

Abstract
Recent genome-wide association studies (GWASs) have identified candidate genes contributing to cancer risk through low-penetrance mutations. Many of these genes were unexpected and, intriguingly, included well-known players in carcinogenesis at the somatic level. To assess the hypothesis of a germline-somatic link in carcinogenesis, we evaluated the distribution of somatic gene labels within the ordered results of a breast cancer risk GWAS. This analysis suggested frequent influence on risk of genetic variation in loci encoding for “driver kinases” (i.e., kinases encoded by genes that showed higher somatic mutation rates than expected by chance and, therefore, whose deregulation may contribute to cancer development and/or progression). Assessment of these predictions using a population-based case-control study in Poland replicated the association for rs3732568 in EPHB1 (odds ratio (OR) = 0.79; 95% confidence interval (CI): 0.63–0.98; Ptrend = 0.031). Analyses by early age at diagnosis and by estrogen receptor α (ERα) tumor status indicated potential associations for rs6852678 in CDKL2 (OR = 0.32, 95% CI: 0.10–1.00; Precessive = 0.044) and rs10878640 in DYRK2 (OR = 2.39, 95% CI: 1.32–4.30; Pdominant = 0.003), and for rs12765929, rs9836340, rs4707795 in BMPR1A, EPHA3 and EPHA7, respectively (ERα tumor status Pinteraction<0.05). The identification of three novel candidates as EPH receptor genes might indicate a link between perturbed compartmentalization of early neoplastic lesions and breast cancer risk and progression. Together, these data may lay the foundations for replication in additional populations and could potentially increase our knowledge of the underlying molecular mechanisms of breast carcinogenesis.
Introduction
With the advent of technical and methodological advances, several GWASs identifying common genetic variation associated with risk of developing cancer have been completed recently [1]. Thus, initiatives such as the National Cancer Institute's Cancer Genetic Markers of Susceptibility (CGEMS) and efforts carried out by deCODE Genetics and the Breast Cancer Association Consortium have led to the identification of breast cancer risk alleles in single nucleotide polymorphisms (SNPs) replicated across populations [2]–[6]. Intriguingly, illustrating the unbiased nature of GWASs, most hits have corresponded to a priori unexpected candidate genes. In this context, the involvement of biological processes beyond the canonical DNA damage response in breast cancer is further suggested by the observed differential influence of low-penetrance risk alleles among BRCA1 and BRCA2 mutation carriers [7]–[9].
A potential common characteristic of the unexpected low-penetrance susceptibility genes is the previously identified contribution to tumorigenesis, but at the somatic level. Common genetic variation in loci encoding for FGFR2 and MAP3K1 influences risk of breast cancer [2], [4], and these genes were previously found to be somatically mutated in diverse neoplasias including breast cancer [10], [11]. In addition, and central to the understanding of cancer progression, common risk alleles showed differential influence according to ERα tumor status [12], and variation in the locus encoding for ERα, ESR1, also influences risk of breast cancer [13], [14]. More recently, additional breast cancer susceptibility loci have been described that include CDKN2A/B as candidates [15]. While these observations suggest a “germline-somatic” link in breast carcinogenesis, an analogous situation may exist for other neoplasias. Variation in loci encoding for CDH1 and SMAD7 influences risk of colorectal cancer [16], [17] and, similarly, these genes were previously identified as inactivated or deregulated in tumors [18]–[21]. Moreover, deregulated germline expression of a paradigmatic proto-oncogene, MYC, may be a common mechanism of tumorigenesis in epithelial tissues [22]–[25]. However, despite some evidence of a germline-somatic link, as yet there is no explicit evaluation of this hypothesis and its potential usefulness in replication studies. Here we present an examination of this link through analysis of the CGEMS GWAS breast cancer dataset and subsequent assessment of the predictions in a case-control study of incident breast cancer in Poland.
