

Abstract
BACKGROUND AND PURPOSE
In 2006, a life-threatening ‘cytokine storm’, not predicted by pre-clinical safety testing, rapidly occurred in all six healthy volunteers during the phase I clinical trial of the CD28 superagonist monoclonal antibody (mAb) TGN1412. To date, no unequivocal explanation for the failure of TGN1412 to stimulate profound cytokine release in vitro or in vivo in species used for pre-clinical safety testing has been established. Here, we have identified a species difference almost certainly responsible for this disparate immunopharmacology.
EXPERIMENTAL APPROACH
Polychromatic flow cytometry and intracellular cytokine staining were employed to dissect the in vitro immunopharmacology of TGN1412 and other therapeutic mAbs at the cellular level to identify differences between humans and species used for pre-clinical safety testing.
KEY RESULTS
In vitro IL-2 and IFN-γ release from CD4+ effector memory T-cells were key indicators of a TGN1412-type response. This mechanism of cytokine release differed from that of other therapeutic mAbs, which can cause adverse reactions, because these other mAbs stimulate cytokine release primarily from natural killer cells. In contrast to humans, CD28 is not expressed on the CD4+ effector memory T-cells of all species used for pre-clinical safety testing, so cannot be stimulated by TGN1412.
CONCLUSIONS AND IMPLICATIONS
It is likely that activation of CD4+ effector memory T-cells by TGN1412 was responsible for the cytokine storm. Lack of CD28 expression on the CD4+ effector memory T-cells of species used for pre-clinical safety testing of TGN1412 offers an explanation for the failure to predict a ‘cytokine storm’ in humans.
Keywords: TGN1412, cytokine storm, CD28, monoclonal antibody, effector memory T-cells, pre-clinical safety testing, pro-inflammatory cytokines
Introduction
TGN1412 is a superagonist CD28-specific monoclonal antibody (mAb) developed as an immunotherapeutic for the purpose of balancing the immune system (TeGenero, 2005). TGN1412 was called a ‘superagonist’ because it could activate T-cells without the need for prior engagement of the T-cell receptor complex (Tacke et al., 1997; Lühder et al., 2003). In rodent models, treatment with CD28 superagonists orthologous to TGN1412 stimulated the release of anti-inflammatory cytokines and the expansion of regulatory T-cells believed to counter autoimmune responses (Rodríguez-Palmero et al., 1999; Lin and Hünig, 2003; Beyersdorf et al., 2005). These CD28 superagonists proved to be an effective treatment of inflammatory autoimmune disease in rodent models (Beyersdorf et al., 2006), and it was believed that a humanized antibody (TGN1412) would have similar potential for human therapy (Elflein et al., 2003). Pre-clinical safety testing of TGN1412 in cynomolgus macaques, at doses of up to 50 mg·kg–1·week–1, was well tolerated with no signs of toxicity or systemic immune stimulation. However, the first-in-man phase I clinical trial of TGN1412, conducted at a clinical trial unit in London, ended in disaster.
A near-fatal systemic inflammatory immune response, termed a ‘cytokine storm’, began 1 h after infusion of TGN1412 in all six healthy recipients, and all became critically ill within 12–16 h (Suntharalingam et al., 2006). Subsequent analysis of the TGN1412 used at the trial showed that it was formulated correctly, complied with its product specification and was free from pro-inflammatory contaminants (Duff, 2006; Stebbings et al., 2007). At the request of an Expert Special Group on phase I clinical trials, appointed by the UK Medicines and Healthcare Products Regulatory Agency, an investigation was carried out by the authors into the biological properties of TGN1412. We found that TGN1412 only stimulated a pro-inflammatory cytokine response in vitro if appropriately presented to human peripheral blood mononuclear cells (PBMCs) (e.g. by immobilization onto a plastic surface), and that rather than receiving a low safe starting dose, trial volunteers had in fact been given a near maximum immunostimulatory dose (Duff, 2006; Stebbings et al., 2007; 2009;). Significantly, PBMCs from cynomolgus macaques were not stimulated in vitro by immobilized TGN1412 in agreement with the failure of in vivo administration at any dose tested to evoke any adverse response (Duff, 2006; Stebbings et al., 2007).
Here, we address three further questions which arose from the initial investigation. First, we demonstrate that the cytokine release syndrome stimulated by TGN1412 is not mediated by the same mechanism(s) as those underlying adverse reactions to other therapeutic mAbs (e.g. Campath-1H and Mabthera). Second, we show that the new in vitro safety procedures are applicable to other therapeutic mAbs if interleukin (IL)-2 and interferon (IFN)-γ are used as key markers for a TGN1412-type response. Third, we identify a species difference in CD28 expression on the CD4+ effector memory T-cell subset as being most likely responsible for the failure of pre-clinical safety testing of TGN1412 in cynomolgus macaques. This is a CD28-specific, rather than a general, difference affecting applicability of such models to the safety testing of new therapeutic mAbs and immunomodulatory biotherapeutic agents.
Methods
In vitro stimulations with therapeutic mAbs
Stimulation with therapeutic mAbs was performed in triplicate in 96-well round bottom polypropylene microtitre plates (Corning Inc., Corning, NY, USA) containing TGN1412 (humanized IgG4κ, anti-CD28 superagonist), Mabthera (chimeric mouse/human IgG1, anti-CD20, rituximab), Campath-1H (humanized IgG1, anti-CD52, alemtuzumab), ANC28.1 (murine IgG1, anti-CD28 superagonist), CD28.2 (murine IgG1, anti-CD28 agonist) and humanized IgG4κ isotype-matched control for TGN1412. Therapeutic mAbs were immobilized onto plates by addition of 1 µg mAb diluted in 60 µL of phosphate-buffered saline (PBS) per well followed by incubation at room temperature for 1–2 h, then washing twice with 200 µL of PBS to remove unbound antibody. For analysis of rapid cytokine expression, 96-well round bottom polystyrene microtitre plates (Corning Inc.) were employed with mAbs immobilized by air drying, as previously described (Stebbings et al., 2007; Findlay et al., 2010). Fc immobilization of TGN1412 was accomplished using a polyclonal chicken anti-human IgG Fc that was first air-dried onto plates (Stebbings et al., 2007; Findlay et al., 2010), followed by two washes and a 1 h incubation with 1 µg of TGN1412 diluted in 60 µL of PBS per well, then washing twice with 200 µL of PBS. Human PBMCs, isolated from the heparinized blood of healthy donors by density gradient separation (Lymphoprep, Axis-Shield, Oslo, Norway) according to manufacturer's instructions, were resuspended in RPMI 1640 culture medium supplemented with 15% fetal calf serum, l-glutamine and antibiotics (penicillin and streptomycin). For functional assays, PBMCs were added to therapeutic mAb-coated plates at 1 × 106 cells·mL–1 of culture medium. For the measurement of cytokine release by elisa, supernatants were harvested after PBMC stimulation for 24, 48 and 72 h in a humidified incubator at 37°C with 5% CO2. For cytokine release assays, lectin phytohaemagglutinin (PHA) at 10 µg·mL–1 was used as a positive control. Where intracellular cytokine staining was measured, the secretion inhibitor brefeldin A was added to cell cultures at 10 µg·mL–1. Early kinetics of cytokine responses to therapeutic mAbs were assessed after 30, 60, 90, 120, 240 and 360 min incubations by intracellular cytokine staining. The mitogens, phorbol myristate acetate (PMA) at 0.05 µg·mL–1 and ionomycin at 0.75 µg·mL–1, were used together as a positive control for intracellular cytokine staining. Effector memory cell enrichment and depletion prior to stimulation were performed using MACS human CD4+ T-cell effector memory isolation kit (Miltenyi Biotech, Surrey, UK), according to the manufacturer's instructions, after first removing sodium azide from all reagents using Amicon Ultra-4 30 kDa centrifugal filter devices (Millipore, Billerica, MA, USA).
