

Abstract
Individuals of the same species are generally thought to have very similar genomes. However, there is growing evidence that structural variation in the form of copy number variation (CNV) and presence–absence variation (PAV) can lead to variation in the genome content of individuals within a species. Array comparative genomic hybridization (CGH) was used to compare gene content and copy number variation among 19 diverse maize inbreds and 14 genotypes of the wild ancestor of maize, teosinte. We identified 479 genes exhibiting higher copy number in some genotypes (UpCNV) and 3410 genes that have either fewer copies or are missing in the genome of at least one genotype relative to B73 (DownCNV/PAV). Many of these DownCNV/PAV are examples of genes present in B73, but missing from other genotypes. Over 70% of the CNV/PAV examples are identified in multiple genotypes, and the majority of events are observed in both maize and teosinte, suggesting that these variants predate domestication and that there is not strong selection acting against them. Many of the genes affected by CNV/PAV are either maize specific (thus possible annotation artifacts) or members of large gene families, suggesting that the gene loss can be tolerated through buffering by redundant functions encoded elsewhere in the genome. While this structural variation may not result in major qualitative variation due to genetic buffering, it may significantly contribute to quantitative variation.
It is generally assumed that the genomes of different individuals of the same species are similar in content. However, there is growing evidence for structural variation among the genomes of different individuals. Structural variation includes rearrangements (inversions and translocations) and copy number variation (CNV). The most extreme form of CNV is presence–absence variation (PAV), in which a particular sequence is present in some individuals and missing in others. While single nucleotide polymorphisms (SNPs) are the most common and most frequently assayed type of intraspecific genetic variation, there is evidence that more nucleotide bases are affected by CNV than by SNPs between any two individuals (Zhang et al. 2009). This structural variation challenges the notion of understanding the genome of a species through the analysis of a single reference sequence from one individual or genotype. CNV and PAV are likely to have functional significance and may explain some variation not captured by SNP-based genome-wide association studies (Manolio et al. 2009). For example, both CNV and PAV can contribute to phenotypic variation for some human diseases (Feuk et al. 2006; Sharp et al. 2006; Beckmann et al. 2007; Cooper et al. 2007; Sebat 2007; Hurles et al. 2008; Bucan et al. 2009; Merikangas et al. 2009; Zhang et al. 2009; Beroukhim et al. 2010; Conrad et al. 2010; Wellcome Trust Case Control Consortium 2010). Specifically, phenotypic variation results from CNV in dosage effect-sensitive genes (Charcot-Marie-Tooth disease), genes influenced by position effect (spastic paraplegia), and genes with a mutant allele unmasked when the functional copy is deleted (for review, see Stankiewicz and Lupski 2010). CNVs underlying complex traits such as Alzheimer disease and Autism spectrum disorders have been detected in human patients (Stankiewicz and Lupski 2010).
Copy number variation has been documented in several species, including the human genome (Sebat et al. 2004; Sharp et al. 2005; Tuzun et al. 2005; Conrad et al. 2006, 2010; Redon et al. 2006; McCarroll and Altshuler 2007; Wong et al. 2007; Kidd et al. 2008; Wellcome Trust Case Control Consortium 2010) and several other mammalian species, including mice (Graubert et al. 2007), rats (Guryev et al. 2008), chimpanzees (Perry et al. 2008), rhesus monkeys (Lee et al. 2008), and canines (Chen et al. 2009). It is difficult to compare the number of CNV in different studies, as the number of observed CNV is heavily influenced by the diversity of individuals that are examined and the technology used for detection. The general consensus is that there are several hundred to over a thousand CNVs between individuals within a species. It should be noted that in most cases these studies include segregating individuals, and many of the CNVs are observed as heterozygotes. Studies of several highly inbred model organisms including C. elegans (Maydan et al. 2010) and Arabidopsis thaliana (Santuari et al. 2010) have also identified numerous CNVs.
Zea mays (maize) is a highly polymorphic species (for review, see Buckler et al. 2006; Messing and Dooner 2006; Springer and Stupar 2007). The recent completion of a reference genome from one genotype, B73, affords the opportunity to assess structural variation and complexity within this species (Schnable et al. 2009). Detailed analyses of specific loci as well as genomic approaches have identified numerous duplications within the maize genome, many of which are located in colinear regions (Schnable et al. 2009) derived from an ancient allopolyploidization event (Gaut and Doebley 1997; Swigonova et al. 2005; Wei et al. 2007). There is also evidence for a high frequency of tandem duplicates within maize (Messing et al. 2004; Emrich et al. 2007; Schnable et al. 2009), including several well-characterized genes affecting pigmentation such as R-r (Robbins et al. 1991), P1 (Zhang and Peterson 2005), and A1-b (Yandeau-Nelson et al. 2006). In addition, there is evidence for many dispersed duplications that are not located within colinear regions, but are instead likely derived from transposition events (Bennetzen 2005; Lai et al. 2005; Morgante et al. 2005; Yang and Bennetzen 2009).
There are many examples of structural variation among different maize genotypes. Cytogenetic studies have provided evidence for structural variation in maize chromosomes (McClintock et al. 1981; Kato et al. 2004). More recent studies have sequenced multiple haplotypes for specific loci and have identified structural variation affecting both repetitive and low-copy sequences (Fu and Dooner 2002; Yao et al. 2002; Brunner et al. 2005; Wang and Dooner 2006). For example, Wang and Dooner (2006) documented that only 25%–84% of bases within a ∼100-kb region were shared among eight haplotypes. The frequency of CNV and PAV between the reference genome (B73) and a second genotype (Mo17) has been assayed using BAC libraries (Morgante et al. 2005) and comparative genomic hybridization (CGH) (Springer et al. 2009; Belo et al. 2010). These scans have identified hundreds of copy number variants as well as several thousand sequences present in the reference genome but absent in Mo17 (PAVs). A proportion of these CNV and PAV identified in Mo17 relative to B73 affect the copy number or content of genes present in these two lines.
In this study, we used a gene-focused microarray to assess the frequency and identity of genes affected by CNV or PAV within a diverse panel of maize and teosinte (Zea mays ssp. parviglumis) genotypes. We included the teosinte lines to evaluate whether extensive structural variation in maize predates or is related to domestication. Over 10% of the ∼32,500 genes surveyed exhibit CNV/PAV relative to the B73 reference genome. The majority of the CNV/PAV events are observed in both maize and teosinte, suggesting that these have not entered the genome during maize domestication or improvement. This study provides evidence for prevalent CNV/PAV within maize and provides an opportunity to characterize the types of genes affected by structural variation.