Results
Distribution of somatic gene sets in ordered breast cancer GWAS results
Previously, analysis of the CGEMS GWAS dataset using the lowest genotypic P value per gene locus suggested true associations in genes annotated with Gene Ontology (GO) biological process terms linked to somatic events [26], [27]. However, since there is a positive correlation between the extension of a given locus and the number of SNPs it may contain (and, therefore, the possibility of significant association results being obtained by chance), an unadjusted GWAS rank is biased at its lowest P values for specific processes in which large gene products frequently participate [26], [28], [29] (Fig. 1A
). Nevertheless, cancer genes tend to expand across large genomic regions [30], and examination of eight genes likely involved in breast cancer through low-penetrance mutations–CASP8, COX11, ESR1, FGFR2, LSP1, MAP3K1, RAD51L1 and TOX3
[2]–[6], [13], [14]–showed a trend for larger genomic loci (mean (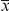 ) genomic extension = 211 kilo bases (kb) and standard deviation (σ) = 283 kb; compared to
) genomic extension = 211 kilo bases (kb) and standard deviation (σ) = 283 kb; compared to 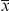 = 66 kb and σ = 128 kb for all annotated genes in the CGEMS GWAS rank).
= 66 kb and σ = 128 kb for all annotated genes in the CGEMS GWAS rank).
Figure 1. GWAS ranks and distribution of cancer somatic gene sets.

A, Original GWAS results ranked according to the lowest genotypic association test P value per gene locus (unadjusted for genomic extension; taken SNPs in defined genomic window of ±10 kb relative to the first and last exons of a given gene). The Y-axis indicates the number of SNPs per gene locus while the X-axis indicates the lowest association P value per gene locus. Bias can be appreciated as the number of SNPs per gene locus increases at lower P values. B, GWAS results ranked according to the lowest association P value per gene locus but adjusted by genomic extension through case-control permutations. Compared to the previous graph, the bias largely disappears. C, Following the rank in B, the Y-axis indicates odds ratios (ORs) of allele effects and density distributions of gene sets (driver kinases correspond to a light lilac curve; the rest of the genome in the GWAS dataset is shown by a dark lilac curve), while the X-axis indicates the log-transformed association P values, previously adjusted by genomic extension. As indicated by the density curves, SNPs mapping to driver kinase loci are relatively more frequent at lower association adjusted P values. This observation is supported by GSEA results using the same CGEMS GWAS adjusted rank; nominal P<0.001 and FDR-adjusted P = 0.010 (Table S2). D, Similarly to the graph in C, distribution of CRGs in the CGEMS GWAS rank adjusted through permutations.
Having identified caveats to the ranking of GWAS results, we performed 10,000 permutations of case-control status and used the null distribution of t statistics from the age-adjusted partial correlation analysis to correct the original rank, which then showed an unbiased distribution (Fig. 1B ). Prior to the evaluation of somatic sets, analysis of GO biological process terms in the GWAS permutation P values rank did not show any significant asymmetry using the Gene Set Enrichment Analysis (GSEA) tool [31] with multiple testing correction by the false discovery rate (FDR) approach [32]. Nonetheless, most processes with nominally significant P values were those previously highlighted, which are associated with somatic events [26], [27] (Table S1). This observation appears to agree with recently described results of pathway-based analysis of the same GWAS dataset [33].
Next, evaluation of somatic sets related to cancer prognosis and treatment response prediction, and to genetic and genomic alterations (see Materials and Methods), revealed significant asymmetrical distribution of “driver kinases” [34], [35]; that is, kinases whose deregulation through frequent somatic mutation contributes to tumor development and/or progression (“driver mutations”). In contrast, “passenger mutations” were defined as essentially neutral and linked to the inherent genetic instability in cancer cells [34], [35]. Thus, the driver kinases set was found to be biased towards the top (nominal significant association results) of the GWAS permutation rank (GSEA nominal P<0.001; FDR-adjusted P = 0.010) (Fig. 1C and Table S2). Among the remaining of somatic sets evaluated, only cooperation response genes (CRGs) to oncogenic mutations [36] showed a trend for a distribution similar to that of driver kinases (GSEA nominal P = 0.080; FDR-adjusted P value = 0.25) (Fig. 1D ), although the intersection between both sets only contained two genes (Table S2). Therefore, in somatic cancer genes, common genetic variation in driver kinase loci might frequently influence risk of breast cancer.