Cytokine responses to therapeutic mAbs
Concentrations of IFN-γ, tumour necrosis factor (TNF)-α, IL-2 and IL-8 in culture supernatants were measured by elisa using the following antibody pairs: anti-human IFN-γ mAb clone NIB42 and clone 4S.B3, anti-human TNF-α antibody clone 357-101-4 and polyclonal anti-TNF-α antibody (H91), anti-human IL-2 mAb clone 5355 and polyclonal anti-human IL-2, affinity-purified polyclonal sheep anti-human IL-8 antibodies for both capture and detection. Streptavidin–HRP conjugate was used to detect and quantify biotinylated antibody in conjunction with tetramethylbenzidine for IL-2 and IFN-γ, and O-phenylenediamine for TNF-α and IL-8.
Polychromatic intracellular cytokine staining was performed as described previously (Stebbings et al., 2007). For surface staining, combinations of the following anti-human fluorochrome conjugates were employed: CD4 PE-Cy7 (clone SK3), CD4 APC (clone RPA-TA), CD4 APC-Cy7 (clone OKT4), CD28 PE-Cy7 (clone CD28.2), CD45RO APC (clone UCHL-1), CD95 PE (clone DX2) and CCR7 FITC (clone 150503). For intracellular cytokine staining, combinations of the following anti-cytokine fluorochrome conjugates were used: TNF-α PE (clone MAb11), IL-2 PE (clone 5344.111), IL-2 FITC (clone 5344.111), IL-2 PerCP Cy5.5 (clone MG1-17H12), IFN-γ PE (clone 4S.B3), IFN-γ Pacific Blue (clone 4S.B3). Acquisition of 30 000 gated lymphocyte events per well was performed using a FACSCanto II flow cytometer equipped with HTS plate reader (BD Biosciences, Oxford, UK) and analysed using FACSDiva software (BD Biosciences). For analysis of memory subsets, differential staining for CD45RO and the chemokine receptor, CCR7, was employed to subdivide human CD4+ T-cells into CD45RO-CCR7+ naive T-cells, CD45RO+CCR7+ central memory T-cells and CD45RO+CCR7- effector memory T-cells (Sallusto et al., 1999; 2004;). For macaque memory subset analysis, differential staining for CD95 and CD28 was employed to subdivide CD4+ T-cells into CD95-CD28+ naive T-cells, CD95+CD28+ central memory T-cells and CD95+CD28- effector memory T-cells (Pitcher et al., 2002; Karlsson et al., 2007). For cross-species effector memory subset comparison, differential staining for CD28 and CCR7 was employed to subdivide CD4+ T-cells into CD28+CCR7+ naive and central memory subsets (both species), CD28+CCR7- human effector memory T-cells, CD28+CCR7- macaque transitional memory T-cells and CD28-CCR7- macaque effector memory T-cells (Picker et al., 2006).
Real-time (RT)-PCR was performed as described previously (Burns et al., 2008). For the measurement of cytokine mRNAs, PCR reactions contained intron-spanning primers specific for cytokine or house-keeping gene (GAPDH). Primer sequences for human GAPDH were forward; 5′-GTC AGT GGT GGA CCT GAC CT-3′, reverse; 5′-CCC TGT TGC TGT AGC CAA AT-3′. Human IL-2 primer sequences were forward; 5′-AAG TTT TAC ATG CCC AAG AAG G-3′, reverse; 5′-AAG TGA AAG TTT TTG CTT TGA GC-3′. Human TNF-α primer sequences were forward; 5′-CAG CCT CTT CTC CTT CCT GAT-3′, reverse; 5′-GCC AGA GGG CTG ATT AGA GA-3′. Human IFN-γ primer sequences were forward; 5′-GGC ATT TTG AAG AAT TGG AAA G-3′, reverse; 5′-TTT GGA TGC TCT GGT CAT CTT-3′. The PCR product size (bp) for TNF-α, IL-2, IFN-γ and GAPDH were 123, 113, 112 and 251 respectively. The amplification product of each primer pair was subjected to melting point analysis and subsequent gel electrophoresis to ensure specificity of amplification.
Pre-clinical non-human primate model T-lymphocytes
Macaques were housed and maintained in accordance with United Kingdom Home Office guidelines for the care and maintenance of non-human primates. Blood from four cynomolgus (Macaca fascicularis) and four rhesus (Macaca mulatta) macaques was obtained under anaesthesia. The following cross-reactive anti-human fluorochrome conjugates were used for whole blood staining; CD4 APC-Cy7 (clone OKT4), CD95 PE (clone DX2), CCR7 FITC (clone 150503) and CD28 PE-Cy7 (clone CD28.2). The extracellular loop of the CD28 molecule is entirely conserved between man and macaques (Bhogal and Combes, 2007), and cross reactivity of CD28.2 for cynomolgus and rhesus macaques is 100% (Sopper et al., 1997; Yoshino et al., 2000). Red blood cells were removed using FACS lysing solution and samples analysed using a FACSCanto II flow cytometer (BD Biosciences).
Statistical analysis
Statistical differences were assessed using the unpaired Student's t-test. Significance was assumed at P < 0.01. Group data are shown as mean ± SEM.
Materials
TGN1412 was obtained from TeGenero AG, Würzburg, Germany; Mabthera from Roche Diagnostics Ltd, Burgess Hill, UK; and Campath-1H from GlaxoWellcome plc now GlaxoSmithKline, Brentford, UK. Tysabri, used as a humanized IgG4κ isotype-matched control for TGN1412, was obtained from Elan Pharmaceuticals, Monksland, Ireland; ANC28.1 was purchased from Axxora UK, Cambridge, UK; Pure CD28.2 from BD Biosciences; and polyclonal chicken anti-human IgG Fc from Abcam plc, Cambridge, UK. PHA, brefeldin A, PMA, ionomycin, tetramethylbenzidine, O-phenylenediamine, RPMI, l-glutamine, penicillin and streptomycin were purchased from Sigma-Aldrich Ltd, Poole, UK. For elisa, anti-human TNF-α antibody pairs were a kind gift from Dr A. Meager (Meager et al., 1987); anti-IFN-γ antibody pairs were purchased from BD Biosciences; anti-IL-2 antibody pairs from R&D Systems, Abingdon, UK; and affinity-purified polyclonal sheep anti-human IL-8 antibodies were produced in-house. Streptavidin–HRP conjugate was purchased from GE Healthcare Life Sciences, Little Chalfont, UK. For flow cytometry, CD4 PE-Cy7, CD4 APC, CD45RO APC, CD95 PE, TNF-α PE, IL-2 PE, IL-2 FITC, IFN-γ PE and FACS lysing solution were purchased from BD Biosciences. IL-2 PerCP Cy5.5 and IFN-γ Pacific Blue were purchased from Cambridge Bioscience, Cambridge, UK; CD28 PE-Cy7 from eBioscience Ltd, Hatfield, UK; and CCR7 FITC from R&D Systems.
The drug/molecular target nomenclature follows Alexander et al. (2009).
Results
TGN1412 stimulates rapid cytokine expression in vitro
To determine if in vitro stimulation with immobilized TGN1412 (Stebbings et al., 2007) induces rapid cytokine expression, with kinetics similar to the ‘cytokine storm’ caused by this mAb in humans, we looked at early responses within the first 6 h following stimulation using intracellular cytokine staining and RT-PCR for cytokine mRNAs. Within 30 min, both immobilized TGN1412 (directly coated onto wells; 3.63% ± 0.90) and Fc-immobilized TGN1412 (captured from aqueous phase by anti-IgG Fc region-specific mAb directly coated onto wells; 3.19% ± 0.29) stimulated significant increases (both P < 0.0001) above those seen with control antibody (1.08% ± 0.10), in the frequency of TNF-α-producing lymphocytes (Figure 1A). After 6 h, the frequency of TNF-α-producing lymphocytes obtained with Fc-immobilized TGN1412 was comparable to PMA + ionomycin stimulation, whereas immobilized TGN1412 and ANC28.1 were less stimulatory (Figure 1A). Both CD28 agonist CD28.2 and control antibody failed to stimulate an increase in the frequency of TNF-α-producing cells above background staining (Figure 1A).
Figure 1.