Results
Identification of genes affected by structural variation
Structural variation can include rearrangements (inversions and translocations), CNV, and PAV. Comparative genomic hybridization (CGH) of DNA samples to microarrays can be used to detect both CNV and PAV. A custom long oligonucleotide microarray was designed using the reference sequence of the B73 maize genotype (Schnable et al. 2009) and was used to perform CGH analyses of 32,487 maize genes (see Methods). High-quality hybridization data was obtained for 33 genotypes, including 19 diverse maize genotypes and 14 teosinte genotypes (listed in Supplemental Table 1). The visualization of log2 signal intensity relative to B73 reveals that many genes have variable signal over scales of the entire genome (Supplemental Fig. 2), single chromosomes (Fig. 1A), or small regions of a chromosome (Fig. 1B). The array-based CGH analysis detected genes with consistently higher (UpCNV) or consistently lower (DownCNV/PAV) signal than in the reference B73 genome (Table 1). Because the array was designed using B73 genomic sequence, the primary biological cause for increased CGH signal for a genotype would be an increase in the copy number of the probe sequence in that genotype relative to B73. In contrast, there are multiple potential causes for significantly negative log2 ratios, including polymorphisms within a probed sequence relative to B73, fewer copies of the gene in the other genotype (DownCNV) or absence of the sequence in the other genotype (PAV). It should be noted that numerous polymorphisms would be required for all probes from a gene to exhibit low signal. Our previous data (Springer et al. 2009) suggest that hybridization intensity is not strongly affected until there are at least four to five SNPs within the probe sequence. This level of polymorphism spread across multiple portions of the coding region would represent a highly divergent allele.
Figure 1.

Structural variation affects many genes. The average log2(other/B73) is plotted for all 2767 genes on chromosome 6 (A) or for 293 genes within a 20-Mb region of chromosome 1 (B) for eight genotypes. (Blue data points) UpCNV with more copies in the other inbred line relative to B73; (red data points) genes with significantly lower signal in the other line relative to B73 and are examples of DownCNV or PAV; (red arrows) several multigene structural variants that are observed in multiple genotypes; (black arrows) the position of several single gene structural variants that are observed in multiple genotypes.
Table 1.
Discovery of structural variation affecting maize genes

aIncludes a set of 420 genes identified by classical genetic studies and curated at CoGe (website).
bInclude all non-histone maize chromatin genes curated by http://www.ChromDB.org.
cIncludes all genes with putative cell wall function identified by Penning et al. (2009).
dIncludes the set of maize genes curated by GRASSIUS.
eIncludes genes that show increased signal in some genotypes and decreased signal in others.
Analysis of the array CGH data identified 479 UpCNV genes and 3410 DownCNV/PAV (Supplemental Table 2). The array CGH analysis cannot distinguish between DownCNV and PAV, as these both exhibit lower hybridization intensities than in the reference samples. However, it is possible to use the B73 reference genome to classify these events as either DownCNV or PAV. In order to be classified as a DownCNV, a gene would need to have multiple copies in the B73 genome. Of the 3410 DownCNV genes, 586 have probes with multiple close matches in the B73 reference genome and were classified as DownCNV candidates, while the remaining 2824 genes are single copy in the B73 reference genome sequence and were classified as likely examples of PAV (Table 1). This is a useful classification scheme to estimate the relative frequency of DownCNV and PAV, but it may result in some false assignments as PAV if additional copies of the sequence reside in the ∼5% of the B73 genome that was not sequenced or in regions of the B73 sequence that were collapsed during assembly. Due to the potential misclassification of DownCNV and PAV, these two classes were grouped together for many subsequent analyses. Interestingly, there are a number of genes that were classified as UpCNV in some genotypes and DownCNV in other genotypes. This suggests that genes that are present in multiple copies in B73 can frequently exhibit either increases or decreases in copy number in other genotypes.
The structural variants were observed throughout the maize genome (Fig. 2). There are more structural variants near the end of the chromosomes than within the central centromeric regions of the maize chromosomes, but this generally mirrors the genic density. Chromosomal regions were classified as high, moderate, or low recombination rates based upon a comparison of the genetic and physical map (Liu et al. 2009). The proportion of genes within each of these regions that exhibit PAV or CNV were determined (Table 2). The CNV exhibit a significantly (χ2, P < 0.0005) different distribution than expected with higher levels of CNV in the low recombination regions. In contrast, the PAV do not show altered rates in high and low recombination regions.
Figure 2.

Distribution and frequency of structural variation throughout the maize genome. The physical locations of the 32,487 genes are plotted along the 10 maize chromosomes. The color of each gene indicates whether structural variation was observed and the type of variation and the y-axis indicates the number of genotypes that contain the structural variant.
Table 2.
Recombination rate affects frequency of CNV

aThe proportion of genes within each class of recombination frequency that are affected by CNV is shown. The observed distribution is significantly (P < 0.0001) different from expected (χ2 test). Both the UpCNV and Down CNV show similar distributions.
bThe observed proportion of genes affected by PAV is not significantly different from the expected proportions.
Validation of structural variants
Several approaches validated the detected structural variants. Primer pairs for 12 genes located within putative PAV were used to perform PCR on the same genotypes used for microarray analysis (Table 2; Supplemental Fig. 3). The presence–absence patterns were largely supported by the PCR analysis with 92% of “absent” calls and 81% of present calls confirmed (Table 3). In some cases, the PCR failed to amplify a band in genotypes that were not predicted to be missing the sequence (Supplemental Fig. 3). These additional failed reactions could be due to polymorphisms within the primer sites or large insertions between the primers. Further PCR-based validation was conducted by using previous data on insertion/deletion polymorphism (IDP) markers (Fu et al. 2006) at 75 CGH predicted PAV in Mo17 or Oh43. The data from Fu et al. (2006) supported the existence of structural variation at ∼85% of the tested loci. Finally, 657 genes identified in this study with structural variation between B73 and Mo17 were also represented (with a minimum of three probes) in a previous high-density CGH analysis of these two lines (Springer et al. 2009), and 96% of these genes exhibit consistent signal changes in the two studies. Many of the same genes (108/180) that were identified in a previous study of B73 and Mo17 (Springer et al. 2009) were identified in the current study.
Table 3.
Validation of multigene PAV events

Distribution of structural variation within maize diversity
The physical positions of genes with structural variants were visualized across the maize chromosomes (Fig. 2). While the majority of structural variants were limited to single genes, there are many examples of larger structural variants that affected multiple nearby genes (Fig. 1A,B; Table 4). These larger structural variants were often observed in numerous maize genotypes. The largest PAV event includes 25 adjacent genes on chromosome 6 (Fig. 1A), which is present in 11 of 25 domesticated maize lines and 3 of 14 teosinte lines, and absent in other genotypes. This region was previously identified as present in B73 and absent in Mo17 (Springer et al. 2009) as well as several other genotypes (Belo et al. 2010). This insertion/deletion variant also segregates among teosinte individuals, suggesting that this large insertion/deletion is not a result of selection or inbreeding that has occurred during maize domestication or improvement. The largest UpCNV event, which includes nine genes located on chromosome 7, is observed in 6/25 domesticated maize lines and 6 of 14 teosinte lines.
Table 4.
