOI] [PubMed] [Google Scholar]
- 28.Tung KS, Hong EJ, Roeder GS. The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis-specific transcription factor Ndt80. Proc Natl Acad Sci U S A. 2000;97:12187–12192. doi: 10.1073/pnas.220464597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roeder GS, Bailis JM. The pachytene checkpoint. Trends Genet. 2000;16:395–403. doi: 10.1016/s0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- 30.Bailis JM, Roeder GS. Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell. 2000;101:211–221. doi: 10.1016/S0092-8674(00)80831-4. [DOI] [PubMed] [Google Scholar]
- 31.Carballo JA, Johnson AL, Sedgwick SG, Cha RS. Phosphorylation of the axial element protein Hop1 by Mec1/Tell ensures meiotic interhomolog recombination. Cell. 2008;132:758–770. doi: 10.1016/j.cell.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 32.Niu H, Wan L, Baumgartner B, Schaefer D, Loidl J, Hollingsworth NM. Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell. 2005;16:5804–5818. doi: 10.1091/mbc.E05-05-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lydall D, Nikolsky Y, Bishop D, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- 34.Grushcow JM, Holzen TM, Park KJ, Weinert T, Lichten M, Bishop DK. Saccharomyces cerevisiae checkpoint genes MEC1, RAD 17 and RAD24 are required for normal meiotic recombination partner choice. Genetics. 1999;153:607–620. doi: 10.1093/genetics/153.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.San-Segundo PA, Roeder GS. Pch2 links chromatin silencing to meiotic checkpoint control. Cell. 1999;97:313–324. doi: 10.1016/s0092-8674(00)80741-2. [DOI] [PubMed] [Google Scholar]
- 36.San-Segundo PA, Roeder GS. Role for the silencing protein Dot1 in meiotic checkpoint control. Mol Biol Cell. 2000;11:3601–3615. doi: 10.1091/mbc.11.10.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong EJ, Roeder GS. A role for Ddc1 in signaling meiotic double-strand breaks at the pachytene checkpoint. Genes Dev. 2002;16:363–376. doi: 10.1101/gad.938102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hochwagen A, Tham WH, Brar GA, Amon A. The FK506 binding protein Fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell. 2005;122:861–873. doi: 10.1016/j.cell.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Wu HY, Burgess SM. Meiotic chromosome metabolism is monitored by two distinct surveillance mechanisms in budding yeast. Curr Biol. 2006;16:2473–2479. doi: 10.1016/j.cub.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shinohara M, Sakai K, Ogawa T, Shinohara A. The Mitotic DNA damage checkpoint proteins Rad17 and Rad24 are required for repair of double-Strand breaks during meiosis in yeast. Genetics. 2003;164:855–865. doi: 10.1093/genetics/164.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joyce EF, McKim KS. Drosophila PCH2 is required for a pachytene checkpoint that monitors double-strand-break-independent events leading to meiotic crossover formation. Genetics. 2009;181:39–51. doi: 10.1534/genetics.108.093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu L, Weiner B, Kleckner N. Meiotic cells monitor the status of the interhomolog recombination complex. Genes Dev. 1997;11:106–118. doi: 10.1101/gad.11.1.106. [DOI] [PubMed] [Google Scholar]
- 43.Niu H, Wan L, Busygina V, Kwon Y, Allen JA, Li X, Kunz RC, Kubota K, Wang B, Sung P, et al. Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol Cell. 2009;36:393–404. doi: 10.1016/j.molcel.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niu H, Li X, Job E, Park C, Moazed D, Gygi SP, Hollingsworth NM. Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol Cell Biol. 2007;27:5456–5467. doi: 10.1128/MCB.00416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martini E, Diaz RL, Hunter N, Keeney S. Crossover homeostasis in yeast meiosis. Cell. 2006;126:285–295. doi: 10.1016/j.cell.2006.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwacha A, Kleckner N. Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell. 1994;76:51–63. doi: 10.1016/0092-8674(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 47.Lao JP, Oh SD, Shinohara M, Shinohara A, Hunter N. Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol Cell. 2008;29:517–524. doi: 10.1016/j.molcel.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Schimenti JC. Mouse pachytene checkpoint 2 (Trip13) is required for completing meiotic recombination but not synapsis. PLoS Genetics. 2007;3:e130. doi: 10.1371/journal.pgen.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zanders S, Alani E. The pch2Δ mutation in baker's yeast alters meiotic crossover levels and confers a defect in crossover interference. PLoS Genet. 2009;5:e1000571. doi: 10.1371/journal.pgen.1000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joshi N, Barot A, Jamison C, Borner GV. Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet. 2009;5:e1000557. doi: 10.1371/journal.pgen.1000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borner GV, Barot A, Kleckner N. Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc Natl Acad Sci U S A. 2008;105:3327–3332. doi: 10.1073/pnas.0711864105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu HY, Burgess SM. Ndj 1, a telomere-associated protein, promotes meiotic recombination in budding yeast. Mol Cell Biol. 2006;26:3683–3694. doi: 10.1128/MCB.26.10.3683-3694.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kosaka H, Shinohara M, Shinohara A. Csm4-dependent telomere movement on nuclear envelope promotes meiotic recombination. PLoS Genet. 2008;4:e1000196. doi: 10.1371/journal.pgen.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wanat JJ, Kim KP, Koszul R, Zanders S, Weiner B, Kleckner N, Alani E. Csm4, in collaboration with Ndj 1, mediates telomere-led chromosome dynamics and recombination during yeast meiosis. PLoS Genet. 2008;4:e1000188. doi: 10.1371/journal.pgen.1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheridan S, Bishop DK. Red-Hed regulation: recombinase Rad51, though capable of playing the leading role, may be relegated to supporting Dmc1 in budding yeast meiosis. Genes Dev. 2006;20:1685–1691. doi: 10.1101/gad.1447606. [DOI] [PubMed] [Google Scholar]
- 56.Henderson KA, Keeney S. Tying synaptonemal complex initiation to the formation and programmed repair of DNA double-strand breaks. Proc Natl Acad Sci U S A. 2004;101:4519–4524. doi: 10.1073/pnas.0400843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007;104:10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cartagena-Lirola H, Guerini I, Manfrini N, Lucchini G, Longhese MP. Role of the Saccharomyces cerevisiae Rad53 checkpoint kinase in signaling double-strand breaks during the meiotic cell cycle. Mol Cell Biol. 2008;28:4480–4493. doi: 10.1128/MCB.00375-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell. 2002;10:373–385. doi: 10.1016/s1097-2765(02)00593-2. [DOI] [PubMed] [Google Scholar]
- 60.Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 61.Perez-Hidalgo L, Moreno S, San-Segundo PA. The fission yeast meiotic checkpoint kinase Mek1 regulates nuclear localization of Cdc25 by phosphorylation. Cell Cycle. 2008;7:3720–3730. doi: 10.4161/cc.7.23.7177. [DOI] [PubMed] [Google Scholar]
- 62.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.