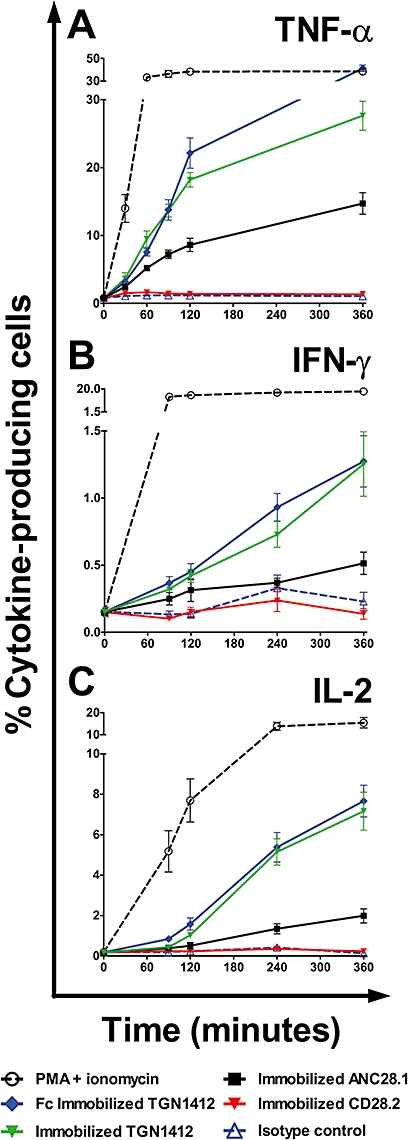
Kinetic analysis of cytokine-producing human PBMCs stimulated in vitro with 1 µg per well Fc-immobilized TGN1412 (IgG4κ), immobilized TGN1412 (IgG4κ), immobilized CD28 superagonist ANC28.1 (murine IgG1), immobilized CD28 agonist CD28.2 (murine IgG1) or immobilized isotype-matched control (IgG4κ). The frequency of cytokine positive cells is expressed as a percentage of total lymphocytes for (A) TNF-α, (B) IFN-γ and (C) IL-2, against time in minutes. Data shown are the mean responses of eight donors obtained from two independent experiments.
Stimulation with Fc-immobilized TGN1412 (0.37% ± 0.05) and immobilized TGN1412 (0.32% ± 0.02) both produced significant increases (above those seen with the control antibody, 0.13% ± 0.03) in the frequency of IFN-γ-producing lymphocytes within 90 min (both P < 0.0001; Figure 1B). After 6 h, Fc-immobilized TGN1412 was no more efficient than immobilized TGN1412 (Figure 1B). However, the frequency of IFN-γ-producing cells stimulated by either immobilized TGN1412 or ANC28.1 was very modest compared with PMA + ionomycin stimulation (Figure 1B). Both CD28 agonist CD28.2 and control antibody failed to stimulate an increase in the frequency of IFN-γ-producing cells above background staining (Figure 1B).
Within 90 min, both immobilized TGN1412 and Fc-immobilized TGN1412 stimulated significant increases (above those seen with the control antibody) in the frequency of IL-2-producing lymphocytes (both P < 0.0001; Figure 1C). After 6 h, the frequency of IL-2-producing cells stimulated by both immobilized and Fc-immobilized TGN1412 was approximately half that achieved with PMA + ionomycin (Figure 1C). The superagonist ANC28.1 did not give a significant increase in the frequency of IL-2-producing cells until 120 min (P = 0.004), and the response at 360 min was smaller than seen with immobilized TGN1412 (Figure 1C). Both control antibody and CD28 agonist CD28.2 failed to stimulate an increase in the frequency of IL-2-producing lymphocytes above background staining (Figure 1C).
Rapid expression of the genes for TNF-α, IL-2 and IFN-γ was confirmed by RT-PCR following in vitro stimulation with immobilized TGN1412 (data not shown). Increased levels of TNF-α gene expression were detected 30 min after stimulation with TGN1412, and increased IL-2 and IFN-γ gene expression at 60 min. The cytokine gene expression that showed the greatest increase following TGN1412 stimulation was TNF-α, followed by IL-2 and then IFN-γ (data not shown).
Substantial IL-2 and IFN-γ release is characteristic of a TGN1412-type response
We compared the in vitro cytokine release caused by TGN1412 with two other therapeutic mAbs that have been associated with adverse reactions in a proportion of patients (i.e. Campath-1H and Mabthera) (Wing et al., 1995; Chung, 2008). Considerable amounts of TNF-α were measured in culture supernatants, by elisa, following 24 h in vitro stimulation with immobilized Mabthera, Campath-1H and TGN1412, but not an isotype-matched control antibody (Figure 2A). Although concentrations of TNF-α stimulated by the three therapeutic mAbs were not markedly different after 24 h, the order of responses was Campath-1H > TGN1412 > Mabthera. TGN1412 showed a time-dependent increase in TNF-α concentration, such that after 72 h the order of responses changed to TGN1412 > Campath-1H > Mabthera (Figure 2A). After 72 h, TNF-α concentrations in response to TGN1412 were relatively close to those elicited by PHA (Figure 2A).
Figure 2.
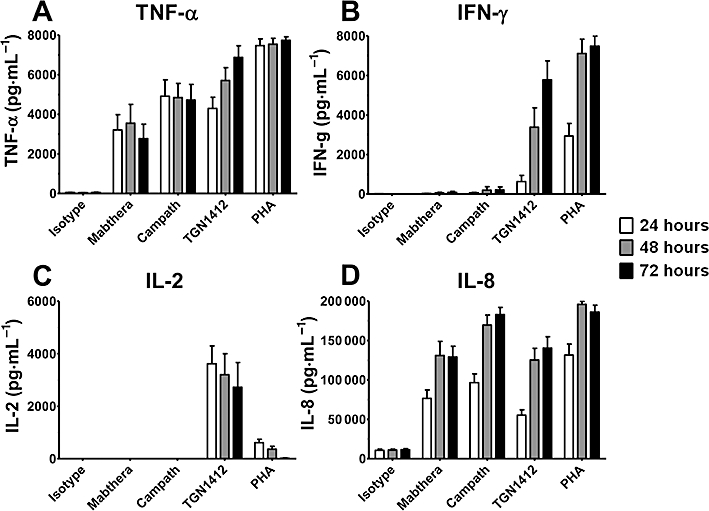
Cytokine release from human PBMCs stimulated in vitro for 24, 48 and 72 h with 1 µg per well immobilized isotype-matched control (IgG4κ), Mabthera (IgG1), Campath-1H (IgG1) and TGN1412 (IgG4κ). The lectin PHA at 10 µg·mL–1 was used as positive control. Cytokine release was measured by elisa of culture supernatants. (A) TNF-α release, (B) IFN-γ release, (C) IL-2 release and (D) IL-8 release are expressed in pg·mL–1. Data shown are mean responses of nine donors obtained from two independent experiments.
Substantial quantities of IFN-γ were detected in culture supernatants following 24 h in vitro stimulation with immobilized TGN1412 and much more after 48 and 72 h (Figure 2B). In contrast, only small amounts of IFN-γ were detected following 24 h in vitro stimulation with control antibody, Mabthera or Campath-1H (Figure 2B). A small increase in IFN-γ concentration was measured following 72 h in vitro stimulation with Mabthera and Campath-1H, but not with control antibody (Figure 2B). After 72 h, IFN-γ concentrations stimulated by TGN1412 were similar to those in response to PHA (Figure 2B).
Substantial concentrations of IL-2 were detected in culture supernatants following 24 h in vitro stimulation with immobilized TGN1412, and these decreased at 48 and 72 h (Figure 2C). In contrast, no IL-2 was detected in culture supernatants following 24, 48 or 72 h in vitro stimulation with Mabthera, Campath-1H or control antibody (Figure 2C). After 72 h, IL-2 concentrations stimulated by TGN1412 were substantially greater than responses to PHA (Figure 2C). Consistent with the release of IL-2, only in vitro stimulation with TGN1412 resulted in a lymphoproliferative response (data not shown), as previously described (Stebbings et al., 2007).
High concentrations of IL-8 were measured in culture supernatants following 24 h in vitro stimulation with immobilized Mabthera, Campath-1H and TGN1412 (Figure 2D). A relatively high background concentration of IL-8 was detected in culture supernatants following 24 h in vitro stimulation with control antibody (Figure 2D), and culture medium alone (data not shown). Following 72 h in vitro stimulation, increases in IL-8 concentration were measured with all three therapeutic antibodies, but not control antibody. The order of responses at 72 h was Campath > TGN1412 > Mabthera (Figure 2D). After 72 h, IL-8 concentrations stimulated by Campath-1H were similar to responses to PHA (Figure 2D).