The set of driver kinases contained a benchmark gene, FGFR2 [2], [4], and a locus recently replicated in an independent study, BMPR1B [37]. Nevertheless, a significant bias was still observed following exclusion of these two loci (GSEA nominal P = 0.001; FDR-adjusted P = 0.048), which suggests that variation at additional driver kinase loci influences risk of breast cancer. Importantly, using the set of non-driver kinases–either the subsequent equivalent set as originally statistically ordered or the total set (n = 344) [35]–did not reveal significant bias (GSEA nominal P = 0.99 and 0.66, respectively), which reinforces the idea of frequent involvement of driver kinases. However, if only the individual statistical data for each locus were considered, most of the driver kinase loci would perhaps not have been selected for replication in other populations.
Independent association results for common variation in driver kinase loci
Given the possible bias in GWAS rank identified above, we examined the top 20 driver kinase variants in the original rank (Table S3, including details of the CGEMS and results below) in a case-control study of incident breast cancer in Szczecin (Poland), previously used in other replications [38]. Applying genotyping quality controls and Hardy-Weinberg equilibrium analysis, 16 SNPs representing an identical number of driver kinase loci (i.e., a single SNP for each locus and representing the strongest potential statistical association) were examined for their association with risk of breast cancer using 880 controls and 1,173 cases (see Materials and Methods). In this analysis, the rs3732568 variant in the ephrin type-B receptor 1 (EPHB1) locus was found to be associated with risk of breast cancer: OR = 0.79, 95% CI: 0.63–0.98; Ptrend = 0.031 (Table 1). Further evaluation of this association through 10,000 case-control permutations in our study gave a similar significance value, Ptrend = 0.034. Importantly, this association was in the same direction and with similar magnitude to the result in the CGEMS GWAS: age-adjusted OR = 0.78, 95% CI: 0.64–0.94; Ptrend = 0.009.
Table 1. Association between genetic variation in EPHB1 and risk of breast cancer in Poland.
| EPHB1, rs3732568 | ||||||
| Controls | Cases | |||||
| n | % | n | % | OR | 95% CI | |
| C/C | 693 | 79.8 | 891 | 83.2 | 1.00 | |
| C/A | 165 | 19.0 | 172 | 16.1 | 0.79 | 0.62–1.00 |
| A/A | 10 | 1.2 | 8 | 0.7 | 0.60 | 0.23–1.55 |
| Total | 868 | 1,071 | ||||
| Trend | 0.79 | 0.63–0.98 | ||||
| Ptrend = 0.031† | ||||||
Adjusted by age.
While deregulated expression or function of EPHs and EPH receptors is thought to play a critical role in the initial stages of epithelial neoplasia [39], [40], recent analysis of early breast cancer expression changes suggests a link between disruption of cell adhesion and extracellular matrix pathways, and the risk of developing breast cancer [41]. Analysis of this recent dataset also revealed an early expression change of EPHB1, between normal breast tissue and atypical ductal hyperplasia (Fig. 2). This alteration consisted of infra-expression in hyperplasia, akin to its potential role in the compartmentalization of early neoplastic lesions [42]. Together, association studies, early expression changes in carcinogenesis and the regulation of cell adhesion suggest the involvement of EPHB1 in risk of breast cancer.
Figure 2. Early change of EPHB1 expression in breast carcinogenesis.

The graphs show expression profiles in histologically normal (HN) breast tissues versus patient-matched atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS) [41]. Results of two EPHB1 microarray probes (names shown at the top) and the corresponding significance P values are shown.