Structural variants affecting multiple genes

The observation of genes affected by structural variation in a diverse set of maize and teosinte lines provided the opportunity to address several questions about the distribution of these events within maize. Individual genotypes differed from B73 at between 21 and 217 (mean = 114) UpCNVs and between 405 and 1375 DownCNV/PAV (mean = 917). As expected, the teosinte lines showed slightly greater divergence, differing by an average of 999 DownCNV/PAV compared with an average of 852 in maize. The majority of structural variants were observed in more than five of the genotypes tested (Fig. 3A). The finding that many of the structural variants are present at common frequencies suggests that these structural variants are tolerated in the homozygous state and, at least for the domesticated lines, are not associated with major fitness costs.
Figure 3.

Enrichment for rare CNV/PAV in teosinte genotypes. (A) The number of genotypes containing each was determined and the percent of events was plotted. Only 10% of structural variants are detected in one or two genotypes, while over 60% of structural variant events are detected in at least six genotypes. (B) The plot shows the allele frequency distribution for structural variant events in teosinte (black) and maize (gray). The proportion of DownCNV/PAV that are observed in one to 16 genotypes is shown. Teosinte has an excess of DownCNV/PAV observed in a single genotype relative to maize genotypes. (C) A similar plot is shown illustrating the distribution of allele frequency for UpCNV in maize (gray) and teosinte (black) genotypes.
We proceeded to assess the frequency of rare events separately in maize and teosinte (Fig. 3B,C). Teosinte has a higher frequency of unique structural variants than maize, possibly reflecting higher levels of diversity in teosinte or structural variants that are tolerated in heterozygotes, but would be deleterious in inbred genotypes. It should be noted that our power to detect structural variants is much lower when they are present as heterozygotes than as homozygotes based on a comparison of the CNV detected in B73xMo17 F1 plants relative to those detected in Mo17. Given this limitation and the fact that the majority (10/14) of teosinte genotypes tested are segregating individuals from wild populations, it is likely that the bias toward rare events in teosinte is even higher than actually observed within our data.
A small proportion of structural variants (3%) are observed only in teosinte, while ∼11% of the structural variants are only observed in domesticated maize lines. The remaining 86% of the variants are observed in both maize and teosinte. It is likely that the identification of fewer teosinte-specific events is due in part to the inclusion of fewer teosinte genotypes. We proceeded to further assess the frequency of structural variants in subpopulations of maize. The reference B73 genome represents the stiff stalk subpopulation of maize. Each of the other genotypes was assigned to one of five other subpopulations based on pedigree or SNP data (Hansey et al. 2010). The subpopulations include nonstiff stalk (n = 4), tropical (n = 5), ex-plant varietal protection (n = 6), inbred teosinte (n = 4), or wild teosinte (n = 10). To visualize the distribution of both UpCNV and DownCNV/PAV within these subpopulations, event frequencies within subpopulations were used for hierarchical clustering (Fig. 4; Supplemental Fig. 4). The clustering identified variants that are restricted to certain subpopulations of maize or those that are present in multiple populations.
Figure 4.
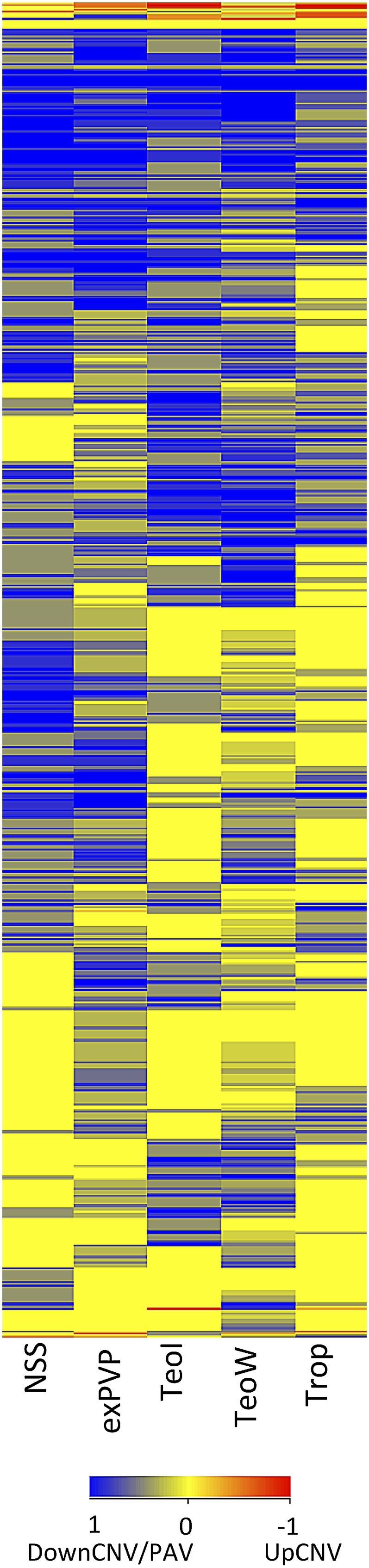
Structural variation haplotype frequencies in subpopulations of maize. Each of the genotypes was assigned to a subpopulation based on pedigree information or structure analysis. The subpopulations are nonstiff stalk (NSS, n = 4), ex-plant varietal protection varieties (exPVP, n = 6), inbred teosinte (TeoI, n = 4), wild teosinte (TeoW, n = 10), or tropical (Trop, n = 5). The frequency of the structural variant within this subpopulation was used to perform hierarchical clustering of both the structural variants and the subpopulations. The color indicates the type and frequency of each structural variant, with blue indicating DownCNV/PAV and red indicating UpCNV. The brighter colors represent higher allele frequencies.
Characterization of genes affected by structural variation
The observation that many maize genes can vary in copy number or presence among genotypes leads to queries about the potential functional impacts. Two observations suggest that genes affected by structural variation are enriched for sequences with low levels of conservation among species. First, genes showing structural variation are significantly enriched (∼1.5-fold) for genes that do not have any Gene Ontology (GO) annotation. Second, all classes of CNV genes are significantly enriched (2.8-fold overall) for maize-specific genes based on homolog clustering with annotated genes of rice, sorghum, and Arabidopsis (Table 5). The 1488 maize-specific genes affected by CNV/PAV include 1097 for which no additional homologs were found within maize and 391 that are in multigene families. The lack of clear homologs in other species is consistent with the prediction that many PAV genes may have nonessential functions, and may indicate that some of these sequences are previously unclassified transposable elements. Indeed, some examples of “gene” content variation among rice subspecies were later identified as transposons, and it can be difficult to identify and eliminate all transposons in genome-wide analyses (Bennetzen et al. 2004).
Table 5.
Gene family sizes and species specificity of CNV genes

aIncludes genes not assigned to families due to failure to cluster (3521 of total and 1096 of CNV genes) and genes assigned to families that lack membership of rice, sorghum, or Arabidopsis (1017 of total and 392 of CNV genes). Deviation from expected values were highly significant (P < 0.0001) for all CNV classes based on χ2 tests.
bDeviation from expected family size distributions were highly significant (P < 0.0001) for all CNV classes except for CNV (Both), which yielded a significant P-value of 0.0241.