Cytokine release from CD4+ T-cells is characteristic of a TGN1412-type response
To determine whether or not pro-inflammatory cytokine responses originate from the same cell types following in vitro stimulation with immobilized TGN1412, Mabthera and Campath-1H, we used intracellular cytokine staining and immunophenotyping in conjunction with flow cytometry. Following 6 h of in vitro stimulation with Mabthera, Campath-1H or control antibody, there was no increase in the frequency of IL-2 producing CD4+ or CD4- lymphocytes (no responses above background staining of 0.1%; data from two representative donors; similar data were achieved with 14 further donors; Figure 3A). In contrast, TGN1412 stimulated a large increase in the frequency of IL-2-producing lymphocytes of which the majority were CD4+ (Figure 3A; data from two representative donors) and a small proportion were CD4-, from the same two representative donors (Figure 3A). Similar to TGN1412, PMA + ionomycin stimulated a large increase in the frequency of IL-2-producing lymphocytes, of which the majority were CD4+ and a smaller proportion CD4- (Figure 3A).
Figure 3.
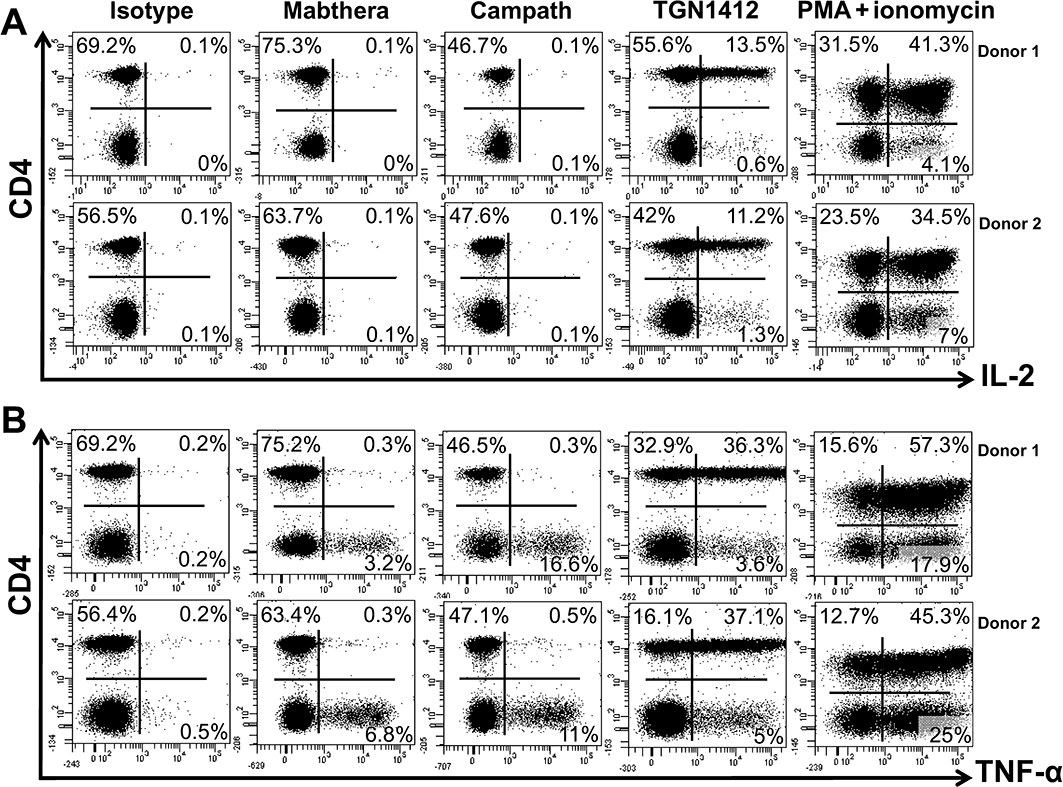
Intracellular cytokine staining of human PBMCs for (A) IL-2 and (B) TNF-α. Prior to staining, PBMCs were stimulated with 1 µg per well immobilized isotype-matched control (IgG4κ), Mabthera (IgG1), Campath-1H (IgG1) or TGN1412 (IgG4κ) for 360 min. PBMCs were counter-stained for surface CD4 expression. The frequency of cytokine positive cells is expressed as a percentage of total lymphocytes. Cells in the right-hand quadrants are positive for cytokine expression. Cells in the upper quadrants are positive for CD4 expression. The mitogens PMA + ionomycin were used as positive control. Data from two representative donors are shown (similar data were obtained with 14 additional donors from three independent experiments).
Following 6 h in vitro stimulation with control antibody, there was no increase above background staining in the frequency of TNF-α-producing lymphocytes within the CD4+ (Figure 3B) or CD4- fractions (Figure 3B). In contrast, stimulation with Mabthera or Campath-1H stimulated increases in the frequency of CD4-, but not CD4+ TNF-α-producing lymphocytes (Figure 3B). Much greater increases in the frequency of TNF-α-producing lymphocytes were seen with Campath-1H compared with Mabthera. TGN1412 stimulated a large increase in the frequency of TNF-α-producing lymphocytes, but the majority of these were CD4+ and a small proportion CD4- (Figure 3B). In comparison, PMA + ionomycin stimulated a large increase in the frequency of CD4+ and CD4- TNF-α-producing lymphocytes (Figure 3B).
TGN1412 stimulates multiple cytokine release from CD4+ effector memory T-cells
Following stimulation with immobilized TGN1412, we identified that human CD4+CD45RO+ memory T-cells, rather than CD4+CD45RO- naive T-cells, were the predominant source of TNF-α, IFN-γ and IL-2 (data from two representative donors; similar data were achieved with six further donors; Figure 4A, B and C respectively). The frequency of TNF-α, IFN-γ and IL-2 producing CD4+ memory T-cells was greater compared with CD4+ naive T-cells (Figure 4A, B and C respectively). Moreover, the overall intensity of intracellular cytokine staining was greater for CD4+ memory T-cells compared to naive T-cells (Figure 4A, B and C). Further subdivision of CD4+ memory T-cells into effector and central memory T-cells, based upon differential CCR7 staining, could only be achieved prior to stimulation as CCR7 expression changes upon activation (Sallusto et al., 1999; 2004; Amyes et al., 2005). We therefore prepared PBMC cultures that were enriched in CD4+ effector memory T-cells and compared them with those that were depleted in CD4+ effector memory T-cells following in vitro stimulation with TGN1412. Human CD4+ effector memory T-cell enrichment produced cells that were on average 88.4% ± 0.64 CD4+CCR7-. Staining for TNF-α, IL-2 and IFN-γ from two representative donors is shown; similar data were obtained with four further donors (Figure 5A,B). Following stimulation with TGN1412, intracellular cytokine staining revealed that few (0.8 and 0.9%) CD4+ T-cells in effector memory-depleted PBMCs produced IFN-γ (Figure 5A) compared with substantial frequencies (14.3 and 16.3%) in effector memory-enriched PBMCs (Figure 5B). The majority of IFN-γ-producing CD4+ T-cells in effector memory-enriched PBMCs also produced IL-2 and TNF-α (Figure 5B). Similarly, the proportion of CD4+ T-cells producing IL-2 in effector memory-enriched PBMCs (55.4 and 49.4%; Figure 5B) was far greater compared with effector memory-depleted PBMCs (both 10.2%; Figure 5A) following stimulation with TGN1412. A less marked difference was seen with the frequency of TNF-α-producing CD4+ T-cells in the effector memory-enriched PBMCs (88.2 and 85.2%; Figure 5B) compared with (49.2 and 64.4%; Figure 5A) in effector memory-depleted PBMCs.
Figure 4.