Next, given accepted models of inherited breast cancer susceptibility [43], we examined associations with risk at early age of diagnosis (≤40 years old). This analysis indicated two additional potential associations: rs6852678 in CDKL2, recessive model OR = 0.32, 95% CI: 0.10–1.00; P = 0.044; and rs10878640 in DYRK2, dominant model OR = 2.39, 95% CI: 1.32–4.30; P = 0.003 (Table 2). Results for rs6852678 appeared to be consistent with CGEMS GWAS analysis; age-adjusted recessive model OR = 0.71, 95% CI: 0.53–0.95; P = 0.019; however, the pattern for rs10878640 might be more complex (CGEMS GWAS ORs = 1.05 and 0.68 for heterozygotes and minor allele homozygotes, respectively).
Table 2. Associations between genetic variation in driver kinase loci and risk of breast cancer at ≤40 years of first age at diagnosis.
| CDKL2, rs6852678 | ||||||
| Controls | Cases | |||||
| n | % | n | % | OR | 95% CI | |
| C/C | 39 | 51.3 | 62 | 51.2 | 1.00 | |
| C/T | 28 | 36.8 | 54 | 44.6 | 1.21 | 0.66–2.23 |
| T/T | 9 | 11.8 | 5 | 4.1 | 0.35 | 0.11–1.12 |
| Total | 76 | 121 | ||||
| Recessive | 0.32 | 0.10–1.00 | ||||
| Precessive = 0.044 | ||||||
Having potential differences by ERα tumor status, we next examined associations in ERα-positive and -negative breast cancer patients. Thus, rs3732568 in EPHB1 showed a similar influence on either type of breast cancer (Table 3)–which is consistent with an overall significant association–and rs12765929 in BMPR1A and rs9836340 in EPHA3 showed a potential major impact on the risk of ERα-negative breast cancer (P for difference in OR (interaction) by ERα status <0.05), while rs4707795 in EPHA7 showed a differential effect between ERα-negative versus ERα-positive breast cancer risk (Pinteraction = 0.007) (Table 3). None of these additional candidates linked to ERα tumor status, or those linked to an early age of diagnosis above, showed significant expression differences at early stages of breast carcinogenesis as EPHB1. On the other hand, the remaining SNPs examined in this study after applying quality controls and Hardy-Weinberg equilibrium analysis (i.e., 10 out of 16), did not show significant associations following CGEMS evidence (Table S3). Together, the gene-set based analysis of GWAS data and the subsequent replication attempt might indicate that common genetic variation in specific driver kinase loci, and particularly in EPH receptor genes, influence risk of breast cancer.
Table 3. Associations of genetic variation in driver kinase loci and risk of breast cancer by ERα tumor status†.
| BMPR1A, rs12765929 | ||||||||||
| Controls | ERα-negative | ERα-positive | ||||||||
| n | % | n | % | OR | 95% CI | n | % | OR | 95% CI | |
| G/G | 514 | 59.1 | 189 | 64.5 | 1.00 | 389 | 58.4 | 1.00 | ||
| G/T | 306 | 35.2 | 96 | 32.8 | 0.87 | 0.65–1.16 | 243 | 36.5 | 1.07 | 0.86–1.33 |
| T/T | 50 | 5.7 | 8 | 2.7 | 0.45 | 0.21–0.98 | 34 | 5.1 | 0.93 | 0.59–1.48 |
| Total | 870 | 293 | 666 | |||||||
| Trend | 0.79 | 0.62–1.00 | 1.02 | 0.86–1.21 | ||||||
| Ptrend = 0.050 | Ptrend = 0.81Pinteraction = 0.024 | |||||||||
Adjusted by age.