The remaining 2317 maize genes affected by CNV/PAV are conserved in other plant species, and among these, 2231 have orthologs identified in rice and/or sorghum. The relative genomic positions of orthologous genes were compared with rice and sorghum to determine how often the structural variant genes are located at syntenic positions. Among all orthologous maize genes (n = 27,550), 85.5% are syntenic. This compares with only 64.9% of orthologous CNV/PAV genes, a significant reduction (χ2, P < 0.0001). Lack of synteny could have resulted from gene movement from its ancestral position or from gene duplication concomitant with movement, thereby leaving an intact ancestral copy. Such cases would be manifested by the existence of syntenic co-orthologs, i.e., genes that are paralogous to CNV genes and having a common rice or sorghum ortholog to which synteny has been maintained. Overall, we detected 1964 nonsyntenic maize genes that have syntenic co-orthologs. Over 21% of these (424) correspond to CNV/PAV genes identified in this study, almost twice that expected by chance (χ2, P < 0.0001). Thus, many of the structural variant genes with orthologs in other grasses are within-species duplicates that have moved from their ancestral positions. No evidence was found that these genes belong to the PACK-MULE or helitron classes of transposons (Schnable et al. 2009), which are known to mediate gene capture and movement in maize (Bennetzen 2005; Lai et al. 2005; Morgante et al. 2005; Schnable et al. 2009; Yang and Bennetzen 2009). Thus, other mechanisms appear to be at play.
The genes affected by structural variation are often part of large gene families in the reference genome and are significantly less likely to be single-copy genes (Table 4). In particular, the genes affected by structural variation are often found within paralogous clusters. Only 18.6% (4263/22,948) of all maize genes within multigene families reside in paralogous clusters, compared with 30.4% of (730/2399) of CNV genes (χ2, P < 0.0001). This observation is consistent with the expectation that paralogous clusters are rapidly evolving and unstable with respect to copy number.
The functional annotations of the CNV/PAV genes were assessed using the biological network gene ontology tool (BiNGO) (Maere et al. 2005) to identify overrepresented genes. There are relatively few functional categories that exhibit over-representation (Supplemental Fig. 5; Supplemental Table 4). The UpCNV genes exhibit enrichment for thylakoid-related genes, which may reflect intraspecific variation for specific chloroplast/mitochondrial DNA insertions as previously noted by Lough et al. (2008). The enrichment for membrane proteins (UpCNV) and genes involved in stress response (DownCNV/PAV) may be a consequence of the enrichment for these types of genes in tandem arrays (Rizzon et al. 2006).
The list of genes affected by structural variation was compared with several manually curated gene lists (Table 1), including 420 genes defined by classical genetics, 269 non-histone chromatin genes, 1198 cell wall genes, and 1723 transcription factors. For each of these lists, the number of genes affected by structural variation was less than expected based on the frequency of all genes affected by structural variation (χ2, P < 0.005). However, a number of genes within these lists do exhibit structural variation. For example, several instances of copy number variation were supported by prior analyses of variation in maize. The pericarp color1 (p1) gene was identified as a putative DownCNV, and previous studies have documented tandem repeats for this gene (Chopra et al. 1998), including 11 tandem repeats in B73 (Goettel and Messing 2009). A qPCR analysis of the copy number for the p1-coding sequence (data not shown) indicates that many of the genotypes with relatively low signal, such as M37W, P39, TIL1, TIL9, TIL17, and TIL15, are likely to have only a single copy of the p1-coding sequence and confirms the relative copy number changes observed by aCGH. The bZIP factor opaque-2 heterodimerizing protein1 (OHP1) was also identified as a DownCNV (Fig. 5). At least two closely related OHP1 sequences are present as tandem duplicates in B73. Copy number variation for OHP1 was previously documented (Pysh and Schmidt 1996) in some maize genotypes, including a single copy of OHP1 in Oh43 and Tx303 and multiple copies in W22. Our data are in agreement for these varieties and additionally provide evidence that OHP1 is present as a single copy in approximately 17 of the genotypes tested, and that there are at least two copies in the other genotypes (Fig. 5). There is also evidence for potential copy number variation from the CGH data (Fig. 5), as well as previous SSR studies (http://maizegdb.org/) for the globulin1 gene. Interestingly, in each of these three examples, the majority of teosinte lines have low copy number for these genes, while many of the domesticated maize genotypes have complex, multicopy alleles.
Figure 5.

Examples of CNV for previously characterized maize genes. The CGH data are summarized for three maize genes. For each genotype the average log2 ratio for all probes from the gene is summarized as the height of the bar, and the standard deviation for the multiple probes is represented by the error bars.
Discussion
The CGH analysis of diverse domesticated maize genotypes as well as teosinte lines revealed pervasive structural variation affecting over 10% of the genes annotated in the B73 reference genome (61% of which have homologs in other grasses). If we restrict our analysis to genes associated with GO annotation terms, we find that 8% of these genes are affected by CNV/PAV. The identification of genes affected by CNV or PAV in a diverse panel of maize genotypes allowed us to characterize the portion of this complex plant genome for which loss is tolerated. In addition, it provided an opportunity to investigate the distribution of structural variant events in domesticated and undomesticated maize and to speculate on potential phenotypic contributions of structural variation in maize.
The presence of substantial structural variation affecting gene content has implications for the application of the reference genome concept and how a reference genome is used to “anchor” next-generation sequence data from DNA or RNA of other individuals. It is worth noting that the number of CNV and PAV identified in this study is likely an underestimate of the actual number of CNV and PAV, since the current analysis could only detect structural variation within genes, and previous studies have found that only about one-third of the variants in low-copy maize DNA included genes (Springer et al. 2009). The actual number of genic CNV/PAV may also be underestimated since relatively strict criteria were used to call variants, and we may not have had sufficient power to detect rare CNV/PAV, particularly in the segregating teosinte individuals for which many variants may be present as heterozygotes, and thus not detected.
Mechanisms of structural variation
A current understanding of evolutionary mechanisms for producing and maintaining copy number variants (specifically gene duplications) is limited. Recombination- and replication-based mechanisms of CNV emergence have been proposed (Innan and Kondrashov 2010). The variation in the frequency of CNV in regions of high and low recombination suggests that recombination-based mechanisms play a role in either creating or maintaining CNV within the maize genome. Interestingly, the low-recombination regions had elevated frequencies of CNV. Both UpCNV and DownCNV show elevated frequencies within the low-recombination central portion of maize chromosomes. It is possible that this reflects a requirement for recombination in order to remove local duplications and eliminate CNV. Alternatively, it is possible that intrachromosomal recombination is elevated in these regions with lower interchromosomal recombination.
In contrast, PAV rates were not influenced by recombination rate and are likely produced by mechanisms different from CNV. Woodhouse et al. (2010) studied the fractionation of genome regions that result from whole-genome duplication events. They found evidence for a short deletion mechanism that utilizes short direct repeats to explain differences in gene content within the duplicated regions of the reference maize genome. This mechanism is likely to also contribute to the high rate of PAV that we observe among maize genotypes.