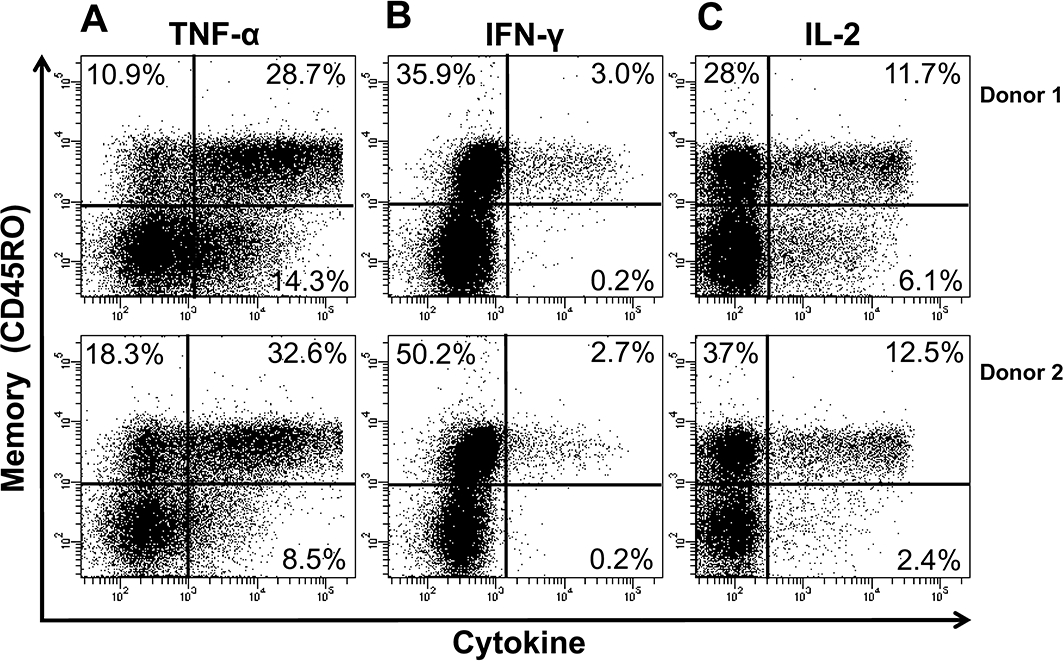
Intracellular cytokine staining of human CD4+ memory T-cells for (A) TNF-α, (B) IFN-γ and (C) IL-2, following stimulation with 1 µg per well immobilized TGN1412. Analysis shown is gated on CD4+ T-cells. Cells in the upper quadrants are CD45RO+ memory T-cells, and cells in the lower quadrants are CD45RO- naive T-cells. Cells in the right-hand quadrants are positive for the indicated cytokines. The frequency of cytokine positive cells is expressed as a percentage of CD4+ T-cells. Data from two representative donors are shown (similar data were obtained with six additional donors from two independent experiments).
Figure 5.
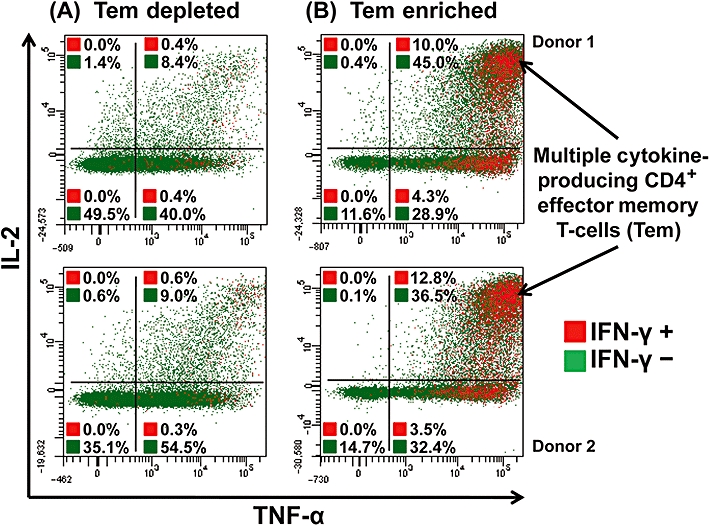
Intracellular cytokine staining of human CD4+ effector memory T-cell (Tem)-depleted (A) and -enriched (B) PBMCs for TNF-α, IL-2 and IFN-γ, following stimulation with 1 µg per well immobilized TGN1412. Tem-depleted (left panels) and Tem-enriched (right panels) PBMCs were surface stained for T-cell markers and intracellular cytokines; analysis shown is gated on CD4+ T-cells. Cells in the right-hand quadrants are positive for TNF-α, cells in the upper quadrants are positive for IL-2, cells shown in red are positive for IFN-γ and cells shown in green are negative for IFN-γ. Cells in the upper right corner in red are positive for TNF-α, IL-2 and IFN-γ. The frequency of cytokine positive cells is expressed as a percentage of CD4+ T-cells. Data from two representative donors are shown (similar data were obtained with four additional donors from two independent experiments).
CD28 is expressed by human, but not macaque, CD4+ effector memory T-cells
Immunophenotyping of human, cynomolgus and rhesus macaque peripheral blood CD4+ T-cell memory subsets was performed using a range of markers (CD95 and CD28 for macaques, CD45RO and CCR7 for humans) as previously described (Sallusto et al., 1999; 2004; Pitcher et al., 2002). Anti-human CD45RO mAbs are not cross-reactive with macaque T-cells, so CD95 expression was used to distinguish CD4+ memory from naive T-cells in macaques (data not shown). For comparative analysis of human and macaque effector memory subsets, the markers CCR7 and CD28 were utilized. Dot plots from two representative individuals from each species are shown (Figure 6A). All human CD4+ T-cell memory subsets (naive, central and effector) expressed high levels of CD28 with the effector memory subset defined by a lack of CCR7 expression (Sallusto et al., 1999; 2004; Figure 6A). Similarly, the naive and central memory CD4+ T-cells of both cynomolgus and rhesus macaques expressed CD28 (Figure 6A). However, we found that the CD4+ effector memory T-cell subsets of both cynomolgus and rhesus macaques did not express CD28, as previously reported for rhesus macaques (Pitcher et al., 2002; Moniuszko et al., 2004; Figure 6A). As for human cells, we found that both cynomolgus and rhesus macaque CD4+ effector memory T-cells lack CCR7 expression; however, a population of CD28+CCR7- were noted that have previously been described as a transitional central memory subset in rhesus macaques (Picker et al., 2006). When stimulated with a CD28 independent agent such as PMA and ionomycin for 4 h, both cynomolgus and rhesus macaque CD4+CD28–CCR7- effector memory T-cells produced pro-inflammatory cytokines, such as IFN-γ, demonstrating their functional ability (Figure 6B).
Figure 6.
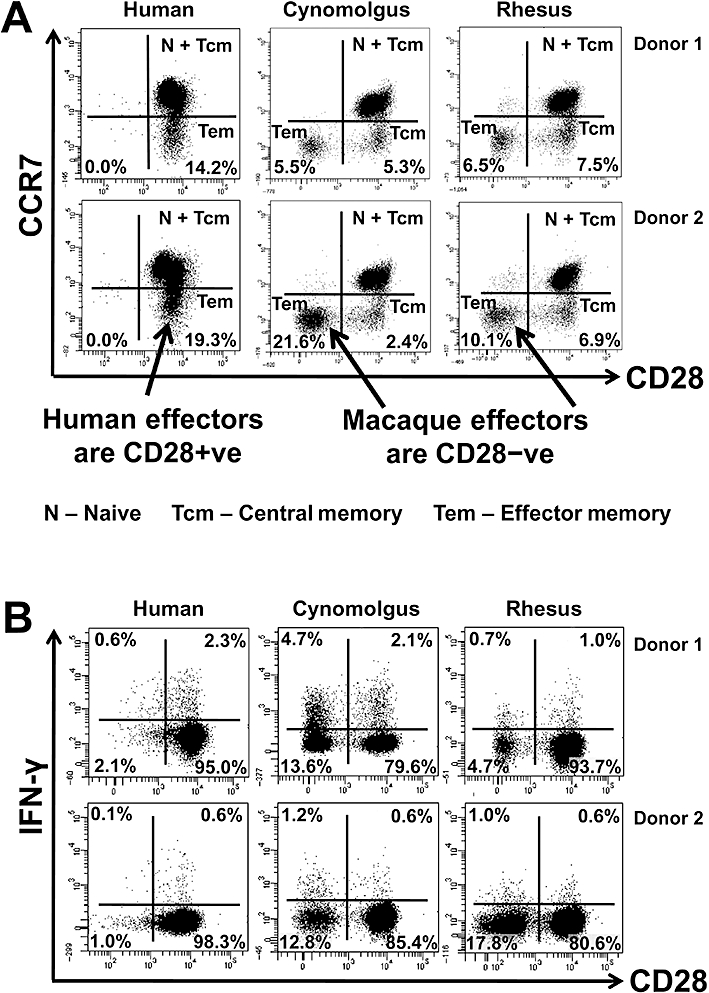
Comparative immunophenotyping of human, cynomolgus and rhesus macaque CD4+ T-cells. All analysis shown is gated on CD4+ T-cells. (A) Analysis of memory markers using CD28 and CCR7 staining. Human, cynomolgus and rhesus macaque naive CD4+ T-cells (CCR7+CD28+) and central memory T-cells (CCR7+CD28+), macaque transitional memory T-cells (CCR7-CD28+) and human effector memory CD4+ T-cells (CCR7-CD28+) all express CD28. In contrast, both cynomolgus and rhesus effector memory T-cells (CCR7-CD28-) do not express CD28. Data from two representative donors are shown (similar data were obtained with two additional rhesus, two cynomolgus and six human donors from two independent experiments). (B) Intracellular IFN-γ staining versus CD28 expression on CD4+ T-cells following PMA and ionomycin stimulation for 4 h. Human CD4+ T-cells constitutively express CD28, so IFN-γ staining stems only from the CD28+ population. Macaque CD4+ T-cells can be subdivided into CD28+ (naive, central and transitional memory T-cell) and CD28- (effector memory T-cell) populations, both of which produce IFN-γ following PMA and ionomycin stimulation for 4 h. A greater fraction of macaque CD4+CD28- T-cells produce IFN-γ compared to CD4+CD28+ T-cells following mitogen stimulation (similar data were obtained with two additional rhesus, two cynomolgus and two human donors).