Discussion
Evaluation of a germline-somatic link in breast carcinogenesis suggests a role for driver kinases and, perhaps to a lesser extent, genes with a synergistic response to oncogenic mutations. This study might be limited by the assignment of the lowest genotypic P value per gene locus within a defined genomic window (i.e., ±10 kb)–thus excluding a large proportion of variation that cannot be assigned to a specific known gene–and by its focus on the additive model of influence of risk alleles when adjusted through case-control permutations. Future analyses taking into account the potential perturbation of germline gene expression by, for example, common variation at distant regulatory regions may improve the identification of susceptibility genes using GWAS complete data. Another limitation in the interpretation of the results presented here may lie in the case-control study designs: the CGEMS addressed breast cancer risk in postmenopausal women, while the Polish study was relatively enriched in early-onset cases. Therefore, studies in additional populations, with diverse designs, are warranted to corroborate the results shown here.
The results of the replication study may be consistent with previously detected somatic genetic alterations and/or functional roles. Somatic mutations in CDKL2 were nonsense and were only detected in breast and ovarian cancer cell lines or tumors [11], [35]. CDKL2 (also known as p56 or KKIAMRE) is the most distant member of the CDC2-related serine/threonine protein kinase family, involved in epidermal growth factor signaling [44], but with a mostly uncharacterized function. DYRK2 was found to be mutated in breast and central nervous system tumors, in nonsense and missense alterations, respectively [11], [35]. The functional role of DYRK2 in the DNA damage response [45] may link to CGEMS GWAS results for RAD51L1 [3]: loss of DYRK2 function alters the activation of apoptosis in response to DNA damage via ATM [45], which may therefore promote carcinogenesis.
Having revealed potential associations linked to known somatic alterations, the most striking results of this study may concern the identification of risk alleles at three EPH receptor loci. EPH-mediated signaling regulates important biological process altered in carcinogenesis, such as cell-to-cell communication, and cell migration and adhesion via the actin cytoskeleton [39], [40]. Thus, through RHO and RAS/MAPK activities [46], this signaling pathway has been implicated in the maintenance of epithelial tissue architectures and is therefore thought to act as a tumor suppressor [39], [40]. These observations may indicate that, similarly to colorectal tumorigenesis [42], EPH-mediated compartmentalization of early breast tissue neoplastic lesions is critical to prevent the subsequent emergence of carcinoma. Therefore, through a germline expression or functional perturbation, EPHB1 may contribute to the observed variability in the transition from an in situ lesion to an invasive carcinoma [47]. While the associations revealed here warrant further replication in other populations, the existing data could potentially increase current knowledge of the genetic basis and molecular mechanisms of breast carcinogenesis.
Materials and Methods
CGEMS dataset
The National Cancer Institute CGEMS initiative has conducted genome-wide association studies to identify common genetic variants and the corresponding functionally affected genes involved in breast cancer and prostate cancer susceptibility. An initial CGEMS whole genome scan was designed to study the main effect of SNPs on breast cancer risk in postmenopausal women [2]. The study involved 1,145 invasive postmenopausal breast cancer cases and 1,142 matched controls from the Nurses' Health Study nested case-control study [48]. Results of the CGEMS GWAS of breast cancer were obtained upon approval of a Data Access Request.
GWAS rank
In our previous analyses [26], [27], ordered CGEMS GWAS results (i.e., ranks) corresponded to the lowest P value per gene for the genotypic test in a genomic region of +/−10 kb at each gene locus, defined by the Ensembl human genome release 57. Assigned SNPs were curated using Ensembl gene annotations. We [26] and others [28] noted that such ranks were biased along with the genomic extension–and therefore with the number of SNPs–per gene locus. To adjust for this bias, several statistical strategies are possible [28], including carrying out permutations of the case-control status to correct the significance of the original statistic. In our analysis, considering typed and informative SNPs in each gene locus, we first chose the maximum absolute value of the t statistic from the age-adjusted partial correlation in the additive model. Next, 10,000 permutations of the same informative SNPs were performed to create a null distribution for this maximum t statistic, which was used to assess its significance corrected by number of SNPs.