Toleration of gene loss in maize inbreds
It is surprising that individuals of the same species can have such variable gene copy number and content. A recent study (Conrad et al. 2010) found that two human individuals have ∼1000 CNV/PAV that affect approximately 600 genes, and that roughly 35% these could be identified as homozygotes. However, there are relatively few examples of CNV/PAV sequences being linked to disease (The Wellcome Trust Case Control Consortium 2010), suggesting that relatively few of these CNV/PAV have major phenotypic consequences. Similarly, in the current study we have identified numerous CNV/PAV within both maize and teosinte genotypes. Given that the maize genotypes we assayed are highly inbred (and therefore homozygous for the CNV/PAV) and have been selected for agricultural productivity, the majority of these CNV/PAV are not likely associated with lethality or major loss of fitness in an agricultural environment. Moreover, the presence of most of these variants in teosinte means that these variants are segregating in natural populations and are therefore unlikely to have strongly negative effects on fitness, at least not as heterozygotes. This leads to a question of how substantial levels of gene loss can be tolerated with relatively low perturbation of phenotype. The types of genes that are affected and the complex structure of the maize genome may provide clues as to how gene loss is tolerated in maize inbred lines.
A subset (∼40%) of the genes subject to structural variation are not found in the genomes of other model plants (Arabidopsis, rice, sorghum). These sequences may be novel transposon sequences or novel transcribed sequences that do not encode functional genes. Many of the remaining CNV/PAV genes that did have annotations and/or orthologs are present in gene families that include members at syntenic positions or within paralogous clusters. The maize genome, which arose from an ancient allopolyploidization event (Gaut and Doebley 1997; Swigonova et al. 2005; Wei et al. 2007), has many gene families with a very high level of redundancy. Gene losses within these gene families may be tolerated if they result in only minor differences in phenotype or fitness. For example, the Gnarley1 (Gn1) locus, a member of the knox gene family, was identified as “absent” in five genotypes. Ectopic expression of Gn1 can result in morphological phenotypes, but loss-of-function alleles of Gn1 do not result in major phenotypic consequences (Foster et al. 1999). Analysis of 16 of the genes affected by PAV that are included on the list of classically defined maize genes (http://synteny.cnr.berkeley.edu/wiki/index.php/Classical_Maize_Genes) reveals that the majority of these (14/16) have duplicates located within the collinear portion of the maize genome.
The observation that many of the genes affected by CNV or PAV are members of gene families has some important implications for the phenotypic consequences of PAV in plant genomes. Many plant genomes have substantial levels of gene duplication that have arisen from whole-genome duplications as well as other mechanisms (Freeling 2009). Even the relatively small genomes of Arabidopsis and rice contain evidence for ancient whole-genome duplications (Blanc et al. 2003; Yu et al. 2005; Paterson et al. 2006). Comparisons of plant genomes have revealed relatively high levels of instability and frequent gene loss that often affects members of gene families (Bennetzen 2007; Freeling 2009; Woodhouse et al. 2010). If we assume that there is redundancy or partial redundancy for function within the gene family, then the effect of losing a single member of a gene family can be genetically buffered by the family members. In effect, this means that within complex, highly duplicated genomes, a PAV is likely to contribute quantitative variation rather than major, qualitative defects. This may result in high levels of structural variation in crop plant genomes that contributes to important quantitative variation. Indeed, there are recent examples of rice quantitative trait loci (QTL) that are caused by deletion of genes (Shomura et al. 2008; Zhou et al. 2009).
Implications of structural variation for heterosis
The concept of partial redundancy within gene families, coupled with high rates of CNV/PAV that affect different inbred varieties, may have implications for heterosis. Generally, heterosis is considered at the level of two alleles that may provide complementation when present in a heterozygote. However, it may be useful to consider each member of a gene family as an “allele” that provides partial to complete functionality for the gene family. Inbred lines show relatively high rates of CNV/PAV that affect the copy number, or presence, of individual members of gene families. The loss of a single member of a gene family may result in a relatively minor loss of the total functionality of the gene family as other family members provide compensatory function. The cumulative effect of many gene families lacking partially redundant members would result in decreased vigor in the inbreds. However, the loss-of-function would be complemented (at a genomic, not allelic) level in the hybrid, resulting in substantial hybrid vigor. The hypothesis that heterosis is the result of restoring full functionally of gene families would suggest that heterosis would be more prevalent in organisms with high levels of gene duplication and variation affecting individual family members.
It has been suggested that variation in gene content among maize inbred lines could contribute to heterosis or hybrid vigor (Fu and Dooner 2002; Springer and Stupar 2007; Springer et al. 2009). High levels of variability in gene content among inbred lines will result in hybrids containing more genes than either inbred parent and, indeed, expression studies have found that hybrids express more genes than either parent (Stupar and Springer 2006; Stupar et al. 2008). Historically, the complementation model of heterosis has been supported by the fact that an inbred line has not been created with all superior alleles (Birchler et al. 2003). Due to the high number of PAVs, it would be very difficult to create an inbred line containing all genes. Many of the maize inbreds were missing 500–1000 genes relative to B73. If we assume that each of these lines contains a similar number of genes that are not in B73, it becomes quite difficult to identify a series of recombination events that would create a chromosome containing all genes. Furthermore, the current complex arrangement of different complements of genes in the two haplotypes of a heterozygote can lead to apparent pseudo-overdominance. This would be a particular problem in the low-recombination centromeric regions of each chromosome. In total, these low-recombination regions include ∼750 PAV genes, and the low rate of recombination events would make it quite difficult to generate ideal haplotypes. Recent analyses of residual heterozygosity suggest that these low-recombination regions may be particularly important for heterosis (McMullen et al. 2009). The allele frequencies that we observed for structural variants suggest that some variants have been entirely removed from certain populations. Maize breeding efforts are often focused on breeding within a heterotic group, or subpopulation, to create inbreds that are crossed to an inbred from another heterotic group. We found evidence for a number of structural variants that are entirely missing from one subpopulation, limiting the potential for improvement on inbred lines through selection within that subpopulation only.
Distribution of structural variation within maize and teosinte
The identification of relatively few rare variants suggests that many of the structural variants represent haplotypes that have been segregating for some time in maize and teosinte populations. While technical aspects (such as the genome used as reference) and statistical power issues (the numbers of lines representing each subpopulation) may influence the ability to discover rare structural events, these are unlikely to completely account for the paucity of rare events observed in this study. The majority (∼86%) of structural variants in this study were observed in both maize and teosinte, suggesting that they are relatively old events in terms of domestication. In addition, the presence of these events in teosinte would indicate that they are tolerated within natural populations and are not an artifact of many generations of artificial selection. A small proportion (∼10%) of the variants were observed only in domesticated maize lines. Interestingly, many of these maize-specific events (252/347) are observed in three or fewer genotypes. Therefore, the maize-specific variants are enriched for rare alleles, and these may represent relatively new events that have arisen within breeding populations.