Discussion
Following the TGN1412 incident, we developed new pre-clinical in vitro procedures using human PBMCs which can be applied to therapeutic mAbs during development, in order to prevent a repeat of the TGN1412 incident (Stebbings et al., 2007; Findlay et al., 2010). However, at the time, we were unaware of whether or not the cytokine release syndrome caused by TGN1412 was stimulated by the same mechanism(s) as the adverse effects caused by other therapeutic mAbs in a proportion of recipients. We also did not understand why TGN1412 bound to human and macaque CD28 with equal affinity (TeGenero, 2005), yet caused a ‘cytokine storm’ only in humans. This difference not only questioned the suitability and validity of existing pre-clinical non-human primate models for the safety testing of therapeutic mAbs, but also for other immunomodulatory biological agents.
When we investigated other therapeutic mAbs, known to be capable of causing adverse reactions, Mabthera and Campath-1H (Wing et al., 1995; Chung, 2008), using our new in vitro procedures, we did not detect lymphoproliferative responses or IL-2 release, the cardinal features of TGN1412 stimulation (Stebbings et al., 2007). Thus, the ‘cytokine storm’ caused by TGN1412 appeared to be mediated by a mechanism of action different from that responsible for the adverse reactions seen with other therapeutic mAbs. Interestingly, very low in vitro levels of IFN-γ were detected with both Mabthera and Campath-1H, compared with TGN1412, which may prove a good correlate for the likelihood and severity of adverse reactions. In contrast, TNF-α and IL-8 responses were less discriminatory as substantial release was seen with TGN1412, Mabthera and Campath-1H. The significance of substantial IL-8 and TNF-α release with Mabthera and Campath-1H in these assay systems is unclear because cross-linking of Campath-1H-opsonized CD4+ T-cells failed to stimulate TNF-α release (Wing et al., 1996). Such release could be caused by an Fc region-mediated mechanism such as ligation of CD16 on natural killer (NK) cells in the case of Campath-1H (Wing et al., 1996), as orientation of immobilized mAb in wells is random. However, it is unlikely that TGN1412-mediated IL-8 and TNF-α release in vitro was predominately an Fc region-mediated mechanism as no responses were seen with the IgG4κ isotype-matched control.
TGN1412-mediated cytokine release predominantly originated from CD4+ memory T-cells, which produced high levels of TNF-α, IL-2 and IFN-γ. In contrast, other therapeutic mAbs that we tested only stimulated CD4- lymphocyte cytokine release that comprised high levels of TNF-α, but very low levels of IFN-γ and no IL-2. Thus, it can be concluded that the cytokine release stimulated by TGN1412 was mediated via a mechanism distinct from that underlying cytokine release stimulated by the other two therapeutic mAbs tested here, because different cell types were involved. We also noted that a small proportion of TGN1412-mediated cytokine release originated from CD4- lymphocytes, but at present it is unclear whether this occurs via the same mechanism underlying the cytokine release mediated by other therapeutic mAbs or not. Within blood, multiple cell types and their subsets can release different cytokines with overlapping repertoires which are not discriminated by elisa assessment of culture supernatants from mixed cell populations stimulated with mAbs, whereas phenotypic analysis of cytokine-producing cells using flow cytometry, as is shown here, has allowed the dissection of different effector mechanisms. The likely candidates for cytokine release from the CD4- leucocytes triggered by different mAbs were CD8+ T-cells, NK cells, neutrophils and B-cells. Overall, these findings demonstrate that the in vitro safety procedures described by Stebbings et al. (2007) are applicable to other therapeutic mAbs, if IL-2 and IFN-γ release are used as biomarkers for a TGN1412-type adverse response.
One of the most striking features of the TGN1412-mediated ‘cytokine storm’ observed in humans was substantial TNF-α release within the first hour of infusion followed by dramatic increases in IFN-γ, IL-2, IL-6, IL-8 and IL-10 plasma concentrations within 4 h (Suntharalingam et al., 2006). The rapid nature of this response suggested direct stimulation of pro-inflammatory cytokine-producing cells, rather than a cascade of cytokine release (with each cytokine produced stimulating the release of another cytokine or cytokines). Similarly, within the first hour of in vitro TGN1412 stimulation, we observed a rapid induction of TNF-α mRNA and protein expression, consistent with the rapid release of this cytokine in the trial volunteers. Concentrations of TNF-α measured by elisa in culture supernatants collected 24 h after in vitro TGN1412 stimulation were also consistent with the peak concentrations reported in trial volunteers, with the considerable caveat that a comparison is being made between an in vitro system and an in vivo response. However, despite similar in vitro expression kinetics for IFN-γ following TGN1412 stimulation, the concentrations of IFN-γ were far lower than TNF-α responses in vitro, and an order of magnitude lower than concentrations of IFN-γ predicted from in vivo responses in trial participants (Suntharalingam et al., 2006). Only after prolonged in vitro stimulations for 48–72 h could high concentrations of IFN-γ, equivalent to levels measured in the plasma of trial volunteers at 4 h, be achieved. This delayed release of high levels of IFN-γ was presumably due to a requirement for expansion of low initial numbers of IFN-γ producing T-cells through differentiation of stimulated T-cells and/or cell proliferation. Similarly, the expression of IL-2 following in vitro TGN1412 stimulation showed similar kinetics to its appearance in humans, but the concentrations in cell supernatants were lower than expected. These apparently disparate results did not seem to support our hypothesis that in vitro TGN1412 stimulation is a model of in vivo responses seen in man. Moreover, it appeared that in vitro TGN1412 stimulation greatly underestimated some in vivo cytokine responses compared with those reported in humans. If the explanation were as simple as differences in cell numbers, then all cytokine responses would have been equally lower, but they were not.
To understand why in vitro levels of IFN-γ in response to TGN1412 stimulation were at least an order of magnitude lower than plasma concentrations reported in humans, we immunophenotyped the cytokine-producing cells. Initial data showed that human CD4+CD45RO+ memory T-cells were the predominant source of cytokines following TGN1412 stimulation, but this did not explain why in vitro IFN-γ levels were low when compared with TNF-α levels. However, when we subdivided CD4+ memory T-cells into CCR7+ central memory and CCR7- effector memory cells, we were able to identify a difference in TGN1412-stimulated cytokine release that could account for low in vitro IFN-γ release. PBMCs depleted of CD4+ effector memory T-cells and stimulated with TGN1412 produced mainly TNF-α, some IL-2 and very little IFN-γ (i.e. responses very similar to the responses of whole PBMCs). In marked contrast, PBMCs enriched for CD4+ effector memory T-cells produced high levels of TNF-α, IL-2 and IFN-γ, when stimulated with TGN1412, with a large proportion of cells simultaneously producing all three cytokines. Differences in the proportions of CD4+ effector memory T-cells producing TNF-α, IL-2 and IFN-γ combinations following TGN1412 stimulation may reflect the heterogeneous nature of this population which contains multiple functional subsets with different cytokine repertoires (e.g. Th1, Th2, Th9, Th17 and Th22) (Annunziato and Romagnani, 2009; Sallusto and Lanzavecchia, 2009). PBMCs would also become enriched in effector memory T-cells following prolonged stimulation as activated naive and central memory T-cells matured into effector memory T-cells gaining their expanded cytokine-secreting potential, including IFN-γ. This would explain why in vitro stimulation with TGN1412 for 24 h initially produced only low levels of IFN-γ compared to very high levels at 72 h.