GSEA application
The distribution of gene sets in ranked GWAS results was examined using the non-parametric algorithm in the GSEA tool, with default values for all parameters [31] except for the set size when appropriated. In GSEA, a pre-defined gene set is mapped to a rank–in our case genes/loci ordered according to the adjusted association statistic–to assess potential bias using an enrichment score that reflects the degree to which this set is overrepresented at the extremes of the entire ranked list. In the interpretation of the results, caution should be taken when considering sets of different size. In our study, different hypotheses were examined independently (i.e., gene sets linked to prognosis, prediction or genetic/genomic somatic alterations), and P values were corrected for multiple testing within each group : 1) genes whose expression in primary breast tumors was associated with patient prognosis and/or metastasis [49]–[55]; 2) genes whose expression in primary breast tumors was associated with patient therapeutic treatment response [56]–[59]; 3) genes whose expression levels differed according to ERα breast tumor status or grade [60], or in response to 17β-estradiol [61]; and 4) genes with somatic genetic and/or genomic somatic alterations (Table S2). This last group was made up of five sets : i/ driver kinases (conditional probability of containing driver mutations >0.70, n = 119 as defined previously [35], of which 95 were uniquely mapped in the GWAS rank); ii/ CRGs to oncogenic mutations [36]; iii/ cancer gene census, somatically-mutated only [62], [63]; iv/ genes affected by somatic chromosomal rearrangements and/or fusions [64]; and v/ amplified and over-expressed cancer genes [65] (Table S2).
Gene expression analysis
Raw expression microarray data on breast cancer progression [41] were downloaded from the Gene Expression Omnibus reference GSE16873 and normalized with robust multiarray average (RMA) [66] and significance analysis was performed using the significance analysis of microarray (SAM) algorithm [67].
Study samples in Poland and association study
A case-control study of unselected invasive breast cancer collected between 1996 and 2003 in Szczecin (Poland) was analyzed. The series included 976 cases of breast cancer unselected for age and an additional group of 367 cases of breast cancer diagnosed at age 50 or below. Therefore, the series was enriched for early-onset cases: mean age of diagnosis was 52.4 years (range 19–88). Subjects were unselected for family history and 15% of cases reported a first- or second-degree relative with breast cancer. The participation rate exceeded 70% among women with breast cancer invited to enroll. Collected information included year of birth, age at diagnosis of breast and/or ovarian cancer, tumor bilaterality, family history (first- and second-degree relatives with breast and/or ovarian cancer) and tumor pathological features in >80% of cases (ERα and progesterone receptor status, and grade). Cases were also examined for BRCA1 founder mutations in Poland [68] and, if positive, excluded from the association study (n = 50). The control group included cancer-free adult women from the same population (920 women with mean age of diagnosis of 56.7, range 20–91) taken from the healthy adult patients of five family doctors practicing in the Szczecin region. These individuals were selected randomly from the patient lists of the participating doctors. The study was carried out with informed consent of the probands and approved by local ethics committees. Genotypes were obtained using Sequenom iPLEX chemistry at the International Hereditary Cancer Center. Quality controls were of >95% calling for each SNP and >90% of calls per sample. Thus, in the set of 16 SNPs, we observed an average concordance rate of 98.7% of genotype calls using 3.3% replicates. Genotypes of 880 controls and 1,173 cases were effectively analyzed using conditional and unconditional logistic regressions (age adjustment using similar strata size; 20–46, 46–56, 56–66, and 66–91 years old).