The wild teosinte individuals used for this study were collected from populations located near the probable location of domestication (Piperno et al. 2009; Ranere et al. 2009). We searched for structural variants potentially associated with domestication by using the relative frequencies within maize and teosinte. We did not find any structural variants that were present in the majority of maize genotypes but not detected in any teosinte genotypes. However, it should be remembered that structural variants were documented based on comparisons to a reference domesticated maize line, and that genes present in teosinte, but not maize, cannot be detected. Therefore, domestication-associated copy number variants would be expected to be present in most teosintes, but in few or no maize lines. There were only four variants that were observed in most (>85%) teosinte lines but in very few (less than three) maize genotypes, and thus there was no evidence for strong effects of domestication on structural variation.
The analysis of structural variation in maize and teosinte provides evidence for widespread genome content variation. This high level of variation could reflect the ancestoral polyploid nature of the maize genome by the fact that maize has high rates of out-crossing, or active transposition and genome contraction processes to create a dynamic genome. In addition, studies on genome content variation within a species can be used to develop an understanding of the core genome (shared by all members of a species) and the non-core genome (“dispensable genome”, as suggested by Morgante et al. 2007). It is likely that these structural variants will be associated with phenotypic diversity within maize, and further research is important to document how these variants affect phenotype. An understanding of which genes are affected by structural variation may provide a valuable resource to probe the function of many maize genes.
Methods
Array design
A custom long oligonucleotide microarray was designed by NimbleGen (Roche NimbleGen) using the 32,540 filtered maize genes predicted from the B73 reference genome (Schnable et al. 2009). Partial-length gene fragments and transposable elements are not included in this filtered gene set. The custom array included three to four probes (45–60 mers) each for 32,487 genes for which probes could be designed, as well as 17,995 control probes that are not present in the maize genome, but exhibit nucleotide frequencies similar to maize. Of the 119,609 genic probes, 114,854 (96%) were unique in the genome and 118,730 (99%) were present no more than two times in the B73 genome. Detailed information about the array format is available at GEO accession no. GPL10846 and this array can be ordered from Roche NimbleGen (product OID24389).
Plant materials
Maize inbred lines were obtained from the USDA North Central Regional Plant Introduction Station. Teosinte inbred lines were provided by John Doebley (University of Wisconsin, Madison). Teosinte accessions (Ames 21809, Ames 21810, and Ames 21814) originally collected from the Guerrero state of Mexico were obtained from the USDA North Central Regional Plant Introduction Station. All genotypes are listed in Supplemental Table 1 along with germplasm accession numbers. These include diverse maize inbred lines (n = 19), inbred teosinte lines (n = 4), and wild teosinte individuals (n = 10). Additional replications of maize inbred lines B73 and Mo17 were repeated multiple times to assess consistency within array measurements.
DNA labeling and microarray hybridization
DNAs were isolated (Saghai-Maroof et al. 1984) from above-ground seedling tissue. DNA (0.5–1 μg) samples were labeled, amplified, and hybridized for 72–96 h at 42°C according to the array manufacturer's protocol (NimbleGen Arrays User's Guide: CGH Analysis v5.1). Post washing, slides were immediately scanned using the GenePix 4000B Scanner (Molecular Devices) according to the array manufacturer's protocol. Array images and data were processed using NimbleScan software. Experimental integrity was verified by evaluation of the signal uniformity across each array and the signal-to-noise ratio of experimental probes. A total of 71 samples (genotypes listed in Supplemental Table 1) provided high-quality data and were used for subsequent analyses; the raw data is available at GEO accession no. GSE23756.
Data normalization
The different genotypes examined are not equally diverged from the B73 reference genome used to develop the probe sequences. For this reason we normalized the data using an approach that does not assume similar distributions of data from each genotype. The implemented normalization approach assumes that, for any genotype, the majority of probes will not exhibit any significant variation relative to B73 and, therefore, the peak of the log2(signal/B73) histogram should be centered at a value of zero (Supplemental Fig. 1). Briefly, the DNAcopy algorithm was used to produce spatially normalized hybridization values for all probes for the 71 samples using NimbleScan (Roche NimbleGen). A robust B73 average (henceforth termed B73avg) was generated from nine replicate samples of B73 hybridization. Subsequently, the log2(signal/B73avg) was calculated for each probe for all 71 samples. The distributions of these ratios were normalized so that the mode of the distribution of log2(signal/B73avg) for each genotype equaled.
Identification of CNV and PAV
For each probe, the log2 ratio (relative to B73) is expected to be near zero if the same sequence is present in both genotypes. Following normalization, the histogram of all log2 ratios (relative to B73) revealed varying distributions of the data (Supplemental Fig. 1). The distribution of the log2 ratios is affected by both measurement error and biological variation. Because the amount of technical variation can vary between hybridizations, we calculated 99th percentile cut-off values for each genotype separately. The cut-off values were determined from the distribution of all data with values over 0, and subsequently used to identify genes with structural variation for each genotype (see Supplemental Fig. 1 for a full description of this process). UpCNV (more copies of a gene in some genotypes relative to B73) were identified as genes for which all probes (three or four) per gene had values above the 99% cut-off value. DownCNV/PAV (fewer copies or no copies of a gene in some genotypes relative to B73) were identified as genes for which all probes exhibited a log2 ratio below the negative value of the 99% cut-off value. It should be noted that the cut-off values for both UpCNV and DownCNV/PAV were determined based on the confidence interval for the subset of data with positive log2 values, since this subset of data will only reflect noise and structural variation, while negative log2 ratios would additionally reflect SNP polymorphism rates (Supplemental Fig. 1). This approach was quite stringent in that it required significant variation to be observed at all probes for a gene. We observed a very low false-positive rate (none to eight genes detected) when this approach was applied to any single B73 replicate. Following the stringent discovery process, a relaxed set of criteria was implemented (>95% cut-offs) to characterize the structural variant across all genotypes.
Functional characterization of genes affected by structural variation
The genomic distribution of genes was assessed using the genomic coordinates from the B73 reference genome for each of the genes. We identified multigene structural variants in cases where two or more adjacent genes exhibit the same type of structural variation (UpCNV or DownCNV/PAV) in a highly similar set of genotypes. The GOslim annotation (http://www.maizesequence.org) of genes that were affected by structural variation were assessed using BiNGO (Maere et al. 2005); a Cytoscape (Shannon et al. 2003) plug-in that maps over-represented functional themes present in a given gene-set onto the GO hierarchy. P-values for enrichment or GOslim terms were calculated using a hypergeometric distribution statistical testing method with false discovery rate (FDR) correction (Benjamini and Hochberg 1995). The maize-specific genes and gene families were identified based on homolog clustering with annotated genes of rice, sorghum, and Arabidopsis using the method of Vilella et al. (2009) as previously described (Schnable et al. 2009). Paralogous clusters were defined as two or more genes belonging to the same gene family that were separated on a chromosome by no more than two nonparalogous intervening genes. Syntenic mapping of maize genes to rice and sorghum was previously described (Schnable et al. 2009). In addition, we examined the frequency of CNV/PAV using several manually curated gene lists, including classically defined maize genes (http://synteny.cnr.berkeley.edu/wiki/index.php/Classical_Maize_Genes), nonhistone chromatin genes (http://www.chromdb.org), transcription factors (http://www.grassius.org), and maize cell wall genes (http://cellwall.genomics.purdue.edu/).