This finding suggests that the ‘cytokine storm’ observed in trial participants likely involved TGN1412 stimulation of effector memory T-cells in peripheral tissues such as the lungs and gut, resulting in secretion of high levels of cytokines into the blood. This notion is also consistent with the known capability of CD4+ T-cell subsets to produce cytokines (i.e. naive and central memory CD4+ T-cell subsets both have limited potential for cytokine expression, whereas the effector memory subset has a considerable capability for cytokine production, with IL-2 being a major component of this) (Sallusto et al., 1999; Amyes et al., 2005). Importantly, IFN-γ expression is largely limited to the CD4+ effector memory T-cells (Sallusto et al., 1999; Amyes et al., 2005). Because CD4+ effector memory T-cells are only a minor subset of peripheral blood lymphocytes compared with CD4+ naive and central memory T-cells, their low numbers in peripheral blood could have explained why in vitro IFN-γ responses were low compared with plasma concentrations observed in trial participants. Conversely, central and effector memory T-cells show a different pattern of distribution within the tissues compared with peripheral blood (Campbell et al., 2001; Picker, 2006). The lymph nodes and tonsils are enriched in central memory CD4+ T-cells, whereas the lungs and gastrointestinal mucosa contain mainly effector memory CD4+ T-cells (Campbell et al., 2001; Picker, 2006). Moreover, peripheral blood contains just 2–5% of all lymphocytes, while the lungs and gastrointestinal mucosa harbour up to 90% of the body's lymphocytes (Cerf-Bensussan and Guy-Grand, 1991; Smit-McBride et al., 1998).
If infusion of TGN1412 caused high concentrations of cytokines to be rapidly released into the blood by the large numbers of CD4+ effector memory T-cells within the tissues, then this would explain why in vitro procedures using peripheral blood containing few of these cells greatly underestimated some cytokine responses such as IFN-γ in comparison with responses observed in vivo. However, it should be noted that immobilizing mAb to a plastic surface is a highly artificial means of stimulation that is unlikely to simulate what is happening inside the body. Nevertheless, this is the only published method currently available to induce substantial cytokine release with TGN1412, and the conclusion that it is the CD4+ effector memory T-cells which make the pro-inflammatory cytokines in response to TGN1412 stimulation, but not in the case of other therapeutic mAbs studied, is likely to be correct.
Identification of the central role of CD4+ effector memory T-cells in TGN1412-mediated cytokine release provides an explanation for the failure of pre-clinical testing using non-human primate models to predict human responses. This is because we and others (Pitcher et al., 2002; Moniuszko et al., 2004) have shown that cynomolgus and rhesus macaque CD4+ effector memory T-cells do not express CD28; therefore, they cannot be stimulated by CD28 superagonists including TGN1412. In contrast, the CD4+ effector memory T-cells of humans express high levels of CD28 on their surface (Sallusto et al., 1999; 2004;), so therefore can be stimulated by TGN1412. It should also be noted that this species difference was only identified through careful dissection of the cellular components of the immune system to look at the individual subsets stimulated by TGN1412. The evolutionary reason for this subtle comparative immunological difference in receptor expression is unclear, but retention of CD28 expression on human CD4+ effector T-cells would allow the required co-stimulatory signals to reduce activation thresholds (Fraser et al., 1991). Effector memory CD4+ T-cells lacking CD28 expression have been reported in humans, but only in association with autoimmune diseases such as rheumatoid arthritis, Crohn's disease or multiple sclerosis (Markovic-Plese et al., 2001; Fasth et al., 2004; Garcia de Tena et al., 2004), and were not observed with the healthy donors used in this study. Similar to human CD4+ effector memory T-cells, macaque CD4+ transitional memory T-cells lack CCR7 expression and express CD28 (Picker et al., 2006). However, macaque CD4+ transitional memory T-cells are functionally different to macaque CD4+ effector memory T-cells; IL-2 preferentially induces macaque transitional memory T-cells to proliferate, whereas macaque effector memory T-cells are poorly responsive and vice versa for IL-15 (Picker et al., 2006). Both macaque and human CD4+ central memory T-cells and macaque CD4+ transitional memory T-cells express CD28, so why did TGN1412 not stimulate these subsets in macaques as well as in humans? Central memory CD4+ T-cells are characterized by strong lymphoproliferative responses (Sallusto et al., 1999; 2004;), which TGN1412 stimulates from human, but not macaque PBMCs (Stebbings et al., 2007). However, we previously reported that immobilized TGN1412 primed macaque CD4+ T-cells and that they underwent marked lymphoproliferation only when exogenous IL-2 was added (Stebbings et al., 2007). Because macaque T-cells do not secrete IL-2 when stimulated with TGN1412 (Stebbings et al., 2007), it could be speculated that production of IL-2 by stimulated CD4+ effector memory T-cells is a missing factor required for in vitro lymphoproliferative responses by macaque CD4+ memory T-cells. Alternatively, it has been suggested that differences in human- and macaque TGN1412-driven responses are due to the expression of inhibitory Siglecs on non-human primate T-cells, which have been lost during human evolution, rendering them less responsive to in vitro stimulation (Nguyen et al., 2006; Waibler et al., 2008). However, it should be noted that macaque T-cells are not ‘less responsive’ to in vitro stimulation with TGN1412, but ‘non-responsive’.
The commercially available antibody ANC28.1 was used as a comparative antibody because it, like TGN1412, is a superagonist anti-human CD28 antibody. However, ANC28.1 is a superagonist in vitro in both aqueous phase and when immobilized, whereas we found that TGN1412 was only a superagonist when immobilized. Therefore, it is likely that ANC28.1 binds to a different region of CD28 from that binding TGN1412, which may explain why lower responses were observed. Moreover, it has been previously published that ANC28.1 induces greater or similar in vitro cytokine release than TGN1412 (Waibler et al., 2008), which is in contrast to our findings here that TGN1412 is far more potent than ANC28.1. Nevertheless, ANC28.1 does stimulate release of the same pattern of cytokines as TGN1412 so is likely to activate the same pro-inflammatory pathways.
The new in vitro procedures described here show that TGN1412-mediated cytokine release evokes a unique mechanism that can be discriminated from the cytokine release observed with the other therapeutic mAbs tested. While much of the immunopharmacology of cytokine release syndrome in humans remains largely unknown, pre-clinical safety testing in non-human primate models will still be useful, but needs to be interpreted in conjunction with subtleties in antigen expression on target cells in pre-clinical models. We now offer an explanation for why pre-clinical testing in non-human primate models failed to predict a ‘cytokine storm’ in humans that is based upon a species difference in CD28 expression on CD4+ effector memory T-cells and that is specific for TGN1412 and other CD28-specific therapeutic mAbs. The likelihood that another mAb or immunomodulatory biotherapeutic agent could stimulate a TGN1412-like adverse response can now be linked to the ability to activate CD4+ effector memory T-cells.
Acknowledgments
We thank Prof Sir Gordon Duff for helpful discussions during the preparation of this paper.
Glossary
Abbreviations
- CCR7
chemokine receptor 7
- IFN-γ
interferon gamma
- IL-2
interleukin-2
- IL-8
interleukin-8
- mAb
monoclonal antibody
- NK
natural killer
- PBMC
peripheral blood mononuclear cell
- TNF-α
tumour necrosis factor alpha
Conflict of interest
The authors have no conflicts of interests.