Supporting Information
(0.02 MB XLS)
(0.05 MB XLS)
(0.03 MB XLS)
Acknowledgments
The authors are indebted to the CGEMS initiative and to Dr. L.A. Emery and colleagues for making their data available. We also wish to thank the study participants in Poland for their generous contribution.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: The work was supported by the Spanish Society of Medical Oncology 2009 grant to JB and MAP, the Ramón Areces Foundation XV and FIS (09/02483) grants, and the Biomedical Research Centre Network for Epidemiology and Public Health group 55 to MAP, and the “Roses contra el Càncer” Foundation and RTICCC RD06/0020/0028 grants to JB. MAP was supported by the “Ramón y Cajal” Young Investigator program of the Spanish Ministry of Science and Innovation and NB by an IDIBELL fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Easton DF, Eeles RA. Genome-wide association studies in cancer. Hum Mol Genet. 2008;17:R109–115. doi: 10.1093/hmg/ddn287. [DOI] [PubMed] [Google Scholar]
- 2.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet. 2009;41:579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2008;40:703–706. doi: 10.1038/ng.131. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet. 2009;41:585–590. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antoniou AC, Sinilnikova OM, McGuffog L, Healey S, Nevanlinna H, et al. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2009;18:4442–4456. doi: 10.1093/hmg/ddp372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antoniou AC, Spurdle AB, Sinilnikova OM, Healey S, Pooley KA, et al. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet. 2008;82:937–948. doi: 10.1016/j.ajhg.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antoniou AC, Sinilnikova OM, Simard J, Leone M, Dumont M, et al. RAD51 135G→C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet. 2007;81:1186–1200. doi: 10.1086/522611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen RM, Goriely A, Wall SA, Roberts IS, Wilkie AO. Fibroblast growth factor receptor 2, gain-of-function mutations, and tumourigenesis: investigating a potential link. J Pathol. 2005;207:27–31. doi: 10.1002/path.1816. [DOI] [PubMed] [Google Scholar]
- 11.Stephens P, Edkins S, Davies H, Greenman C, Cox C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Closas M, Chanock S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin Cancer Res. 2008;14:8000–8009. doi: 10.1158/1078-0432.CCR-08-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng W, Long J, Gao YT, Li C, Zheng Y, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–328. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunning AM, Healey CS, Baynes C, Maia AT, Scollen S, et al. Association of ESR1 gene tagging SNPs with breast cancer risk. Hum Mol Genet. 2009;18:1131–1139. doi: 10.1093/hmg/ddn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 18.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, et al. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 2001;48:367–371. doi: 10.1136/gut.48.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 21.Richards FM, McKee SA, Rajpar MH, Cole TR, Evans DG, et al. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999;8:607–610. doi: 10.1093/hmg/8.4.607. [DOI] [PubMed] [Google Scholar]
- 22.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009;41:885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- 24.Solé X, Hernández P, de Heredia ML, Armengol L, Rodríguez-Santiago B, et al. Genetic and genomic analysis modeling of germline c-MYC overexpression and cancer susceptibility. BMC Genomics. 2008;9:12. doi: 10.1186/1471-2164-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, et al. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107:3001–3005. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonifaci N, Berenguer A, Díez J, Reina O, Medina I, et al. Biological processes, properties and molecular wiring diagrams of candidate low-penetrance breast cancer susceptibility genes. BMC Med Genomics. 2008;1:62. doi: 10.1186/1755-8794-1-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medina I, Montaner D, Bonifaci N, Pujana MA, Carbonell J, et al. Gene set-based analysis of polymorphisms: finding pathways or biological processes associated to traits in genome-wide association studies. Nucleic Acids Res. 2009;37:W340–344. doi: 10.1093/nar/gkp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraft P, Raychaudhuri S. Complex diseases, complex genes: keeping pathways on the right track. Epidemiology. 2009;20:508–511. doi: 10.1097/EDE.0b013e3181a93b98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanley SM, Bailey TL, Mattick JS. GONOME: measuring correlations between GO terms and genomic positions. BMC Bioinformatics. 2006;7:94. doi: 10.1186/1471-2105-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furney SJ, Higgins DG, Ouzounis CA, López-Bigas N. Structural and functional properties of genes involved in human cancer. BMC Genomics. 2006;7:3. doi: 10.1186/1471-2164-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995;57:289–300. [Google Scholar]