Distribution of genes affected by structural variation
The distribution of CNVs and PAVs was compared within each of the 10 maize chromosomes (Table 2). Regions of high, moderate, and low recombination were determined based on the integrated physical-genetic map generated by Liu et al. (2009). The high-recombination regions are toward the ends of the chromosomes, while the low-recombination regions surround the centromeres. The distribution of CNVs and PAVs within the high- and low-recombination regions of all chromosomes were tested and P-values were produced from the χ2 analysis (Table 2).
Validation of CNV/PAV
PCR primers were designed to amplify genomic sequence for 12 genes located within putative PAVs (Table 3). PCR and gel electrophoresis were conducted on the same samples and genotypes from the microarray experiment as per previously published methods (Haun and Springer 2008) using 60°C as the annealing temperature.
Acknowledgments
We thank John Doebley who very graciously provided stocks of the inbred teosinte lines, and several anonymous reviewers who provided very helpful suggestions for analyses. Peter Hermanson helped with DNA isolation and microarray hybridization. The Minnesota Supercomputing Institute provided access to software and user support for data analyses. This project was supported by USDA Hatch funds, the Microbial and Plant Genomics Institute, and a grant from the National Science Foundation to N.M.S. (IOS-0922095), and by grants from the NSF (DBI-0527192) and USDA (1907-21000-030-00) to D.W.
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE23756.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.109165.110.
References
- Beckmann JS, Estivill X, Antonarakis SE 2007. Copy number variants and genetic traits: Closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet 8: 639–646 [DOI] [PubMed] [Google Scholar]
- Belo A, Beatty MK, Hondred D, Fengler KA, Li B, Rafalski A 2010. Allelic genome structural variations in maize detected by array comparative genome hybridization. Theor Appl Genet 120: 355–367 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Bennetzen J 2005. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev 15: 621–627 [DOI] [PubMed] [Google Scholar]
- Bennetzen JL 2007. Patterns in grass genome evolution. Curr Opin Plant Biol 10: 176–181 [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Coleman C, Liu R, Ma J, Ramakrishna W 2004. Consistent over-estimation of gene number in complex plant genomes. Curr Opin Plant Biol 7: 732–736 [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J, Auger D, Riddle N 2003. In search of the molecular basis of heterosis. Plant Cell 15: 2236–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Hokamp K, Wolfe KH 2003. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res 13: 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A 2005. Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17: 343–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP, et al. 2009. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet 5: e1000536 doi: 10.1371/journal.pgen.1000536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler E, Gaut B, McMullen M 2006. Molecular and functional diversity of maize. Curr Opin Plant Biol 9: 172–176 [DOI] [PubMed] [Google Scholar]
- Chen WK, Swartz JD, Rush LJ, Alvarez CE 2009. Mapping DNA structural variation in dogs. Genome Res 19: 500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra S, Athma P, Li XG, Peterson T 1998. A maize Myb homolog is encoded by a multicopy gene complex. Mol Gen Genet 260: 372–380 [DOI] [PubMed] [Google Scholar]
- Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK 2006. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet 38: 75–81 [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Nickerson DA, Eichler EE 2007. Mutational and selective effects on copy-number variants in the human genome. Nat Genet 39: S22–S29 [DOI] [PubMed] [Google Scholar]
- Emrich SJ, Barbazuk WB, Li L, Schnable PS 2007. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res 17: 69–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW 2006. Structural variation in the human genome. Nat Rev Genet 7: 85–97 [DOI] [PubMed] [Google Scholar]
- Foster T, Yamaguchi J, Wong BC, Veit B, Hake S 1999. Gnarley1 is a dominant mutation in the knox4 homeobox gene affecting cell shape and identity. Plant Cell 11: 1239–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M 2009. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol 60: 433–453 [DOI] [PubMed] [Google Scholar]
- Fu H, Dooner HK 2002. Intraspecific violation of genetic colinearity and its implications in maize. Proc Natl Acad Sci 99: 9573–9578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Wen TJ, Ronin YI, Chen HD, Guo L, Mester DI, Yang Y, Lee M, Korol AB, Ashlock DA, et al. 2006. Genetic dissection of intermated recombinant inbred lines using a new genetic map of maize. Genetics 174: 1671–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, Doebley JF 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc Natl Acad Sci 94: 6809–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettel W, Messing J 2009. Change of gene structure and function by non-homologous end-joining, homologous recombination, and transposition of DNA. PLoS Genet 5: e1000516 doi: 10.1371/journal.pgen.1000516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert TA, Cahan P, Edwin D, Selzer RR, Richmond TA, Eis PS, Shannon WD, Li X, McLeod HL, Cheverud JM, et al. 2007. A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS Genet 3: e3 doi: 10.1371/journal.pgen.0030003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, Saar K, Adamovic T, Verheul M, van Heesch SA, Cook S, Pravenec M, Aitman T, Jacob H, Shull JD, et al. 2008. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet 40: 538–545 [DOI] [PubMed] [Google Scholar]
- Hansey CN, Johnson JM, Sekhon RS, Kaeppler SM, de Leon N 2010. Genetic diversity of a maize association population with restricted phenology. Crop Sci (in press). doi: 10.2135/cropsci2010.03.0178 [Google Scholar]
- Haun WJ, Springer NM 2008. Maternal and paternal alleles exhibit differential histone methylation and acetylation at maize imprinted genes. Plant J 56: 903–912 [DOI] [PubMed] [Google Scholar]
- Hurles ME, Dermitzakis ET, Tyler-Smith C 2008. The functional impact of structural variation in humans. Trends Genet 24: 238–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H, Kondrashov F 2010. The evolution of gene duplications: Classifying and distinguishing between models. Nat Rev Genet 11: 97–108 [DOI] [PubMed] [Google Scholar]
- Kato A, Lamb JC, Birchler JA 2004. Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identification in maize. Proc Natl Acad Sci 101: 13554–13559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Li Y, Messing J, Dooner HK 2005. Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci 102: 9068–9073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Gutierrez-Arcelus M, Perry GH, Vallender EJ, Johnson WE, Miller GM, Korbel JO, Lee C 2008. Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum Mol Genet 17: 1127–1136 [DOI] [PubMed] [Google Scholar]
- Liu S, Yeh CT, Ji T, Ying K, Wu H, Tang HM, Fu Y, Nettleton D, Schnable PS 2009. Mu transposon insertion sites and meiotic recombination events co-localize with epigenetic marks for open chromatin across the maize genome. PLoS Genet 5: e1000733 doi: 10.137l/journal.pgen.1000733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lough AN, Roark LM, Kato A, Ream TS, Lamb JC, Birchler JA, Newton KJ 2008. Mitochondrial DNA transfer to the nucleus generates extensive insertion site variation in maize. Genetics 178: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M 2005. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21: 3448–3449 [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. 2009. Finding the missing heritability of complex diseases. Nature 461: 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydan JS, Lorch A, Edgley ML, Flibotte S, Moerman DG 2010. Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans. BMC Genomics 11: 62 doi: 10.1186/1471-2164-11-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Altshuler DM 2007. Copy-number variation and association studies of human disease. Nat Genet 39: S37–S42 [DOI] [PubMed] [Google Scholar]