Supporting Information
Supporting Information: Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes E, McMichael AJ, Callan MF. Human CD4+ T cells are predominantly distributed among six phenotypically and functionally distinct subsets. J Immunol. 2005;175:5765–5773. doi: 10.4049/jimmunol.175.9.5765. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Romagnani S. Heterogeneity of human effector CD4+ T cells. Arthritis Res Ther. 2009;11:1–8. doi: 10.1186/ar2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyersdorf N, Gaupp S, Balbach K, Schmidt J, Toyka KV, Lin CH, et al. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med. 2005;202:445–455. doi: 10.1084/jem.20051060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyersdorf N, Hanke T, Kerkau T, Hünig T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmun Rev. 2006;5:40–45. doi: 10.1016/j.autrev.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Bhogal N, Combes R. Immunostimulatory antibodies: challenging the drug testing paradigm. Toxicol In Vitro. 2007;21:1227–1232. doi: 10.1016/j.tiv.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Burns CJ, Silva MCG, Gray E, Robinson CJ. Quantitative RT-PCR as an alternative to late-stage bioassays for vascular endothelial growth factor. J Pharm Biomed Anal. 2008;47:460–468. doi: 10.1016/j.jpba.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Campbell JJ, Murphy KE, Kunkel EJ, Brightling CE, Soler D, Shen Z, et al. CCR7 expression and memory T cell diversity in humans. J Immunol. 2001;166:877–884. doi: 10.4049/jimmunol.166.2.877. [DOI] [PubMed] [Google Scholar]
- Cerf-Bensussan N, Guy-Grand D. Intestinal intraepithelial lymphocytes. Gastroenterol Clin North Am. 1991;20:549–576. [PubMed] [Google Scholar]
- Chung CH. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist. 2008;13:725–732. doi: 10.1634/theoncologist.2008-0012. [DOI] [PubMed] [Google Scholar]
- Duff G. Expert group on phase one clinical trials: final report. 2006. Available at: http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_063117 (accessed 8 July 2010)
- Elflein K, Rodriguez-Palmero M, Kerkau T, Hünig T. Rapid recovery from T lymphopenia by CD28 superagonist therapy. Blood. 2003;102:1764–1770. doi: 10.1182/blood-2002-11-3586. [DOI] [PubMed] [Google Scholar]
- Fasth AE, Cao D, van Vollenhoven R, Trollmo C, Malmström V. CD28nullCD4+ T cells: characterization of an effector memory T-cell population in patients with rheumatoid arthritis. Scand J Immunol. 2004;60:199–208. doi: 10.1111/j.0300-9475.2004.01464.x. [DOI] [PubMed] [Google Scholar]
- Findlay L, Eastwood D, Stebbings R, Sharp G, Mistry Y, Ball C, et al. Improved in vitro methods to predict the in vivo toxicity in man of therapeutic monoclonal antibodies including TGN1412. J Immunol Methods. 2010;352:1–12. doi: 10.1016/j.jim.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- Garcia de Tena J, Manzano L, Leal JC, San Antonio E, Sualdea V, Alvarez-Mon M. Active Crohn's disease patients show a distinctive expansion of circulating memory CD4+CD45RO+CD28null T cells. J Clin Immunol. 2004;24:185–196. doi: 10.1023/B:JOCI.0000019784.20191.7f. [DOI] [PubMed] [Google Scholar]
- Karlsson I, Malleret B, Brochard P, Delache B, Calvo J, Le Grand R, et al. Dynamics of T-cell responses and memory T cells during primary simian immunodeficiency virus infection in cynomolgus macaques. J Virol. 2007;81:13456–13468. doi: 10.1128/JVI.01619-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Hünig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol. 2003;33:626–638. doi: 10.1002/eji.200323570. [DOI] [PubMed] [Google Scholar]
- Lühder F, Huang Y, Dennehy KM, Guntermann C, Müller I, Winkler E, et al. Topological requirements and signalling properties of T cell-activating, anti-CD28 antibody superagonists. J Exp Med. 2003;197:955–966. doi: 10.1084/jem.20021024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest. 2001;108:1185–1194. doi: 10.1172/JCI12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meager A, Parti S, Leung H, Peil E, Mahon B. Preparation and characterisation of monoclonal antibodies directed against antigenic determinants of recombinant human tumour necrosis factor (TNF) Hybridoma. 1987;6:305–311. doi: 10.1089/hyb.1987.6.305. [DOI] [PubMed] [Google Scholar]
- Moniuszko M, Fry T, Tsai WP, Morre M, Assouline B, Cortez P, et al. Recombinant interleukin-7 induces proliferation of naive macaque CD4+ and CD8+ T cells in vivo. J Virol. 2004;78:9740–9749. doi: 10.1128/JVI.78.18.9740-9749.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec expression on T lymphocytes during human evolution. Proc Natl Acad Sci U S A. 2006;103:7765–7770. doi: 10.1073/pnas.0510484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picker LJ. Immunopathogenesis of acute AIDS virus infection. Curr Opin Immunol. 2006;18:399–405. doi: 10.1016/j.coi.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Picker LJ, Reed-Inderbitzin EF, Hagen SI, Edgar JB, Hansen SG, Legasse A, et al. IL-15 induces CD4+ effector memory T cell production and tissue emigration in nonhuman primates. J Clin Invest. 2006;116:1514–1524. doi: 10.1172/JCI27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, et al. Development and homeostatis of T cell memory in rhesus macaque. J Immunol. 2002;168:219–243. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Palmero M, Hara T, Thumbs A, Hünig T. Triggering of T cell proliferation through CD28 induces GATA-3 and promotes T helper type 2 differentiation in vitro and in vivo. Eur J Immunol. 1999;29:3914–3392. doi: 10.1002/(SICI)1521-4141(199912)29:12<3914::AID-IMMU3914>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur J Immunol. 2009;39:2076–2082. doi: 10.1002/eji.200939722. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- Smit-McBride Z, Mattapallil JJ, McChesney M, Ferrick D, Dandekar S. Gastrointestinal T lymphocytes retain high potential for cytokine responses but have severe CD4 (+) T-cell depletion at all stages of simian immunodeficiency virus infection compared to peripheral lymphocytes. J Virol. 1998;72:6646–6656. doi: 10.1128/jvi.72.8.6646-6656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopper S, Stahl-Hennig C, Demuth M, Johnston IC, Dörries R, ter Meulen V. Lymphocyte subsets and expression of differentiation markers in blood and lymphoid organs of rhesus monkeys. Cytometry. 1997;29:351–362. [PubMed] [Google Scholar]
- Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, et al. ‘Cytokine storm’ in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve pre-clinical testing of immunotherapeutics. J Immunol. 2007;179:3325–3331. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- Stebbings R, Poole S, Thorpe R. Safety of biologics, lessons learnt from TGN1412. Curr Opin Biotechnol. 2009;20:673–677. doi: 10.1016/j.copbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- Tacke M, Hanke G, Hanke T, Hünig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur J Immunol. 1997;27:239–247. doi: 10.1002/eji.1830270136. [DOI] [PubMed] [Google Scholar]
- TeGenero AG. TGN1412 investigator's brochure. 2005. Available at: http://www.circare.org/foia5/tgn1412investigatorbrochure.pdf (accessed 8 July 2010)
- Waibler Z, Sender LY, Merten C, Hartig R, Kliche S, Gunzer M, et al. Signaling signatures and functional properties of anti-human CD28 superagonistic antibodies. Plos One. 2008;3:e1708. doi: 10.1371/journal.pone.0001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing MG, Waldmann H, Isaacs J, Compston DA, Hale G. Ex-vivo whole blood cultures for predicting cytokine-release syndrome: dependence on target antigen and antibody isotype. Ther Immunol. 1995;2:183–190. [PubMed] [Google Scholar]
- Wing MG, Moreau T, Greenwood J, Smith RM, Hale G, Isaacs J, et al. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest. 1996;98:2819–2826. doi: 10.1172/JCI119110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino N, Ami Y, Terao K, Tashiro F, Honda M. Upgrading of flow cytometric analysis for absolute counts, cytokines and other antigenic molecules of cynomolgus monkeys (Macaca fascicularis) by using anti-human cross-reactive antibodies. Exp Anim. 2000;49:97–110. doi: 10.1538/expanim.49.97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.