- 33.Menashe I, Maeder D, Garcia-Closas M, Figueroa JD, Bhattacharjee S, et al. Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade. Cancer Res. 2010;70:4453–4459. doi: 10.1158/0008-5472.CAN-09-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McMurray HR, Sampson ER, Compitello G, Kinsey C, Newman L, et al. Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature. 2008;453:1112–1116. doi: 10.1038/nature06973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saetrom P, Biesinger J, Li SM, Smith D, Thomas LF, et al. A risk variant in an miR-125b binding site in BMPR1B is associated with breast cancer pathogenesis. Cancer Res. 2009;69:7459–7465. doi: 10.1158/0008-5472.CAN-09-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wokolorczyk D, Gliniewicz B, Sikorski A, Zlowocka E, Masojc B, et al. A range of cancers is associated with the rs6983267 marker on chromosome 8. Cancer Res. 2008;68:9982–9986. doi: 10.1158/0008-5472.CAN-08-1838. [DOI] [PubMed] [Google Scholar]
- 39.Merlos-Suárez A, Batlle E. Eph-ephrin signalling in adult tissues and cancer. Curr Opin Cell Biol. 2008;20:194–200. doi: 10.1016/j.ceb.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Vaught D, Brantley-Sieders DM, Chen J. Eph receptors in breast cancer: roles in tumor promotion and tumor suppression. Breast Cancer Res. 2008;10:217. doi: 10.1186/bcr2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emery LA, Tripathi A, King C, Kavanah M, Mendez J, et al. Early dysregulation of cell adhesion and extracellular matrix pathways in breast cancer progression. Am J Pathol. 2009;175:1292–1302. doi: 10.2353/ajpath.2009.090115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortina C, Palomo-Ponce S, Iglesias M, Fernández-Masip JL, Vivancos A, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat Genet. 2007;39:1376–1383. doi: 10.1038/ng.2007.11. [DOI] [PubMed] [Google Scholar]
- 43.Claus EB, Risch NJ, Thompson WD. Using age of onset to distinguish between subforms of breast cancer. Ann Hum Genet. 1990;54:169–177. doi: 10.1111/j.1469-1809.1990.tb00373.x. [DOI] [PubMed] [Google Scholar]
- 44.Taglienti CA, Wysk M, Davis RJ. Molecular cloning of the epidermal growth factor-stimulated protein kinase p56 KKIAMRE. Oncogene. 1996;13:2563–2574. [PubMed] [Google Scholar]
- 45.Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J Biol Chem. 2010;285:4909–4919. doi: 10.1074/jbc.M109.042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest. 2008;118:64–78. doi: 10.1172/JCI33154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schnitt SJ. The transition from ductal carcinoma in situ to invasive breast cancer: the other side of the coin. Breast Cancer Res. 2009;11:101. doi: 10.1186/bcr2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colditz GA, Hankinson SE. The Nurses' Health Study: lifestyle and health among women. Nat Rev Cancer. 2005;5:388–396. doi: 10.1038/nrc1608. [DOI] [PubMed] [Google Scholar]
- 49.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 50.Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006;3:e47. doi: 10.1371/journal.pmed.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U S A. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 53.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 54.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 56.Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–2293. doi: 10.1200/JCO.2004.05.166. [DOI] [PubMed] [Google Scholar]
- 57.Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, et al. Patterns of resistance and incomplete response to docetaxel by gene expression profiling in breast cancer patients. J Clin Oncol. 2005;23:1169–1177. doi: 10.1200/JCO.2005.03.156. [DOI] [PubMed] [Google Scholar]
- 58.Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5:607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 59.Wang XD, Reeves K, Luo FR, Xu LA, Lee F, et al. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8:R255. doi: 10.1186/gb-2007-8-11-r255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 61.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 62.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- 66.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 67.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorski B, Byrski T, Huzarski T, Jakubowska A, Menkiszak J, et al. Founder mutations in the BRCA1 gene in Polish families with breast-ovarian cancer. Am J Hum Genet. 2000;66:1963–1968. doi: 10.1086/302922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(0.02 MB XLS)
(0.05 MB XLS)
(0.03 MB XLS)