- McClintock B, Yamakake TAK, Blumenschein A 1981. Chromosome constitution of races of maize. Its significance in the interpretation of relationships between races and varieties in the Americas. Colegio de Postgraduados, Chapingo, Mexico [Google Scholar]
- McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun Q, Flint-Garcia S, Thornsberry J, Acharya C, Bottoms C, et al. 2009. Genetic properties of the maize nested association mapping population. Science 325: 737–740 [DOI] [PubMed] [Google Scholar]
- Merikangas AK, Corvin AP, Gallagher L 2009. Copy-number variants in neurodevelopmental disorders: Promises and challenges. Trends Genet 25: 536–544 [DOI] [PubMed] [Google Scholar]
- Messing J, Dooner H 2006. Organization and variability of the maize genome. Curr Opin Plant Biol 9: 157–163 [DOI] [PubMed] [Google Scholar]
- Messing J, Bharti AK, Karlowski WM, Gundlach H, Kim HR, Yu Y, Wei F, Fuks G, Soderlund CA, Mayer KF, et al. 2004. Sequence composition and genome organization of maize. Proc Natl Acad Sci 101: 14349–14354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgante M, Brunner S, Pea G, Fengler K, Zuccolo A, Rafalski A 2005. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet 37: 997–1002 [DOI] [PubMed] [Google Scholar]
- Morgante M, De Paoli E, Radovic S 2007. Transposable elements and the plant pan-genomes. Curr Opin Plant Biol 10: 149–155 [DOI] [PubMed] [Google Scholar]
- Paterson AH, Chapman BA, Kissinger JC, Bowers JE, Feltus FA, Estill JC 2006. Many gene and domain families have convergent fates following independent whole-genome duplication events in Arabidopsis, Oryza, Saccharomyces and Tetraodon. Trends Genet 22: 597–602 [DOI] [PubMed] [Google Scholar]
- Penning BW, Hunter CT III, Tayengwa R, Eveland AL, Dugard CK, Olek AT, Vermerris W, Koch KE, McCarty DR, Davis MF, et al. 2009. Genetic resources for maize cell wall biology. Plant Physiol 151: 1703–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, Lee AS, Hyland C, Stone AC, Hurles ME, Tyler-Smith C, et al. 2008. Copy number variation and evolution in humans and chimpanzees. Genome Res 18: 1698–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno DR, Ranere AJ, Holst I, Iriarte J, Dickau R 2009. Starch grain and phytolith evidence for early ninth millennium B.P. maize from the Central Balsas River Valley, Mexico. Proc Natl Acad Sci 106: 5019–5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysh LD, Schmidt RJ 1996. Characterization of the maize OHP1 gene: Evidence of gene copy variability among inbreds. Gene 177: 203–208 [DOI] [PubMed] [Google Scholar]
- Ranere AJ, Piperno DR, Holst I, Dickau R, Iriarte J 2009. The cultural and chronological context of early Holocene maize and squash domestication in the Central Balsas River Valley, Mexico. Proc Natl Acad Sci 106: 5014–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzon C, Ponger L, Gaut BS 2006. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput Biol 2: e115 doi: 10.1371/journal.pcbi.0020115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TP, Walker EL, Kermicle JL, Alleman M, Dellaporta SL 1991. Meiotic instability of the R-r complex arising from displaced intragenic exchange and intrachromosomal rearrangement. Genetics 129: 271–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW 1984. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci 81: 8014–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuari L, Pradervand S, Amiguet-Vercher AM, Thomas J, Dorcey E, Harshman K, Xenarios I, Juenger TE, Hardtke CS 2010. Substantial deletion overlap among divergent Arabidopsis genomes revealed by intersection of short reads and tiling arrays. Genome Biol 11: R4 doi: 10.1186/gb-2010-11-1-r4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. 2009. The B73 maize genome: Complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Sebat J 2007. Major changes in our DNA lead to major changes in our thinking. Nat Genet 39: S3–S5 [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. 2004. Large-scale copy number polymorphism in the human genome. Science 305: 525–528 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T 2003. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. 2005. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77: 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. 2006. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38: 1038–1042 [DOI] [PubMed] [Google Scholar]
- Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M 2008. Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet 40: 1023–1028 [DOI] [PubMed] [Google Scholar]
- Springer NM, Stupar RM 2007. Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res 17: 264–275 [DOI] [PubMed] [Google Scholar]
- Springer NM, Ying K, Fu Y, Ji T, Yeh CT, Jia Y, Wu W, Richmond T, Kitzman J, Rosenbaum H, et al. 2009. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet 5: e1000734 doi: 10.1371/journal.pgen.1000734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR 2010. Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437–455 [DOI] [PubMed] [Google Scholar]
- Stupar RM, Springer NM 2006. Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173: 2199–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupar RM, Gardiner JM, Oldre AG, Haun WJ, Chandler VL, Springer NM 2008. Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant Biol 8: 33 doi: 10.1186/1471-2229-8-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swigonova Z, Bennetzen JL, Messing J 2005. Structure and evolution of the r/b chromosomal regions in rice, maize and sorghum. Genetics 169: 891–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732 [DOI] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E 2009. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19: 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Dooner HK 2006. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc Natl Acad Sci 103: 17644–17649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S, et al. 2007. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet 3: e123 doi: 10.1371/journal.pgen.0030123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium 2010. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464: 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, et al. 2007. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet 80: 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse MR, Schnable JC, Pedersen BS, Lyons E, Lisch D, Subramaniam S, Freeling M 2010. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol 8: e1000409 doi: 10.1371/journal.pbio.1000409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandeau-Nelson MD, Xia Y, Li J, Neuffer MG, Schnable PS 2006. Unequal sister chromatid and homolog recombination at a tandem duplication of the a1 locus in maize. Genetics 173: 2211–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Bennetzen JL 2009. Distribution, diversity, evolution and survival of Helitrons in the maize genome. Proc Natl Acad Sci 106: 19922–19927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Zhou Q, Li J, Smith H, Yandeau M, Nikolau BJ, Schnable PS 2002. Molecular characterization of meiotic recombination across the 140-kb multigenic a1-sh2 interval of maize. Proc Natl Acad Sci 99: 6157–6162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wang J, Lin W, Li S, Li H, Zhou J, Ni P, Dong W, Hu S, Zeng C, et al. 2005. The Genomes of Oryza sativa: A history of duplications. PLoS Biol 3: e38 doi: 10.1371/journal.pbio.0030038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Peterson T 2005. Comparisons of maize pericarp color1 alleles reveal paralogous gene recombination and an organ-specific enhancer region. Plant Cell 17: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR 2009. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 10: 451–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhu J, Li Z, Yi C, Liu J, Zhang H, Tang S, Gu M, Liang G 2009. Deletion in a quantitative trait gene qPE9-1 associated with panicle erectness improves plant architecture during rice domestication. Genetics 183: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]